
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Artemisinin (ART) is a natural anti-malarial sesquiterpene lactone with anticancer properties, but its application is limited because of its low water solubility. To increase the bioavailability and water solubility of ART, we synthesized three series of poly (ɛ-caprolactone)–poly (ethylene glycol)–poly (ɛ-caprolactone) (PCL–PEG–PCL) tri-block copolymers. The structure of the copolymers was characterized by HNMR, FTIR, DSC and GPC techniques. ART was encapsulated inside micelles by a nanoprecipitation method which leading to the formation of ART/PCL–PEG–PCL micelles. The obtained micelles were characterized by DLS and AFM technique. The results showed that the average size of micelles was about 83.22 nm. ART was encapsulated into PCL–PEG–PCL micelles with encapsulation efficacy of 89.23 ± 1.41%. In vivo results demonstrated that this formulation significantly increased drug accumulation in tumours. Pharmacokinetic study in rats revealed that in vivo drug exposure of ART was significantly increased and prolonged by intravenously administering ART-loaded micelles when compared with the same dose of free ART. The MTT assay showed that bare PCL–PEG–PCL micelles is non-toxic to MCF7 and 4T1 cancer cell lines whereas the ART/PCL–PEG–PCL micelles showed a specific toxicity to both cancer cell lines. Therefore, the polymeric micellar formulation of ART based copolymer could provide a desirable process for ART delivery.
Introduction
Cancer is the most important reason of death in industrial countries and the second-leading death reason in developing countries [Citation1]. Plant natural products have a major role in the health care in many countries [Citation2–4]. An effective anticancer drug must have selective toxicity on cancer cells without significant toxicity on normal cells [Citation5,Citation6]. Natural products with anticancer activity and partial toxicity are receiving more notification in medical studies. Artemisinin (ART) is a natural anti-malarial sesquiterpene lactone with anticancer properties. ART and its derivatives which commonly used in malaria therapy have also potent anticancer activity. Recently, many researches have studied on the mechanisms underlying the action and response to artemisinin-like drugs [Citation7–10]. ART has 1, 2, 4-tiroxanering system which usually extracted from the Artemisia annua [Citation11,Citation12]. Despite ART efficacy, the prototype drugs, artemisinin, has pharmacokinetic limitations [Citation13]. Obviously, ART has low solubility in water and oil, poor bioavailability and a short half-life in vivo (∼2.5 h) [Citation14]. It has been suggested that ART may be a therapeutic alternative in aggressive cancers with fast dissemination without developing drug resistance [Citation15–17]. They also show synergism with other antitumor drugs with no increased toxicity to normal cells [Citation18]. It revealed that semisynthetic ART derivatives have much higher antitumor activity than their monomeric complements through apoptosis, arrest of cell cycle and oxidative stress [Citation18,Citation19]. The molecular basis of these anticancer effects and precise activation approach are not fully described. ART can regulate key factors such as nuclear factor-kappa B, surviving, NOXA, hypoxia-inducible factor-1α and BMI-1, involving multiple pathways that may affect drug resistance, drug reaction, drug interfaces and associated factors upon normal cells. For overthrow the poor water solubility of lipophilic drugs, nanotechnology has many advantages. Hydrophobic drugs encapsulation into nanoparticles is an expectant improvement to formulate the drug intravenously injectable [Citation20,Citation21]. The amphiphilic nanoparticles are great nominees for loading the hydrophobic drugs [Citation22]. PCL–PEG–PCL micelles are a tri-block PCL–PEG–PCL copolymer were self-assembled into nanoparticles with core–shell structure: a hydrophobic PCL core and a hydrophilic PEG shell [Citation23–25]. An addition, PCL–PEG–PCL copolymers, setup with hydrophobic drugs, possibly will increase systemic distribution and fidelity of drugs and it can release drugs in a sustained time for the finest range of drug concentration [Citation26–30]. Moreover, PCL–PEG–PCL copolymers are biocompatible, recyclable and simple to construct which showing encouraged uses in drug delivery systems [Citation31]. In this study, to support an aqueous formulation for ART, we organized ART encapsulated PCL–PEG–PCL micelles. We expected that ART/PCL–PEG–PCL micelles is able to slowly release ART and restrain the growth of cancer cells in vitro and in vivo.
Materials and methods
Materials
Dimethylthiazol-2-yl)-3,5-diphenylformazan, Thiazolyl blue formazan (Aldrich, St. Louis, MO, CAS. 57360–69-7), PEG (Mn = 6000 Da) (Aldrich, St. Louis, MO, CAS.81323), 2-ethyl-hexanoate [Sn(Oct)2] (Aldrich, St. Louis, MO, CAS. 301100), ɛ-caprolactone (98%purity) (Acros, New Jersey, CAS.502443), ART (Merck, Darmstadt, Germany, Art No. 820354) (other chemicals and solvent were from chemical lab purity grades, purchased from Emertatchimi, Iran).
Synthesis of PCL–PEG–PCL copolymers
PCL–PEG–PCL tri-block copolymers were prepared according to our previous report. Tri-block copolymers were synthesized by a ring opening polymerization of ɛ-caprolactone with PEG as an initial molecule and Sn(Oct)2 as the catalyst [Citation8,Citation27]. In brief, ɛ-caprolactone (0.5, 1, 2 g), PEG (1 g) and Sn (Oct)2 (0.01 mmol) were heated at 120 °C to begin polymerization. After 12 h, the resulting copolymers were cooled to room temperature, dissolved in chloroform and precipitated in cold diethyl ether. The copolymer was dried up under vacuum at room temperature for 24 h.
