
Abstract
Systemic and uncontrolled administration of erlotinib hydrochloride (ETB) is associated with severe toxicity. A novel targeted and extended release nanosponge (NS) was synthesized from glutathione (GHS) by a one-step reaction between β-cyclodextrin and pyromellitic dianhydride at room temperature for delivery of ETB in lung cancer. Characterization studies were performed using sophisticated instruments. In-vitro release study was performed in the presence of incremental concentrations of GHS which was analyzed using HPLC. Cell cytotoxicity study was evaluated on human lung cancer (A549) cell lines. In-vivo tumour inhibition and biodistribution of ETB-loaded GHS-NS (ETB-NS) were performed on BALB/c mice. NS obtained was spherical, size 212 ± 2.45 nm and high drug entrapment (92.34 ± 5.31%) (p < .001). In-vitro extended drug release (76.89 ± 0.1% release at 168 h), which was directly proportional to the concentration of GHS, demonstrated tumour targeting. There was enhanced in-vitro cytotoxicity and 97.5% inhibition in tumour growth on administering NS when compared to plain ETB (48% inhibition) indicating targeting of NS to the tumour site. Biodistribution study and in-vivo tumour growth inhibition study revealed drug release to the cancerous cell, thus preventing unnecessary drug exposure. ETB-NS exhibits extended drug release proportional to the external GSH concentration.
Introduction
Cancer is now the leading cause of deaths in the world, responsible for millions of life between 2005 and 2015 estimated by the World Health Organisation [Citation1]. Among which lung cancer is a principle cause of cancer-related deaths globally, with a 5-year survival rate in approximately 14% of cases, because most patients are diagnosed with advanced inoperable stage where chemotherapy is the only option for treatment [Citation2,Citation3]. Histologically, there are two types of lung cancer, small cell lung cancer (SCLC), comprising 15% of cases, and nonsmall cell lung cancer (NSCLC) which represents 85% of the total cases [Citation4–6].
Currently, cancer is being treated most commonly by chemotherapy and radiation, but it carries many undesired effects, primarily because of its inefficiency in tumour targeting and toxic effects on healthy tissue. The survival rate (17%) of lung cancer patient has not greatly increased despite tremendous research, thus prompting the need for the development of novel targeted therapeutic approach.
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that is expressed in 60% of NSCLC, emerged as new targets for treatment of various types of cancer [Citation6,Citation7]. Erlotinib hydrochloride (ETB) is a USFDA-approved drug (previously known as OSI-774 and CP-358774), for NSCLC [Citation8]. ETB chemically is [6,7-bis(2-methoxy-ethoxy)-quinazolin-4-yl]-(3-ethynylphenyl) amine hydrochloride, a human epidermal growth factor type/epidermal growth factor receptor (HERl/EGFR) tyrosine kinase inhibitor [Citation9–11]. It promotes cell cycle arrest, apoptosis leading to inhibition of angiogenesis and cell invasion by binding to the intracellular tyrosine kinase domain of EGFR, thereby inhibiting receptor autophosphorylation and blocking downstream signal transduction. ETB is used particularly in the treatment of pancreatic and NSCLC [Citation9,Citation12]. ETB has also been reported to be effective in the management of glioma, ovarian, and head and neck cancer, as well [Citation13].
ETB has poor oral bioavailability, mainly due to its poor solubility, instability in gastrointestinal environment and extensive first-pass metabolism. There are many severe and fatal toxicities, such as skin rash, diarrhoea and haematological side effects, such as anaemia, thrombocytopenia and neutropenia, Stevens–Johnson syndrome, gastrointestinal perforations, ocular lesions and skin rash [Citation14]. There is growing concern regarding development of resistance to EGFR-targeted therapies [Citation15]. Thus, there is an urgent requirement for the development of targeted anticancer formulation.
The therapeutic efficacy of drug molecules can be increased by entrapping them within the internal cavity of nanocarrier system, which improve the solubility, targeting efficiency by passive mechanism and biocompatibility as well as controls the rate of ETB release for the treatment of NSCSL [Citation16].
This paper explores the ability of glutathione nanosponge (GHS-NS) in targeting ETB to cancerous cell. GSH-NS can be used for site-specific delivery of drugs, reducing unnecessary exposure of drug to tissue, thus potentially reducing systemic side effects and causing reduction in therapeutic doses [Citation17,Citation18] ().
Figure 1. Schematic diagram of ETB-GHS-NS synthesis, release and its activity on tumour cells.
The blank GSH nanosponge (GHS-NS) has good swelling property and did not show any cytotoxic effects on several cancer cell lines [Citation19]. Nanosponge (NS) shows good biocompatibility and negligible biotoxicity when evaluated in mice following the injection of doses of 500–5000 mg/kg; the mice show no signs of toxicity or any adverse reactions. Their oral administration has also been evaluated in mice, with no apparent side effects noted [Citation16].
GSH is a thiol-containing tripeptide, and its intracellular concentration (2–10 mM in cytoplasm and cell nucleus) is 100–1000 times higher than that in the extracellular fluids and circulation (2–20 µM). The concentration of GSH in some cancer cells is about fourfold higher than in normal cells [Citation20]. With these significant characteristics, GSH is an excellent biogenic stimulus and can be used for developing site-specific intracellular (cancerous cell) drug delivery system [Citation21]. Introduction of a disulphide bond into complex formulation (GSH-responsive controlled release systems) has been widely reported [Citation22], including polymersomes [Citation23], self-assembled micelles [Citation24], inorganic nanomaterials, nanocapsule [Citation25], nanoparticles [Citation20,Citation26] and nanosponge [Citation27].
Materials and methods
Erlotinib hydrochloride (ETB) was a kind gift from Mylan Pharmaceuticals (India) Pvt. Ltd. (Hyderabad, India). β-Cyclodextrin (β-CD) was a gift sample from Medley Pharmaceuticals Ltd. (Mumbai, India). Pyromellitic dianhydride (PMDA), ethylenediaminetetraacetic acid (EDTA), triethylamine (TEA), dimethyl sulphoxide (DMSO) and 2-hydroxy ethyl disulphide (2-HEDS) were purchased from Sigma-Aldrich Co. (St. Louis, MO). A549 (human lung cancer adenocarcinoma) cell lines were purchased from NCCS (Pune, India). Dulbecco’s modified Eagle’s medium, membrane filter (MWCO 12000 Da) was purchased from HiMedia Laboratories Pvt. Ltd. (Mumbai, India). Sodium lauryl sulphate (SLS), sodium hydroxide, potassium hydrogen phosphate, disodium hydrogen phosphate, potassium dihydrogen phthalate and potassium dihydrogen phosphate and acetic acid were acquired from Merck Life Science Pvt. Ltd. (Mumbai, India). High-performance liquid chromatography (HPLC) grade reagents such as acetonitrile were obtained from S D Fine-Chem Ltd. (Mumbai, India) and methanol was obtained from Merck Life Science Pvt. Ltd. (Mumbai, India). Ammonium acetate was purchased from Merck & Co., Inc. (Mumbai, India). All other chemicals used were of analytical grade.
Preparation of GHS-ETB nanosponge
NS was prepared by a method reported by Trotta et al. 2015 [Citation27] with some modifications. One gram (0.881 mM) of β-CD was solubilized in 4 ml of DMSO in a water bath at room temperature and then 0.05 gm (0.324 mM) of 2-HEDS was added to the above mixture and stirred for 10 min. TEA (catalyst) (0.25 ml) was added under vigours stirring, and 1.625 gm (7.454 mM) of PMDA was added as a cross-linker [Citation17]. The reaction completed in 24 h. The powder obtained was Soxhlet extracted with acetone for 14 h to remove by-products and unreacted reagents. Nanosponge was stored at 25 °C under nitrogen atmosphere. The formation of NS was confirmed by solubility studies FTIR.
Drug loading
Drug was loaded by suspending drug in 20 ml of HPLC grade water on a magnetic stirrer, and ETB was added in a w/w ratio of 1:2, 1:4 and 1:6 (drug: NS), respectively. Further, this mixture was sonicated for 10 min followed by stirring for 24 h. Subsequently, the NS suspension was purified by dialysis to remove the uncomplexed drug.
Characterization
Particle size, polydispersity index (PDI) and zeta potential
The mean particle size, polydispersity index (PDI) and zeta potential of NS were measured by dynamic light scattering (DLS) technique on a Malvern Zetasizer™ Nano ZS90 (Malvern Instruments Ltd., Malvern, UK) at room temperature. Samples were suitably diluted with HPLC grade water before measurement.
Fourier transform infrared spectroscopy (FTIR)
The FTIR spectra of ETB, GHS-NS, physical mixture and ETB-NS were obtained using a Jasco-2000 spectrophotometer, to understand interaction between ETB and NS. The spectra were obtained on KBr pellets in the region from 4000 cm−1 to 400 cm−1.
Differential scanning colorimetry (DSC)
Thermal analysis was performed for plain ETB, β-CD, GHS-NS and ETB-NS using DSC instrument (Mettler Toledo DSC 822e) in the range of ±350 mW at RT with heating rate of 10 °C/min in 25–500 °C temperature range. Standard aluminium sample pans were used; an empty pan was used as reference standard. Analysis was performed in triplicate on 10 mg samples under nitrogen purge.
Powder X-ray diffraction studies (PXRD)
Powder X-ray diffraction (PXRD) patterns of plain ETB and ETB-NS were recorded with an X-ray diffractometer (D8 Advance Diffractometer, Bruker AXS GmbH, Karlsruhe, Germany) employing Cu Kα (wavelength 1.5406 Å, tube operated at 40 kV, 40 mA) at room temperature. Data were collected over an angular range from 4 to 40° 2θ at a step size of 0.01° and scan rate of one second. The obtained diffractograms were analyzed with DIFFRAC plus EVA (ver. 9.0) diffraction software.