Characterization of PCL–PEG–PCL copolymers
The chemical structure of copolymers was known by Fourier transform infrared spectroscopy (FT-IR) (Bruker, Tensor 27) and proton nuclear magnetic resonance spectroscopy (1H NMR) in CDCl3 at 400 MHz (Bruker, Avance 400). Differential scanning calorimetry (DSC) (Mettler-Toledo, model Star SW 9.30) was applied for thermal analysis of the synthesized copolymers. Samples were heated at a rate of 10 °C min−1 and the records were recorded from 0 to 200 °C. The average molecular weight and distribution of the PCL–PEG–PCL copolymers were determined by gel permeation chromatography (GPC) (Knauer, Berlin, Germany) set with differential refractometer detector and an ultra-style gel column (4.6 × 30 mm) (Waters, Milford, MA, model HR 4E). The mobile phase was tetrahydrofuran (THF) with a flow-rate of 1 ml/min and the injection volume was 50 μl of stock solutions (0.1–0.5 w/v %). Polymers were characterized by relative elution time to polystyrene mono-disperse standards in the range of 1500–35,500 Da (Varian Palo Alto, CA) using the calibration curve obtained previously to measurements.
Preparation of ART-loaded micelles
ART-loaded micelles were prepared according to our previous report [Citation31]. Nano precipitation method was used for the preparation of micelles. The acetone was used as the solvent to this method. Briefly, PCL–PEG–PCL copolymer (20 mg), and ART (1, 5, 10 and 20 mg) were dissolved in 2 ml of acetone. The solution was injected drop wise during a syringe (G = 22) into 25 ml of distilled water under certain combination rates and stirred magnetically at room temperature a position the whole vanishing of the organic solvent which caused the amphiphilic copolymers to self-assemble to form the micelles. After eliminating of acetone using rotary vacuum evaporation at 35 °C, the resultant aqueous solution was filtered through a 0.45 μm filter membrane to remove the unloaded ART. The resulting molecules by centrifuging at 20,000 g for 20 min were separated and freeze-dried under a pressure of 14 Pa at −78 °C until two separate all the residual solvents and to harvest the final dried form of ART loaded micelles.
Characterization of the micelles
Particle morphology
The morphology of micelles was assigned using atomic force microscopy (AFM) (JPK, Berlin, Germany, model Nano Wizard). For AFM sample preparation, micelles were diluted with water and a droplet of 2 μl was placed onto a freshly cleaved mica substrate (1 cm2) and air-dried. AFM measurements were done in sporadic contact mode, and morphologies of the NPs were observed using scanning electron microscopy (SEM) (Hitachi S-4800 II, Japan).
Determination of particle size
The particle size distribution of the organized micelles was assigned by DLS using a nano/zeta sizer (Malvern Instruments, Worcestershire, UK, model Nano ZS).
Stability of micelles
For physical stability assessment of micelles, the micelles particle size distribution was examined in phosphate-buffered saline (PBS) and preserved in room temperature in 0, 1, 2, 3, 4 and 6 month after instruction using the method defined in the above section.
Determination of loading efficiency
Two parameters including the drug loading and encapsulation efficiency were determined for determination the drug loading of the drugs in the micelles. Drug loading (DL) was determined as:
(1)
(1)
DL% is the drug loading, Wdrug and Wmicelles represent the weight of loaded drug and l weight of polymersomes, respectively. The drug loading of ART was determinate by high performance liquid chromatography (HPLC) [Citation32,Citation33]. The mobile phase composed of methanol and 5% (w/v) acetic acid at the volume ratio of 70:30. It and was delivered at a flow rate of 1.0 ml/min using a double-reciprocating pump. The analysis wavelength was at 420 nm (Waters, MA, model Breeze). The sample was injected through a 20 μl sample loop. A C8 analytical column (150 mm ×4.6 mm, particle size 5 μm; Perfect still, MZ-Analysen technik, Germany) operational by a guard column of the same packing was applied. The calibration curve for ART was linear over the concentration range of 1–20 μg/ml. Entrapment efficiency was calculated in reference to loading ratio and total dried nano dispersion weight obtained by the following equation:
(2)
(2)
EE % is the efficiency of entrapment. Wdrug in micelle and Winitial drug show the total mass of powders obtained after freeze-drying and the drug fed initially in the micelle preparation step, respectively.
DSC analysis
DSC analysis was applied to make available further information on the drug–copolymer relationship and possible drug–polymer interaction (s) as well as the physical changes happened on the drug or polymer can be measured by the thermal analysis. DSC analysis was carried out on pure drug and drug-loaded micelles. Samples were heated at a rate of 10 °C min−1 and the data were recorded from 0 to 200 °C.
Drug release study
This investigation was approved out to estimate the release of ART from micelles. Briefly, 5 mg of freeze-dried nanoparticles were dispersed in 2 ml phosphate-buffered saline (PBS) containing 2% (v/v) tween 80 and the follow-on suspension was sited within a dialysis sac (12 Mw kDa) and incubated at 37 °C while immersed in 15 ml of PBS. Then, at programmed time intervals, 2 ml of the dialysate was taken out and replaced by 2 ml new PBS. The concentration of ART in the dialysate was indomitable by HPLC. All the release studies were carried out in triplicate. To facilitate study the pH-dependency of the drug release, the experiments were also carried out, as specified earlier, using PBS at a pH of 5.5. To investigate the drug release performance of carriers while incubated in real human plasma, the release experiments were repeated in plasma of a healthy male volunteer in the same condition except that the samples were incubated in plasma instead of PBS. As controls, the release of free ART was studied in PBS with pH= 7.4, pH= 5.5 and human plasma.
Cell cultures and in vitro experiments
Experiments carried out on mice breast adenocarcinoma cell lines (4T1) and human breast adenocarcinoma cell line (MCF-7) that buy from Pasteur Institute (Tehran, Iran). The cells were cultured according to our previous reports [Citation34], in RPMI-1640 (GIBCO, USA) containing % 10 Fetal Bovine Serum (FBS) (Gibco, Germany), 2 mM l-glutamine, penicillin (50 IU/ml) and streptomycin (50 μg/ml) and incubated at 37° C in a humidified incubator with 5% CO2 atmosphere. The cells were then harvested with 0.4 g/l trypsin (Gibco Laboratories) and 0.3 g/l EDTA (Gibco Laboratories) after 70% confluence for 5 min in 37 °C.
Cell viability by MTT assay
To complete the MTT assay, 96 well plates were utilized, cells were seeded onto plates and permitted to adhere and grow overnight in 200 μl medium with 104 cells per well concentration. The cancer cells were then incubated with fresh medium including serial concentrations (2.5–160 μM) of ART and equivalent doses of ART/PCL–PEG–PCL nano particle and PCL–PEG–PCL nanoparticle alone in the solutions form for 48 and 72 h and a group without treatment for control. Then the MTT solution at appropriate concentrations (15 μl of 6 mg/ml MTT solution in each 100 μl media) was added to each well and the plates were then incubated at 37 °C in a 5% CO2 for 4 h. remaining MTT solution was removed and 100 μl of DMSO was added to each well to dissolve the formazan crystals. The plates were shaken for 15 min on a plate shaker to ensure adequate solubility. Absorbance readings of each well were performed at 570 nm. All experiments were arranged in triplicates and repeated twice for statistical analysis. Results were expressed as mean ± SD.
In vivo anticancer activity in the 4T1 breast cancer model
Six-week-old female BALB/c mice were obtained from the Animal House of Pasteur Institute, Tehran, Iran. The mice were kept under standard housing conditions with a regular 12-h dark–light cycle at 24 °C ± 2 °C. The mice were fed with standard diet pellets and clean tap water. All of the experiments using animals were conducted under the regulations set by the University medical zanjan ethics committee’s guidelines for the care of laboratory animals. In Vivo Tumor Growth Studies with ART, PCL–PEG–PCL micelles and ART/PCL–PEG–PCL micelles. The provided 4T1 cells at logarithmic growth phase and the mouse female inbred BALB/c tumour model was developed by injecting 0.2 ml of 4T1 cell suspension (∼107 cells/ml) into the right flank of a female BALB/c mice. To allow tumour formation, the injected mice were kept in animal house of medical sciences of Zanjan University. After 14 days, when the tumours grew to 200 mm3 (tumours volume was measured by a digital vernier caliper), the animals were randomized and selected for various treatment groups (day 0). 1: Control—PBS, 2: PCL–PEG–PCL micelles, 3: ART and 4: ART/micelles. In each group, 7-animals (total 28 animals) were chosen for treatment. Until the tumour volume reached approximately 200 mm3 then, the mice were given 15 intravenous injections, one every other day of 100 μl ART (60 mg/kg) or ART/PCL–PEG–PCL (ART 60 mg/kg) in sterile saline. An untreated control group was injected with sterile PBS. At various time intervals, mice were weighed and tumour size was measured by the vernier calipers. Tumor volume was determined as TV (cm3) = L × W2/2, where “L” is the tumour length and “W” the tumour width. The survival rate and body weight change of the mice were also measured.
Pharmacokinetic study
Male adult Wistar rats with a mean body weight of 220–270 g were fasted overnight prior to the experiments, with free admission to water. The experimental protocol was accepted by animal care and treatment was talented in conformity with the guideline of animal care and research committee of medical sciences of Zanjan University, which is in obedience with the guide for the care and use of laboratory animals. The rats were separated randomly into two groups (n = 7). The aqueous suspension of ART and dispersion of the ART/micelles were injected into the rats via the tail vein at a dose of 25 mg/kg. The micelles were discrete in ultrapure water. Blood samples (500 μl) were withdrawn from the tail vein into heparinized micro tubes at the following times: 0.25, 0.5, 1, 1.5, 2, 4, 8, 12 and 24 h after dosing. The blood samples were centrifuged at 3000 × g for 12 min. The supernatant was together, transferred to tightly potted plastic tubes and store at −20 °C until analysis by HPLC.
Standard preparation
A stock solution of 1 mg/ml ART in methanol were prepared, from which the concentrations of 1000, 2000, 3000, 4000, 5000, 7500, 10 000, 15 000 and 20 000 ng/ml for ART were prepared by serially diluting this solution with the proper amount of rat plasma.
Sample preparation and extraction procedure
To 100 μl of rat plasma samples, 50 μl acetonitrile and 50 μl methanol were added. The mixtures were vortex mixed for 20 s. After centrifugation at 14 000g in an eppendorf micro centrifuge tubes for 20 min. An aliquot of 20 μl was injected into the HPLC system.
Results
Synthesis and characterization of PCL–PEG–PCL copolymer
Using the ring-opening polymerization of caprolactone in the presence of PEG, PCL–PEG–PCL tri-block copolymer was produced. Tri-block copolymers with different molecular weights were obtained by controlling the ɛ-CL: PEG ratio. Characteristics of the synthesized copolymers (P1–P3) are shown in . Copolymer P3 was characterized in detail and used for future applications. The structure and composition of the synthesized PCL–PEG–PCL tri-block copolymer were determined by HNMR spectroscopy in CDCl3. HNMR spectrum of copolymer P3 was shown in . The presence of methylene (CH2) in PCL was observed around 1.28, 1.75, 2.38 and 4.18 ppm, the methylene protons in methylene (CH2) groups of PEG were about 3.68. FT-IR spectrum of copolymer P3 was shown in . The intense bands at 1725.34 cm−1 and 1111.25 cm−1 from spectrum to the presence of carboxylic ester (C=O) and ether (C–O) groups, thereby indicating that the formation of PCL–PEG–PCL copolymer has occurred successfully. GPC results showed that the weight-based average molecular weights of copolymers from 10 200 until 15 961 Da (). To DSC thermograms copolymer P3 the endothermic peak (52.34 °C) including two merged peaks of PEG and PCL presented in .