Scanning electron microscopy (SEM)
The surface morphology of GHS-NS and ETB-NS was examined using scanning electron microscope (JEOL Model JSM-6390LV). Samples were placed on an aluminium stub by a bioadhesive carbon tape and sputter-coated to minimize the surface charging.
Transmission electron microscopy (TEM)
Particle shape and size were evaluated using TEM (JEOL/JEM 2100). The GHS-NS suspensions were sprayed on Formvar-coated copper grid and air-dried before observation.
Stability studies
ETB-NS was stored in an aluminium stoppered vial and kept for stability testing at 60 °C/75% RH for 3 months. The changes in particle size, FTIR peaks, DSC thermogram and encapsulation efficiency of the formulation were calculated by analyzing the samples every month for 3 months (data not shown) [Citation28].
In-vitro drug release
Multicompartment rotating cells with a dialysis membrane (MWCO 12,000 Da) was used. ETB-NS was placed in a donor phase containing a fixed amount of drug (1 ml). Ammonium acetate buffer (pH 4) with 0.5%w/v SLS (1 ml) was added to the receiving compartment to maintain proper sink condition. At pre-determined time intervals, the receiving phase was completely replaced with fresh medium. The aliquots were analyzed by HPLC. Release studies were also carried out in the presence of increasing amounts of GHS in the receiving compartment, ranging from 0 mM to 50 mM (0, 2, 5, 10, 25 and 50 mM).
In-vitro cell culture study
Cell culture
Human lung carcinoma cells (A549 cells) were obtained from the National Centre for Cell Science (Pune, India). All cells were maintained at 37 °C in an incubator with 95% humidity and 5% CO2 in 70% confluence in Dulbecco’s modified Eagle’s medium (HiMedia Laboratories Pvt. Ltd., Mumbai, India) which was supplemented with 10% foetal bovine serum and 1% antibiotic/antimycotic (Medley pharmaceuticals Pvt. Ltd., Mumbai, India) [Citation29].
Cell cytotoxicity study
To assess the biological effectiveness of NS as vehicles for anticancer drugs, the antiproliferative activity of ETB-NS, free drug and GHS-NS was determined using MTT assay in A549 (human lung cancer adenocarcinoma) cell lines with different GSH concentrations. Briefly, cells were grown in Dulbecco’s modified essential medium, accompanied with sodium bicarbonate, Earle's salts, 10% FBS, l-glutamine, nonessential amino acids, 1% sodium pyruvate, 100 U ml−1 penicillin, 100 mg/ml streptomycin and maintained under 5% CO2 atmosphere at 37 °C and 95% relative humidity for 24 h. Cells were harvested by using 0.25% w/v trypsin–EDTA solution, subcultured in 96-well culture plate (Thermo Scientific Pierce Protein Research Products, Rockford, IL) at a density of 1000 cells/well and incubated with different equivalent concentrations (1, 2.5, 5, 10 and 20 μM) of ETB-NS, ETB and GHS-NS. The conversion of MTT to purple-coloured formazan by metabolically active cells indicates cell viability, which is then solubilized in DMSO. The optical density of the released and solubilized formazan reagent was measured at 540 nm spectrophotometrically [Citation30].
In-vivo pharmacokinetic study
Antitumour efficacy in a xenograft model of lung cancer
A lung cancer model was developed by xenografting A549 cells into BALB/c nude mice. Male and female BALB/c nude mice were purchased from the Wockhardt Pvt. Ltd., (Aurangabad (M.S), India). A549 cells were injected subcutaneously to the right side of axilla of animals. All animal experiments adhered to the principles of care and use of laboratory animals and were approved by the Institutional Animal Ethical Committee (IAEC). Mice were divided into three groups each containing 10 animals: (a) a control group (physiological saline), (b) an ETB group (10 mg/kg plain ETB) and (c) an ETB-NS group (10 mg/kg ETB). When the size of tumour masses reached the volume of 150 mm3, drug was injected intraperitoneally once a week for 4 weeks. The mice were then sacrificed on the 28th day after inoculation. The mice were weighed and the tumours were measured with a calliper every four days. The tumour volume was calculated as (length × width2/2) [Citation30].