Figure 1. HNMR spectrum of PCL–PEG–PCL copolymer.
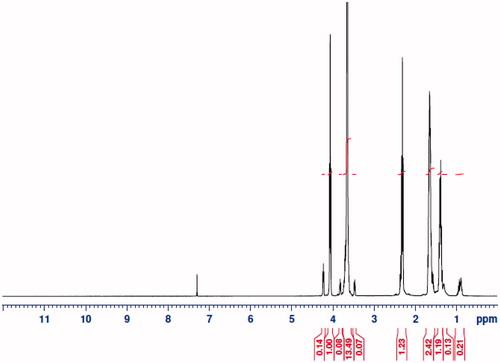
Figure 2. FT-IR spectrum of PCL–PEG–PCL copolymer.
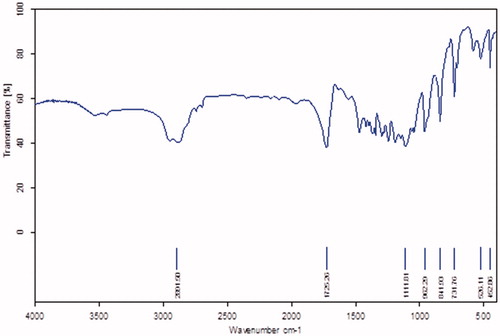
Figure 3. DSC thermogram of PCL–PEG–PCL copolymer.
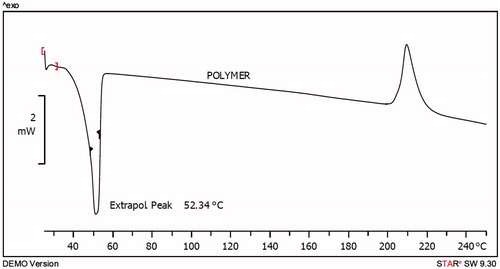
Table 1. Molecular characteristics of the synthesized copolymers.
Preparation and characterization of co-polymeric micelles
To optimize the progression parameters and impending into the impact of the drug/copolymer ratio on loading and encapsulation efficiency, different ART/PCL–PEG–PCL mass ratios in the feed were studied in our work, and results are presented in . The drug loading and encapsulation efficiencies increased with the increase in ART/PCL–PEG–PCL mass ratios and also, particle size and poly dispersity index (PDI) increased, but the stability of the ART/PCL–PEG–PCL micelles decreased. When the ART/PCL–PEG–PCL mass ratio was 1, the obtained ART/PCL–PEG–PCL micelles were not stable in water (micelles be likely to aggregate); thus, the ART/PCL–PEG–PCL mass ratio at 0.25 (M3) was chosen in our study (). The interpretation can be given to conditions of insolubility of ART in the aqueous solution, which could guide more drugs at high concentration to be adsorbed on the external of micelles. Micelle M3 was characterized in detail and used for future applications. The formation of micellar nanostructures was confirmed by AFM apparently, PCL–PEG–PCL micelles (M3) showed a homogeneous spherical morphology, as expected (. The size of nanoparticles was measured by dynamic light scattering technique. As shown in , the-average and zeta potential of ART/PCL–PEG–PCL micelles (M3) were about 83.22 nm with zeta potential −15.4 mV, with their corresponding PDI being 0.220. The micelle size observed by AFM was about 70 nm, smaller than determined by DLS. The micelle diameter determined by DLS represents the hydrodynamics diameter while that obtained by AFM is related to the collapsed micelles after water evaporation. The loading ratio and encapsulation efficiencies of ART loaded to PCL–PEG–PCL micelles (M3) were 18.62 ± 0.42 and 89.23 ± 1.41%, respectively.
Figure 4. AFM image of ART loaded spherical core shell micelles.
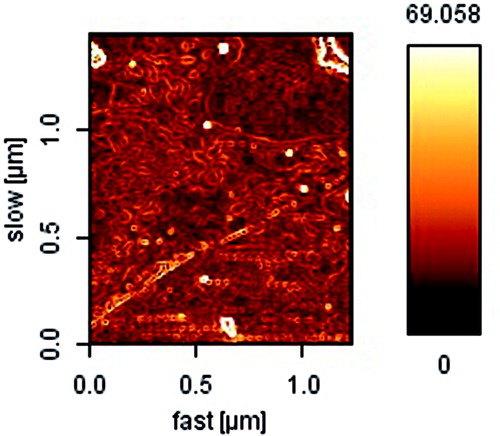
Figure 5. Particle size distribution and zeta potential of ART/PCL–PEG–PCL.
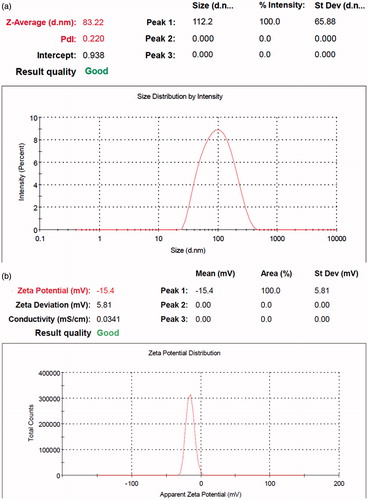
Table 2. Properties of ART/PCL–PEG–PCL micelles.
Impact of copolymer composition on drug entrapment efficiency
To develop an ideal origination of higher DL or EE, the effects of the ratio of CL and PEG on DL and EE were studied. Micelles with fixed PEG length, the DL and EE increased with the increase of feed weight ratio of PEG: ɛ-CL (from 1:0.5 to 1:2). Amphiphilic PCL–PEG–PCL copolymer formed micelles with a core-shell structure in water. The hydrophobic drug could be encapsulated into a hydrophobic core by a hydrophobic interaction of PCL and ART and with the interaction between water and the hydrophilic shell of PEG. The increase of the DL and EE of ART in PCL–PEG–PCL may be caused by the strong hydrophobic interaction between the longer hydrophobic PCL block chain and hydrophobic drugs. This result was expected because incorporation of ART into the hydrophobic cores increased the volume of the micelles.
Stability assay of nano-micelles by determination of size
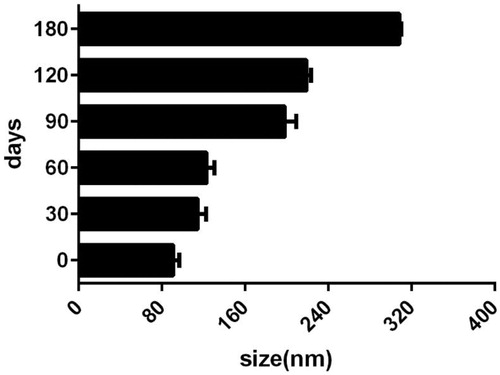
In the clinical aspect of nanoparticle dispersions, the sizes and volume stability of the nano-carriers is very important. Micelles planned for the use as a drug-delivery system must be stable enough to provide a necessary time for drug-delivery and accumulation in the target zone [Citation35]. In the present study, the particle size stability was monitored over a 6 month course. The sizes of micelles variation as a role of incubation time is shown in . The size of all micelles was increased to some extent during the measurement period. This observation cannot be a symbol of aggregation, which commonly leads to numerous fold increases. Some copolymers swelling and/or hydration (as a result of presence of the hydrophilic PEG portions in micelles surfaces) can be responsible for this happening.
Figure 6. The average hydrodynamic diameter of PCL–PEG–PCL copolymer.
DSC analysis
The thermogram of PCL–PEG–PCL copolymer displayed an endothermic peak at 52.34 °C which is indicative for the melting of the crystalline PCL segment of copolymer, the thermogram of ART displayed an endothermic peak at 154.35 °C and micelles displayed endothermic peak at 45.08 °C which stands of the melting of copolymers associated in the form of micelles. This endothermic peak of micelles presumably confirms a physical interaction between copolymer and ART upon loading of the drug in micelles, since the melting point of PCL–PEG–PCL micelles was lower than melting point of copolymer (52.34 °C). The characteristic peak of ART was not observed in micelles. It could be concluded that the ART in the micelles was in an amorphous or disordered crystalline phase or in a solid solution state.
In vitro release of ART
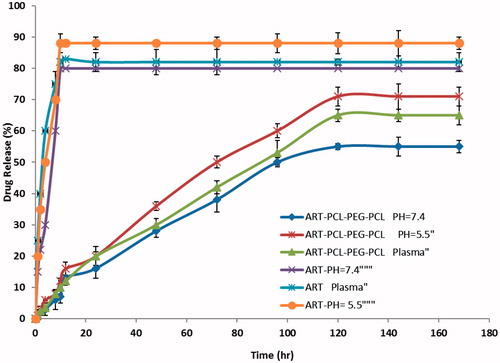
In order to study the effect of the chemical and biochemical features on the release of ART from micelles, the release study was done on drug-loaded micelles in neutral (pH = 7.4) and acidified PBS solution (pH = 5.5) as well as the freshly prepared human plasma with drawn from a volunteer. As controls, the release of free ART was studied to verify that the dispersion of drug molecules through the dialysis membrane was not a rate-limiting step during the release procedure. Free ART was observed to be rapidly released and reached its peak of 80, 88 and 82% of the total in the first 10 h at pH 7.4, 5.5, and plasma, respectively. shows the release profiles of ART from the drug-loaded micelles, at pH 7.4, 5.5, and plasma. Obviously, no significant initial burst ART release was detected from the micelles. As shown in , the ratio of ART released from the micelles increased equally the pH value decreased from 7.4 to 5.5. For example, after 72 h incubation, the quantities of ART released in the media with pH values of 7.4, 5.5 and human plasma were about 38, 50 and 42%, respectively. The reason behind this occurrence lies in the pH sensitivity of the release rate of ART from the micelles because the copolymer is degradable in acidic condition by hydrolysis. Also, the release is faster at acidic pH than in neutral. This behaviour is a highly desirable characteristic in many applications especially in anticancer drug delivery where the micro-environments of extracellular spaces of tumours, intracellular lysosomes and endosomes are acidic, which can possibly enable the drug release from obtained micelles. The results shown that the maximum drug releases were 55, 71 and 65% respectively for PBS pH = 7.4, pH = 5.5 and plasma after a period of 120 h. The sustained release of ART can be attributed to the trap of ART in the micelles core. Therefore, the obtained co-polymeric micelles can be considered as highly attractive nano-carriers for time-controlled drug delivery for hydrophobic drugs to the success of different therapeutic objectives.
Figure 7. The release profiles of ART from ART/PCL–PEG–PCL micelles in different release media (pH = 7.4, plasma, pH = 5.5).