Drug quantification and biodistribution study
The pharmacokinetic and biodistribution studies were performed on BALB/c mice (n = 10) bearing s.c. A549 tumours: A549 cells (2 × 105 cells/50 ml media) were s.c. inoculated into the shaved right lateral flank of BALB/c mice, and the study was initiated when the tumours reached a 300–500 mm3 volume. Mice were treated by tail vein i.v. injection with ETB and ETB-NS at a matched dose of 20 mg ETB/kg, and blood samples were collected from isoflurane-anesthetized mice by cardiac puncture at selected time points (3–240 h). Blood samples were collected in vacutainers (EDTA-containing tubes) and centrifuged at 2500 rpm for 10 min to isolate plasma and diluted with HPLC grade water and frozen at −80 °C until further analysis. At predetermined time points (3, 7, 14, 28 days) following injections, tumours were excised and homogenized.
After blood collection, organs (heart, lung, liver, spleen and kidney) and tumour tissue were harvested, rinsed in buffer and frozen at −80 °C until analysis. Tissue (w = 100 mg), 2 stainless steel homogenizer beads and 0.9 ml water were combined in screw-cap homogenizer vials and subjected to 3 × 30 s cycles at 3600 rpm on a homogenizer. Tissue homogenate (200 ml) was centrifuged at 14,000 g for 5 min, and 300 ml was transferred to a glass LC vial [Citation29,Citation31].
The blood samples were centrifuged at 10,000 rpm for 10 min and the plasma obtained was stored at –20 °C, until further analysis. The samples were vortexed for 30 s, added a protein precipitating agent composed of methanol and acetonitrile (1:1) (200 μl) and vortexed for further 3 min. The mixture was centrifuged at 12,000 rpm for 10 min. The supernatant was filtered and the ETB concentration was quantified by HPLC (Waters 2695, Milford, MA) and Grace® C18 column (250 mm ×4.6 with 5.0 μm particle size) at 25 °C. The mobile phase consisted of a mixture of methanol and pH 4 ammonium acetate buffer (73:27 v/v) at 1.2 ml/min. ETB was quantified at 247 nm using atorvastatin as an internal standard [Citation29,Citation31].
Molecular modelling studies
PyMOL and Discovery Studio 4.1 software were used to model the β-CD, GHS-NS and ETB entrapment into the cavity of β-CD and GHS-NS. The three-dimensional structures of ETB, β-CD and hypothetical GHS-NS were drawn and optimized using the Discovery Studio 4.1. ETB was manually docked into the β-CD and GHS-NS cavities.
Statistical analysis
All the results are given, as mean ± standard deviation (SD). Data were analyzed using Student's t-test or one-way ANOVA (GraphPad InStat software demo, La Jolla, CA). A value of p < .05 was considered significant and p values < .0001 were considered as statistically extremely significant.
Results and discussion
Surgery is the major treatment for most of the malignant tumours, but metastasis often reoccurs postsurgery. Systemic chemotherapy can control the recurrence and metastasis effectively, improve the life quality and prolong the survival time of patients with advance cancers. However, traditional chemotherapy kills and damages both cancerous and normal cells, resulting in bone marrow suppression, liver and kidney dysfunction, gastrointestinal reactions, decreased immune function and other side effects. All these problems can be eliminated by adopting targeted approaches. A bioresponsive NS was developed for site-specific delivery of ETB.
Preparation of GHS-NS
In this study, we have synthesized targeted ETB-NS for delivery of drug to lung cancer site. The NS was synthesized in a single-step reaction among β-CD, PMDA and 2-HEDS using TEA as a catalyst (). The reaction was fast and gave high yield within a few minutes at room temperature (< 95%).
Drug loading
The loading efficiency of ETB in NS complex was considerably high (92.34 ± 5.31%) which confirms the high inclusion phenomenon between ETB and GHS-NS.
Characterization
Particle size, polydispersity index and zeta potential
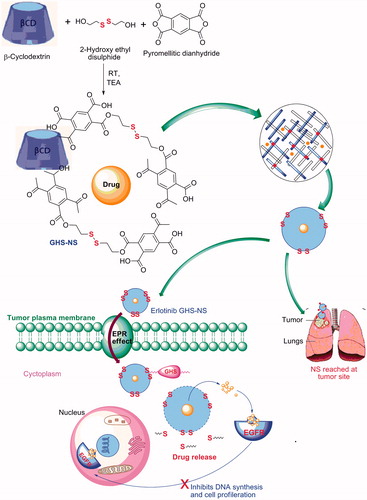
The average particle size of selected ETB-NS was 212 ± 2.45 nm (p > .001) and a relatively narrow size distribution having a polydispersity index of 0.05 (). The zeta potential was determined to measure the surface charges. The zeta potential of the GHS-NS was found to be −30.21 ± 0.47 mV, which indicates high formulation stability (). Particle size and surface charge of a drug carrier are one of the most important factors that determine the fate of both in-vitro and in-vivo NS drug delivery systems [Citation32,Citation33]. Negatively charged carrier systems often exhibit high physical stability [Citation34,Citation35], prolonged circulation [Citation36] and avoid nonspecific cellular uptake [Citation37]. Particle sizes between 100 and 200 nm have a favourable EPR (enhanced permeability and retention) effect on the tumour vasculature [Citation38]. The mean sizes of ETB-NS were within this range for optimal EPR effects [Citation30].
Figure 2. (A) Zeta potential of GHS-NS. (B) Particle size of GHS-NS.
Fourier transform infrared spectroscopy (FTIR)
The FTIR spectral peaks of ETB, GHS-NS, physical mixture and ETB-NS are reported in . The spectra of the GHS-NS were characterized by the strong absorption bands at 3421 cm−1. β-CD-based structure was confirmed by the presence of peak at 1731 cm−1. Peak at 520 cm−1 confirms the presence of S–S bond and a strong C–S stretching vibration at 683 cm−1, thus demonstrating the formation of GHS-NS.
Table 1. FTIR spectral peaks of GHS-NS.
Differential scanning colorimetry (DSC)
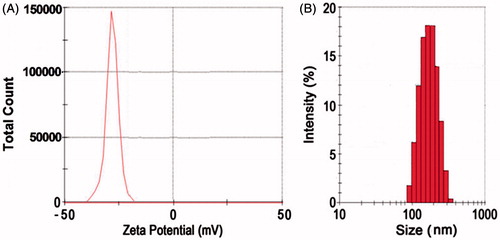
DSC thermograms of ETB and ETB-NS are given in . The thermogram of drug shows a sharp endothermic peak at 234.62 °C, which indicates the melting point of the drug. Absence of peaks in thermogram of ETB-NS indicates formation of inclusion complex between ETB and GHS-NS. Change in the thermograph of drug and ETB-NS is due to change in crystallinity of the NS. The DSC thermogram of β-CD shows a sharp endothermic peak at 260.19 °C, which indicates the melting point of β-CD (). Absence of sharp peaks of β-CD in thermogram of GHS-NS indicates formation of NS due to the change in crystallinity of β-CD after completion of the reaction.
Figure 3. (I) DSC thermogram of (A) erlotinib drug and (B) ETB-loaded GHS-NS. (II) DSC thermogram of (C) β-CD and (D) GHS-NS.
Powder X-ray diffraction studies (XRPD)
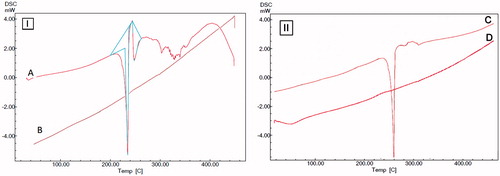
The XRPD pattern of ETB shows the presence of intense, sharp peaks at 6.16, 19.91, 22.15, 22.61, 23.98, 24.21 and 24.73 on 2θ scale which confirms its crystalline structure (). After loading drug into the NS, all sharp peaks are subdued due to change into an amorphous state, suggesting complete drug entrapment ().
Figure 4. (A) PXRD pattern of pure ETB. (B) PXRD pattern of ETB-loaded GHS-NS.
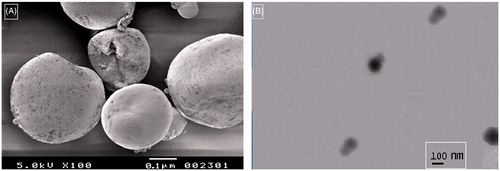
Scanning electron microscopy (SEM) and TEM
SEM and TEM images of NS show porous, spherical particles (∼200–300 nm) ). Use of freeze drying for solvent removal aided in obtaining a porous structure.
Figure 5. (A) SEM image of GHS-NS. (B) TEM image of GHS-NS.
Stability study
ETB-NS was found to be stable over a period of 3 months. Critical parameters such as encapsulation efficiency, particle size and in-vitro drug release remained unaffected.
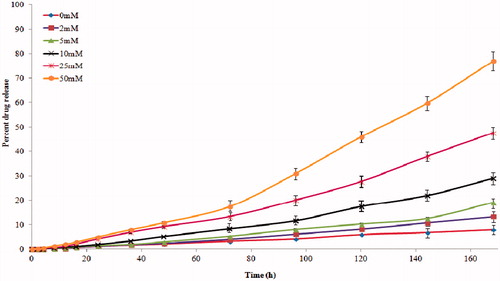
In-vitro drug release
Drug release study was carried out in the absence and in the presence of GHS at different concentrations, 0, 2, 5, 10, 25 and 50 mM (). The drug release from the NS was consistently slow and prolonged over time. The initial drug release was slow due to di-sulphide linkages. It was observed that the drug release proportionally increased with increase in the concentration of GHS. Drug release is induced by interaction of disulphide linkage with the externally added glutathione. Specifically, in the presence of 50 mM GSH, there was about a sixfold increase in the amount of ETB released. These results demonstrated the responsiveness of the NSs to external GSH, hence ensuring drug targeting to cancer cells containing higher concentration of GHS.
Figure 6. In-vitro percent drug release in the presence and absence of GHS.