Study on cancerous cell’s viability after ART/PCL–PEG–PCL-system treatment
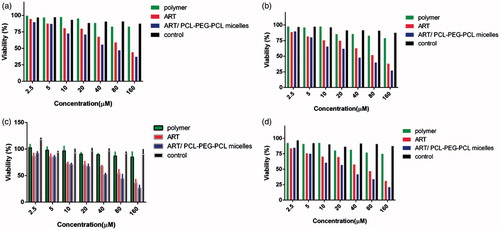
The in vitro anticancer effects ART/PCL–PEG–PCL micelles against mice breast adenocarcinoma (4T1) and human breast adenocarcinoma (MCF7) were estimated using a MTT assay (. The data exhibit that cell toxicity is directly commensurate to ART/PCL–PEG–PCL micelles concentration. Moreover, the PCL–PEG–PCL micelles did not show noticeable discrepancy anticancer effect at different treatment times (48 and 72 h) in all of cancer cell lines. These studies specify that ART/PCL–PEG–PCL micelles have a very remarkable anticancer effect, for breast cancer cell lines. The concentrations leading to 50% cytotoxicity (IC50) were about 62 and 39 μM for 4T1 after 48 and 72 h while it was 55 and 33 μM for MCF-7 after 48 and 72 h, respectively. The IC50 values indicate that the MCF-7 cells are more sensitive to ART treatment than the 4T1 cells. Also the results of this assessment show no toxicity for bare nanoparticle in various concentrations. Relative cell toxicity was calculated by EquationEquation (3)(3)
(3) .
(3)
(3)
Figure 8. MTT Assay of ART, polymer, ART/micelles and control (PBS) on (a) 4T1 cell line after 48 h; (b) 4T1 after 72 h (c) MCF7 after 48 h and (d) MCF7 after 72 h.
Effect of ART/PCL–PEG–PCL micelles in tumour development
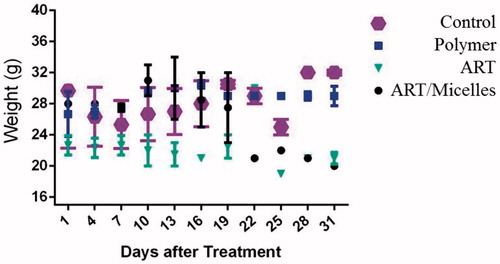
To assess the in vivo anti-tumour efficacy of ART/PCL–PEG–PCL micelles, we tested the conjugate in mice bearing 4T1 murine breast cancers. The tumour volume increase in control mice compared to the treated animals. After the 20th day, tumour volumes of ART treated mice reached to about 500 mm3 while ART/micelles reached under 27 mm3 (). Measurement of tumour growth profile as a function of time showed that tumour size on day 20 in non-treated mice reached to about 2150 mm3 similar to PCL–PEG–PCL micelles. In agreement with the pharmacokinetic results, the much greater anti-tumour efficacy of ART/PCL–PEG–PCL micelles, compared with the same dose of free ART, was likely attributable to prolonged drug circulation in plasma and increased drug accumulation in the tumour. We also measure body weight changes in all mouse groups (. The weight loss was observed in mice given intravenous dose (60 mg/kg) of void ART, compared with control, saline-treated animals. The weight loss observed in polymer treated mice and control group (PBS treated group) while there was not considerably weight loss in ART/micelles treated group. The lack of significant toxicity in ART/micelles treated group was likely attributable to the prolonged half-life in blood and the enhanced tumour localization of conjugate-associated ART.
Figure 9. Effect of ART, polymer, ART/micelles and control (PBS) on the evolution of tumour volume in mice.
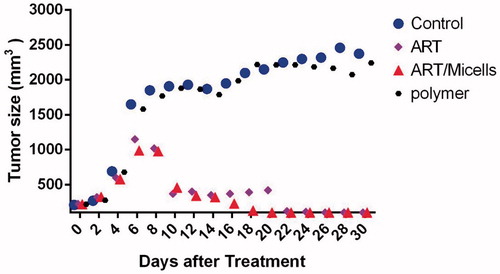
Figure 10. Body weight measurements of mice (seven per group) bearing 4T1 allografts which treated with fifteen intravenous injections, every other day, of ART 60 mg/kg, PCL–PEG–PCL, ART/PCL–PEG–PCL (60 mg ART/kg) and PBS (control).
Pharmacokinetics study
The mean ART plasma concentration–time profiles after injection of 25 mg/kg of ART aqueous suspension and ART-loaded micelles exposed in . summarizes the relevant pharmacokinetic parameters. Plasma ART levels in the ART aqueous group rapidly declined to 2541 ng/ml after 2 h (h). Plasma ART levels declined obviously in the first 0.5 h, while a slow decrease was observed subsequently. In addition, plasma concentrations in the micelles were obviously higher than those in the ART aqueous group at each time point. Even at 24 h after dosing, plasma concentrations of ART in the micelles were still above the 3120 ng/ml. Therefore, the ART plasma concentration in ART aqueous group decreased abruptly, as the drug was distributed and rapidly metabolized, resulting in a high Cl and short t1/2, ∼1.02 h. A sustained release of ART over 24 h was observed when it was carried by the micelles. The distribution and metabolism of ART were decreased when it was carried by micelles (p < .01). Thus, the t1/2 of ART from the ART loaded micelles increased to 3.51 h, while for ART aqueous group the t1/2 was 1.02 h which suggested that entrapping ART into micelles could prolong in vivo residence time, thereby achieving a long-term therapeutic effect. The micelle mean AUC0–∞ value was 16-fold higher than that of ART aqueous group which indicated a higher ART exposure yielded by the micelles formulation, and an increase in the ART systemic bioavailability in vivo.
Figure 11. Comparison of in vivo plasma concentration versus time profiles of the different. ART formulations. All values reported are the mean ± SD (n = 7).
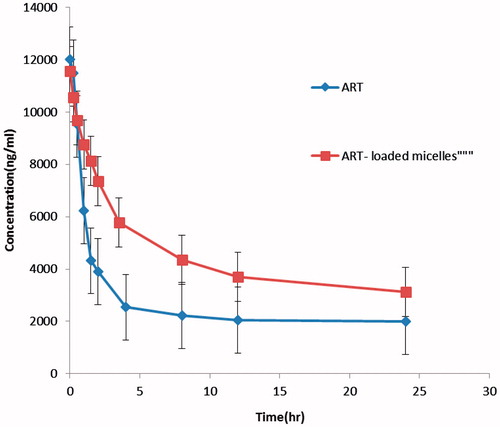
Table 3. Pharmacokinetic parameters of ART following single dose injection of ART aqueous solution and ART/micelles, in rats (n = 7).
Discussion
ART has been reported to be hydrophobic and is presently available only in the form of oily intra muscular injection. The poor physicochemical/pharmaceutical properties of ART limit development of suitable parenteral formulations. ART is a lipophilic drug and insoluble in water, which limits its clinical application. Encapsulation by nanoparticles looks to be a brilliant advance to overcome the hydrophobicity of ART. There are many encapsulation systems, such as spray-drying, solvent removal, etc., to encapsulate ART into micelles or liposomes. In this study, we used PCL–PEG–PCL micelles to encapsulate ART. In the preparation process, ART and PCL–PEG–PCL tri-block copolymer mixture was first dissolved in acetone solution, followed by evaporating the organic solvent. Then, the amphiphilic PCL–PEG–PCL copolymers self-assembled in to supra molecular preparations possessing a hydrophobic inner core and a hydrophilic shell in water, and ART self-assembled into the hydrophobic core of the micelles because of its hydrophobicity. The preparation procedure was simple and easy to scale up. These PCL–PEG–PCL micelles are biodegradable, biocompatible, amphiphilic, stable in blood, non-toxic, non-immunogenic, non-inflammatory [Citation36] and small in size; this makes PCL–PEG–PCL micelles an excellent candidate for drug-delivery systems. To defeat the poor water-solubility of ART, we encapsulated ART into PCL–PEG–PCL micelles by a self-assembly method producing ART/PCL–PEG–PCL micelles. The ART/PCL–PEG–PCL micelles were about 70 nm in diameter. In addition, ART/PCL–PEG–PCL micelles showed drug loading of 18.62 ± 0.42% and a high encapsulation efficiency of and 89.23 ± 1.41%, the ART/PCL–PEG–PCL micelles had a minor negative surface charge of −15.4 mV, which increased the circulation time of the drug. Surface charge is significant indecisive whether the nanoparticles will cluster in blood flow or will stick to or interact with oppositely charged cell membrane. The plasma and blood cells regularly had a negative charge; nanoparticles with slight negative surface charge can minimize nonspecific contact with these components through electrostatic interactions. The ART/PCL–PEG–PCL micelles were capable to slowly release ART, indicating that ART/PCL–PEG–PCL micelles may improve systemic circulation t½ of ART in vivo. The sustained release of ART from PCL–PEG–PCL micelles might be due to the diffusion of ART from micelles and the degradation or hydrolysis of micelles. In summary, PCL–PEG–PCL micelles were used to encapsulate ART, creating ART/PCL–PEG–PCL micelles. The ART/PCL–PEG–PCL micelles improved the water solubility of ART and may have potential application in cancer treatment [Citation37,Citation38]. In addition, the in vitro and in vivo experiments demonstrate that the ART/PCL–PEG–PCL has lower toxicity to normal cells and tissues than free ART, which is likely the major reason for the low systemic toxicity. Hence, all results confirm that the ART/PCL–PEG–PCL has better therapeutic efficacy on tumour cells and lower systemic toxicity than the free ART.
Conclusion
PCL–PEG–PCL micelle-encapsulated ART was prepared with the goal of improving the water insolubility of ART. These ART/PCL–PEG–PCL micelles can slowly release the ART, enhance systemic circulation t½ of ART in vivo and inhibit the growth of cancer cells in vitro, and in vivo by showing promising application in cancer treatment. The in vitro cytotoxicity study revealed that the micelle was safe and low cytotoxic. Our study showed that these micelles provided a suitable and appropriate system for delivery of ART to breast cancer cells. This developed nano-drug delivery system can also be used as a promising base to design further micelles systems to improve the therapeutic effect of ART in the future.
Acknowledgement
This work was supported by the deputy of research of Zanjan University of Medical Sciences (Grant No. A-12–430-16 and A-12–430-11).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
Related Research Data
References
- Asghari F, Samiei M, Adibkia K, et al. Biodegradable and biocompatible polymers for tissue engineering application: a review. Artif Cells Nanomed Biotechnol. 2017;45:185–192.
- Manjili HK, Jafari H, Ramazani A, et al. Anti-leishmanial and toxicity activities of some selected Iranian medicinal plants. Parasitol Res. 2012;111:2115–2121.
- Manjili HK, Sharafi A, Danafar H, et al. Poly (caprolactone)–poly (ethylene glycol)–poly (caprolactone)(PCL–PEG–PCL) nanoparticles: a valuable and efficient system for in vitro and in vivo delivery of curcumin. RSC Adv. 2016c;6:14403–14415.
- Porbarkhordari E, Foladsaz K, Hoseini SH, et al. The hypoglycemic effects of an ethanol extract of peganum harmala in streptozotocin-induced diabetic rats. Iran J Pharm Sci 2014;10:47–54.
- Ma'mani L, Nikzad S, Kheiri-Manjili H, et al. Curcumin-loaded guanidine functionalized PEGylated I3ad mesoporous silica nanoparticles KIT-6: practical strategy for the breast cancer therapy. Eur J Med Chem. 2014;83:646–654.
- Manjili HK, Ma’mani L, Tavaddod S, et al. D,L-Sulforaphane loaded Fe 3 O 4@ gold core shell nanoparticles: a potential sulforaphane delivery system. PLoS One 2016b;11:e0151344.
- Chen Y, Lin X, Park H, et al. Study of artemisinin nanocapsules as anticancer drug delivery systems. Nanomed Nanotechnol Biol Med. 2009;5:316–322.
- Danafar H, Sharafi A, Kheiri Manjili H, et al. Sulforaphane delivery using mPEG-PCL co-polymer nanoparticles to breast cancer cells. Pharm Dev Technol. 2017b;22:642–651.