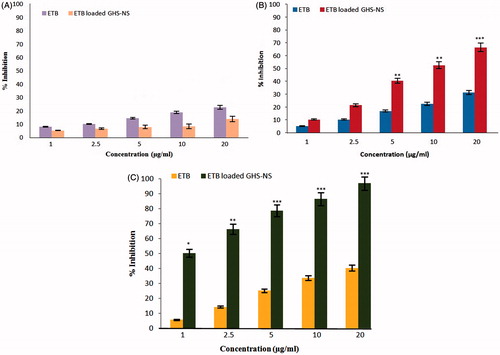
Cell cytotoxicity study
We compared the ability of ETB-NSs and plain ETB to inhibit the growth of A549 cells in the presence of GSH at different concentrations (). Tumour cells having a high intracellular GSH level are more susceptible to receive ETB by NS. Results indicated that there was no difference between control and GHS-NS. There was dose- and time-dependent inhibition in proliferation of A549 cells. shows the cell line study and gives the percent inhibition on proliferation at varying concentrations and at different time intervals. A significant difference was observed between the effects of ETB-NS and plain ETB at 48 h of the study. ETB-NS produced a high inhibitory effect at low dose and for a longer period than plain ETB, which might be due to the retention of an intracellular reducing environment, which permits gradual drug release. This suggests that NS causes internalization of ETB and protects the drug from inactivation or extrusion, thus rendering the low drug doses more effective for a longer time. It can be interpreted that ETB-NS displayed more apparent effects than the plain drug in cell lines having increased concentrations of GSH level [Citation22,Citation27].
Figure 7. (A) Cell line study: percent inhibition of A549 cell lines at different concentration at 24 h. (B) Cell line study: percent inhibition of A549 cell lines at different concentration at 48 h. (C) Cell line study: percent inhibition of A549 cell lines at different concentration at 72 h.
In-vivo pharmacokinetic study
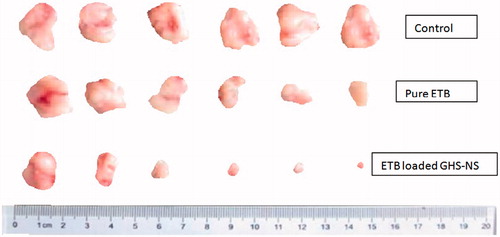
Antitumour efficacy in a xenograft model of lung cancer
The tumour volumes were monitored at regular intervals (). Overall, the tumour volume decreased significantly more on administration of the ETB-NS than with plain ETB. Internalization of drug in the tumour tissues significantly decreases tumour volume. There was 48.51% and 97.73% reduction in tumour volume on administering plain ETB and ETB-NS, respectively, calculated on the 28th day. These results show that ETB-NS may be a more effective cancer therapy and may reduce the dosage of anticancer drugs, ensuring the avoidance of undesired effects [Citation19]. This is the prominent finding of the research.
Figure 8. Antitumour efficacy in a xenograft model of lung cancer.
Drug quantification and biodistribution study
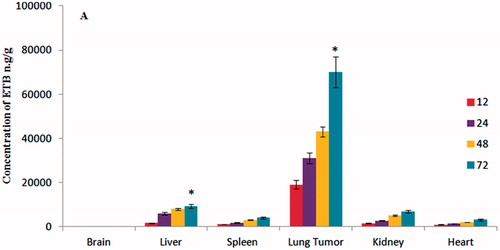
The targeting ability of ETB-NS was determined by comparing the tissue distribution of ETB into various organs such as liver, lung, heart, brain, spleen and kidney with plain ETB. ETB-NS exhibited increased ETB accumulation in tumour tissues while concentrating minimally in the heart, liver, kidney and spleen (). After 72 h of injection of ETB-NS, the ETB concentrations in the tumour tissue were 15.81 (p < .05), 9.04 (p < .05), 7.01 (p < .05) and 15.09 (p < .05) times greater than those in heart, liver, kidney and spleen . ETB was not found in brain, demonstrating inability of ETB to cross the blood–brain barrier. The formulation also exhibited a much stronger and more extensive distribution of drug in tumour foci, i.e. the EPR effect. We believe that the accumulation of ETB-NS was due to the targeting ability of the GHS linkage in the formulation. These results demonstrate that NS can be selectively used to deliver anticancer drugs to lung tumour tissues [Citation22].
Figure 9. Tissue biodistribution of erlotinib after administration of ETB-loaded GHS-NS at different time intervals (12, 24, 48 and 72 h).
Molecular modelling studies
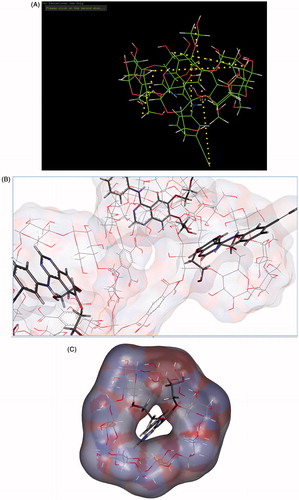
The structure of ETB consists of two aromatic regions (quinazoline ring and phenyl ring). The β-CD cavity is composed of hydrophobic interior of diameter 8–8.5 Aº and width 5–6 A°. Structural investigations of optimized structure of ETB revealed that the length of hydrophobic quinazolinone ring with methoxymethyloxy side chain is approximately 7.3 Aº, where the length of hydrophobic part of ETB occupying β-CD cavity is 6.7 Aº. The length of terminal atoms of ETB in its extended structure is 15.8 Aº. The rim and bottom of β-CD-NS are lipophilic in nature and the oxygen atoms in methoxymethyloxy side chain occupy the rim and amino group at C-4 and occupy the bottom lipophilic surface. The aromatic quinazoline ring and part of phenyl ring accommodate into the hydrophobic GHS-NS cavity (), thus confirming the suitability of GHS-NS for loading of ETB.
Figure 10. (A) Molecular modelling data of ETB-loaded GHS-NS. (B) Molecular modelling data of ETB and GHS-NS interaction. (C) Molecular modelling data of drug entrapment in the GHS-NS.
Conclusion
We have synthesized GHS-NS by a one-step synthetic route which can be used for targeting drugs to nonsmall cell lung cancers. Nanosponge can encapsulate the anticancer drug with high efficiency and exhibit extended release, which is proportional to the external GSH concentration. ETB-NS displays a higher antiproliferative effect than free drug in A549 cell lines having higher GSH concentrations, which might represent a useful approach to treat tumours more resistant to the induction of oxidative stress. From this point of view, the use of GSH-responsive nanosponges might be a promising strategy for the site-specific delivery of ETB, reducing the exposure of nontarget tissue to the drug, thus potentially reducing systemic side effects and therapeutic doses. The bioresponsive NS exhibited excellent cellular drug uptake, extended drug release, in-vivo antitumour efficacy and biodistribution, all these factors make it a promising targeting strategy for cancer therapy. Molecular modelling data also confirm the interaction between ETB and GHS-NS, and its suitability to form a stable complex.
Acknowledgements
The authors are thankful to Cipla Pvt. Ltd. (Mumbai, India) for providing gift samples of erlotinib hydrochloride. Authors are thankful to Ms. Fatima Rafiq Zakaria, Chairman of Maulana Azad Educational Trust, Aurangabad (India) for providing the necessary infrastructure facilities. Special thanks to Dr. Ayaz Ali, Associate Professor, Y.B Chavan College of Pharmacy, Aurangabad (India) for his assistance in performing pharmacokinetic and biodistribution studies.
Disclosure statement
Authors declare no conflict of interest. All authors have contributed in the research.
References
- He Y, Su Z, Xue L, et al. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Controlled Release. 2016;229:89–92.
- Kim ST, Lee J, Kim JH, et al. Comparison of gefitinib versus erlotinib in patients with nonsmall cell lung cancer who failed previous chemotherapy. Cancer. 2010;116:3025–3033.
- Wang Y, Schmid-bindert G, Zhou C. Erlotinib in the treatment of advanced non-small cell lung cancer: an update for clinicians. Ther Adv Med Oncol. 2011;4:19–29.
- Nawaz K, Webster RM. The non-small-cell lung cancer drug market. Nat Rev Drug Discov. 2016;15:229–230.
- Mandal B, Mittal NK, Balabathula P, et al. Development and in vitro evaluation of core–shell type lipid–polymer hybrid nanoparticles for the delivery of erlotinib in non-small cell lung cancer. Financial Res Lett. 2015;81:162–171.
- Sechler M, Cizmic AD, Avasarala S, et al. Non-small-cell lung cancer: molecular targeted therapy and personalized medicine: drug resistance, mechanisms, and strategies. Pharmgenom Pers Med. 2013;6:25–36.
- Qi W, Cooke LS, Stejskal A, et al. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumour growth in prostate cancer. BMC Cancer. 2009;9:142.
- Dora CP, Trotta F, Kushwah V, et al. Potential of erlotinib cyclodextrin nanosponge complex to enhance solubility, dissolution rate, in vitro cytotoxicity and oral bioavailability. Carbohydr Polym. 2015;137:339–349.
- Dowell J, Minna JD, Kirkpatrick P. Erlotinib hydrochloride. Nat Rev Drug Discov. 2005;4:13–14.
- Liversidge G, Jenkins S. Nanoparticulate erlotinib formulations. EP 1871345 B1; 2012.
- Yang KM, Shin IC, Park JW, et al. Nanoparticulation improves bioavailability of erlotinib. Drug Develop Indus Pharm. 2017;43:1557–1565.