- Nakase I, Gallis B, Takatani-Nakase T, et al. Transferrin receptor-dependent cytotoxicity of artemisinin-transferrin conjugates on prostate cancer cells and induction of apoptosis. Cancer Lett. 2009;274:290–298.
- Van Nijlen T, Brennan K, Van den Mooter G, Blaton N, Kinget R, Augustijns P. Improvement of the dissolution rate of artemisinin by means of supercritical fluid technology and solid dispersions. Int J Pharm. 2003;254:173–181.
- Mirzaee H, Sharafi A, Sohi HH. In vitro regeneration and transient expression of recombinant sesquiterpene cyclase (SQC) in Artemisia annua L. South Afr J Bot. 2016;104:225–231.
- Sharafi A, Hashemi Sohi H, Sharafi A, et al. Tissue culture and regeneration of an antimalarial plant, Artemisia sieberi Besser. Res J Pharm. 2014;1:15–20.
- Woodrow C, Haynes R, Krishna S. Artemisinins. Postgrad Med J. 2005;81:71–78.
- Li QG, Peggins JO, Fleckenstein LL, et al. The pharmacokinetics and bioavailability of dihydroartemisinin, arteether, artemether, artesunic acid and artelinic acid in rats. J Pharm Pharmacol. 1998;50:173–182.
- Safari J, Zarnegar Z. Advanced drug delivery systems: nanotechnology of health design a review. J Saudi Chem Soc. 2014;18:85–99.
- Shutava TG, Balkundi SS, Vangala P, et al. Layer-by-layer-coated gelatin nanoparticles as a vehicle for delivery of natural polyphenols. ACS Nano. 2009;3:1877–1885.
- Wang T, He N. Preparation, characterization and applications of low-molecular-weight alginate-oligochitosan nanocapsules. Nanoscale 2010;2:230–239.
- Crespo-Ortiz MP, Wei MQ. Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. BioMed Res Int. 2011;2012:247597.
- Danafar H, Rostamizadeh K, Davaran S, et al. Drug-conjugated PLA–PEG–PLA copolymers: a novel approach for controlled delivery of hydrophilic drugs by micelle formation. Pharm Dev Technol 2015; 1–11. DOI: 10.3109/10837450.2015.1125920.
- Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112:630–648.
- Wang X, Yang L, Chen ZG, et al. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58:97–110.
- Nomani A, Nosrati H, Manjili HK, et al. Preparation and characterization of copolymeric polymersomes for protein delivery. Drug Res 2017. DOI http://dx.doi.org/10.1055/s-0043-106051.
- Kheiri Manjili H, Sharafi A, Attari E, et al. Pharmacokinetics and in vitro and in vivo delivery of sulforaphane by PCL–PEG–PCL copolymeric-based micelles. Artif Cells Nanomed Biotechnol. 2017;1–12. doi.org/10.1080/21691401.2017.1282501.
- Manjili H, Malvandi H, Mousavi M-S, et al. Preparation and physicochemical characterization of biodegradable mPEG-PCL coreshell micelles for delivery of artemisinin. Pharm Sci. 2016a;22:234–243.
- Manjili HRK, Ghasemi P, Malvandi H, et al. Pharmacokinetics and in vivo delivery of curcumin by copolymeric mPEG-PCL micelles. Eur J Pharm Biopharm. 2017;116:17–30.
- Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–476.
- Danafar H. Applications of copolymeric nanoparticles in drug delivery systems. Drug Res (Stuttg). 2016;66:506–519.
- Gharebaghi F, Dalali N, Ahmadi E, et al. Preparation of wormlike polymeric nanoparticles coated with silica for delivery of methotrexate and evaluation of anticancer activity against MCF7 cells. J Biomater Appl. 2017;31:1305–1316.
- Xie W, Zhu W, Shen Z. Synthesis, isothermal crystallization and micellization of mPEG–PCL diblock copolymers catalyzed by yttrium complex. Polymer. 2007;48:6791–6798.
- Yodthong B. Surfactant-free nanospheres of m/PEG-PCL for controlled release of ibuprofen. J Appl Sci. 2009;9:2278–2279.
- Danafar H, Manjili H, Najafi M. Study of copolymer composition on drug loading efficiency of enalapril in polymersomes and cytotoxicity of drug loaded nanoparticles. Drug Res (Stuttg). 2016;66:495–504.
- Danafar H, Hamidi M. Pharmacokinetics and bioequivalence study of amlodipine and atorvastatin in healthy male volunteers by LC-MS. Pharm Sci. 2015;21:167–174.
- Danafar H, Hamidi M. A rapid and sensitive LC–MS method for determination of ezetimibe concentration in human plasma: application to a bioequivalence study. Adv Pharm Bull Chromatogr. 2013;76:1667–1675.
- Manjili HK, Naderi-Manesh H, Mashhadikhan M, et al. The effect of iron-gold core shell magnetic nanoparticles on the sensitization of breast cancer cells to irradiation. J Paramed Sci. 2014;5:85–90.
- Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56:1273–1289.
- Wei X, Gong C, Gou M, et al. Biodegradable poly (ɛ-caprolactone)–poly (ethylene glycol) copolymers as drug delivery system. Int J Pharm. 2009;381:1–18.
- Gou M, Wei X, Men K, et al. PCL/PEG copolymeric nanoparticles: potential nanoplatforms for anticancer agent delivery. Curr Drug Targets. 2011;12:1131–1150.
- Danafar H, Rostamizadeh K, Hamidi M. Polylactide/poly (ethylene glycol)/polylactide triblock copolymer micelles as carrier for delivery of hydrophilic and hydrophobic drugs: a comparison study. J Pharm Invest. 2017a. doi: 10.1007/s40005-017-0334-8.