- Vrignaud S, Hureaux J, Wack S, et al. Design, optimization and in vitro evaluation of reverse micelle-loaded lipid nanocarriers containing erlotinib hydrochloride. Int J Pharm. 2012;436:194–200.
- Parthasaradhi RB, Rathnakar RK, Raji RR, et al. Erlotinib hydrochloride polymorph Form A substantially free of polymorph Form. B. EP221873A1; 2010.
- Cui Y, Dong H, Cai X, et al. Mesoporous silica nanoparticles capped with disulfide-linked PEG gatekeepers for glutathione-mediated controlled release. ACS Appl Mater Interfaces. 2012;4:3177–3183.
- Shepherd FA, Pereira JR, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132.
- Darandale SS, Vavia PR. Cyclodextrin-based nanosponges of curcumin: formulation and physicochemical characterization. J Incl Phenom Macrocycl Chem. 2013;75:315–322.
- Sherje AP, Darvyaker BR, Kadam D, et al. Cyclodextrin-based nanosponges: a critical review. Carbohydr Polym. 2017;173:37–49.
- Caldera F, Tannous M, Cavalli R, et al. Evolution of cyclodextrin nanosponges. Int J Pharm. 2017. Doi: 10.1016/j.ijpharm.2017.06.072
- Swaminathan S, Cavalli R, Trotta F. Cyclodextrin-based nanosponges: a versatile platform for cancer nanotherapeutics development. WIREs Nanomed Nanobiotechnol. 2016;8:579–601.
- Yang X, He D, He X, et al. Glutathione-mediated degradation of surface-capped MnO 2 for drug release from mesoporous silica nanoparticles to cancer cells. Part Part Syst Charact. 2015;32:205–212.
- Wu G, Fang Y, Yang S, et al. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492.
- Daga M, Ulllio C, Argenziano M, et al. GSH-targeted nanosponges increase doxorubicin induced toxicity ‘in vitro’ and ‘in vivo’ in cancer cells with high antioxidant defences. Free Rad Biol Med. 2016;97:24–37.
- Anajafi T, Mallik S. Polymersome-based drug-delivery strategies for cancer therapeutics. Ther Deliv. 2015;6:521–534.
- Xu Z, Wang D, Xu S, et al. Preparation of a camptothecin prodrug with glutathione-responsive disulfide linker for anticancer drug delivery. Chem Asian J. 2014;9:199–205.
- Wang J, Sun X, Mao W, et al. Tumour redox heterogeneity-responsive prodrug nanocapsules for cancer chemotherapy. Adv Mater. 2013;25:3670–3676.
- Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627.
- Trotta F, Caldera F, Dianzani C, et al. Glutathione bioresponsive cyclodextrin nanosponges. Chempluschem. 2016;81:439–443.
- Shende PK, Gaud RS, Bakal R, et al. Effect of inclusion complexation of meloxicam with β-cyclodextrin- and β-cyclodextrin-based nanosponges on solubility, in vitro release and stability studies. Colloids Surfaces B Biointerfaces. 2015;136:105–110.
- Hariri G, Edwards AD, Merrill TB, et al. Sequential targeted delivery of paclitaxel and camptothecin using a cross-linked ‘nanosponge’ network for lung cancer chemotherapy. Mol Pharmaceutics. 2014;11:265–275.
- Jiang Y, Yang N, Zhang H, et al. Enhanced in vivo antitumour efficacy of dual-functional peptide-modified docetaxel nanoparticles through tumour targeting and Hsp90 inhibition. J Control Release. 2016;221:26–36.
- Ernsting MJ, Tang W, Maccallum NW, et al. Preclinical pharmacokinetic, biodistribution, and anti-cancer efficacy studies of a docetaxel-carboxymethylcellulose nanoparticle in mouse models. Biomaterials. 2012;33:1445–1454.
- Alexis F, Pridgen E, Molnar LK, et al. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515.
- Mailänder V, Landfester K. Interaction of nanoparticles with cells. Biomacromolecules. 2009;10:2379–2400.
- Shandiz SAS, Ardestani AS, Shahbazzadeh D, et al. Novel imatinib-loaded silver nanoparticles for enhanced apoptosis of human breast cancer MCF-7 cells. Artif Cells Nanomed Biotechnol. 2017;45:1–10.
- Mehdizadeh M, Rouhani H, Sepehri N, et al. Biotin decorated PLGA nanoparticles containing SN-38 designed for cancer therapy. Artif Cells Nanomed Biotechnol. 2016;45:495–504.
- Cho EC, Xie J, Wurm PA, et al. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009;9:1080–1084.
- Xiao W, Chen W-H, Xu X-D, et al. Design of a cellular-uptake-shielding plug and play template for photo controllable drug release. Adv Mater Weinheim. 2011;23:3526–3530.
- Perrault SD, Walkey C, Jennings T, et al. Mediating tumour targeting efficiency of nanoparticles through design. Nano Lett. 2009;9:1909–1915.