
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Proto-oncogene non-receptor tyrosine protein kinase c-Src has been involved in the development, progression and metastasis of a variety of human cancers. This protein contains two self-binding peptide (SBP) sites separately between the SH3 domain and polyproline-II (PPII) helix and between the SH2 domain and C-terminal phosphorylatable tail (CTPT), which are potential targets of anticancer drugs to regulate the kinase activity. Here, we described an integrated protocol to systematically investigate the structural basis, energetic property and dynamics behaviour of PPII binding to SH3, and to rationally design potent peptide ligands to target the SBP site of SH3–PPII interaction. Our study found that the PPII peptide is a non-typical binder that can only interact effectively with its cognate SH3 domain when it is integrated into the full-length c-Src kinase protein; stripping the peptide from the protein would considerably impair SH3 affinity by increasing entropy penalty upon the domain–peptide binding, suggesting that the protein context plays an essential role in the SBP’s biological function. Next, we identified that the PPII peptide binds to SH3 domain in a class II manner and, on this basis, we derived a series of modified versions of the wild-type PPII peptide using a structure-based rational strategy. These modified peptide mutants have been structurally optimized with respect to their molecular flexibility and interaction potency with SH3 domain, in order to minimize indirect entropy penalty and to maximize direct binding enthalpy simultaneously. Consequently, several rationally designed peptides were obtained, including PPIIm2 (TSKPQTPGRA), PPIIm5 (KPPTPPRA), PPIIm6 (FPPPPPRA) and PPIIm7 (YPPLPPRA), which exhibit a moderately or considerably increased affinity (Kd = 72, 34, 15 and 5.7 μM, respectively) relative to the wild-type PPII (TSKPQTQGLA) (Kd = 160 μM). These peptides can be used as lead molecular entities to further develop new anticancer therapeutics to regulate c-Src kinase activity by targeting the SBP site of SH3–PPII interaction.
Introduction
Intramolecular interactions are established across biomolecular interior and serve as the primary chemical forces leading to protein folding and stabilizing protein globule in solution and crystal state. The intramolecular interactions in monomeric proteins are commonly observed as domain–domain interaction and domain–peptide interaction [Citation1]; the former is formed between two structural domains packed against each other, known as domain-mediated intramolecular interactions (DMIntraIs), while the latter is fulfilled by binding of a flexible peptide stretch to a rigid globular domain, namely peptide-mediated intramolecular interactions (PMIntraIs) [Citation2]. Unlike to those peptide-mediated intermolecular interactions (PMInterIs) they are widely existed in cellular signalling network and ideal for mediating interactions that are easily formed and disrupted, and required as fast response to stimuli [Citation3], PMIntraIs are normally static and functioned as structural stabilizers to maintain and shape protein architecture.
In contrast to the widely distrusted static PMIntraIs, we have described a novel dynamic PMIntraIs, termed as self-binding peptides (SBPs) [Citation4], which represent those short peptide segments in monomeric proteins that can fulfil biological functions by dynamically binding to/unbinding from their cognate domains in the same monomers. In a SBP system, peptide segment, on the one hand, specifically recognizes and interacts with its cognate target to establish a dynamic balance between the bound adduct and unbound state; on the other hand, the segment is integrated to the target in primary sequence via a flexible polypeptide linker. Previously, we demonstrated that, in structural and energetic points of view, SBPs are almost a binding phenomenon, although they are traditionally considered as folding owing to their sequence connectivity with target [Citation4]. We also proposed a two-step binding mechanism for SBP–domain recognition to explain the binding dynamics of the protein intramolecular interaction [Citation5]. Based on these theoretical works, the SBPs have recently been raised as a new and promising class of druggable targets; it is suggested that disrupting SBPs can be used as a feasible strategy to regulate the biological activity and function of oncogenic protein c-Src [Citation6], a non-receptor tyrosine protein kinase that is closely associated with a variety of malignant solid tumours such as colon, liver, lung, breast and pancreas [Citation7].
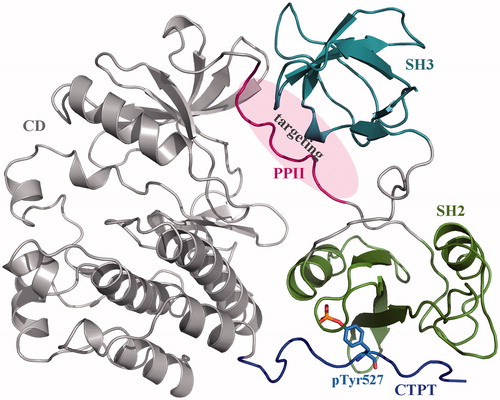
The human c-Src is a monomeric protein consisting of two peptide-recognition domains SH3 and SH2 as well as a catalytic domain CD. The protein also contains two SBPs: one is a polyproline-II (PPII) helical peptide between the SH2 and CD domains, and another is the C-terminal phosphorylatable tail (CTPT) that has to complex with SH2 domain once its Tyr527 residue is phosphorylated (pTyr527). The complex can further induce PPII bound tightly to SH3 domain and, consequently, the CD domain is locked in an autoinhibitory form () [Citation8]. Therefore, it is expected that disruption of SH3–PPII recognition and interaction can influence c-Src packing state and then regulate the kinase activity. Here, we attempted to target the PMIntraI between c-Src SH3 domain and its SBP (i.e. PPII) with rationally designed peptide ligands, and to systematically investigate the structural basis, energetic property and dynamics behaviour of c-Src kinase underlying the disruption. The effect of protein context on PPII binding to SH3 domain was also examined in detail.
Figure 1. Crystal structure of human c-Src kinase (PDB: 2SRC). c-Src is a monomeric protein consisting of three domains (a catalytic domain CD and two peptide-recognition domains SH3 and SH2) and two SBPs (polyproline-II helical peptide PPII and C-terminal phosphorylatable tail CTPT). Phosphorylation of Tyr527 residue (pTyr527) results in CTPT binding to SH2 domain, which further induces SH3–PPII interaction. Consequently, the CD domain is locked in an autoinhibitory form.
Materials and methods
Dynamic and energetic analyses
MD simulations were carried out using AMBER ff03 force field [Citation9] implemented in the AMBER package [Citation10]. A truncated octahedral box of TIP3P water molecules was added with a 12 Å buffer around the investigated systems. Counterions of Na+ were placed based on the Columbic potential to keep the whole systems electroneutral. The steepest descent and conjugate gradient algorithm energy minimizations were performed to remove bad contacts between protein and water molecules. After the minimizations, systems were heated to 300 K over 500 ps followed by constant temperature equilibration at 300 K for 1 ns. Subsequently, MD equilibrium simulations were carried out in an isothermal isobaric ensemble with periodic boundary conditions, during the last production phase a total of 500 snapshots were evenly saved for subsequent energetic analysis. An integration step of 2 fs was used for the MD simulations and the particle mesh Ewald (PME) method [Citation11] was employed to analyse the long-range electrostatic energy of a unit cell in a macroscopic lattice of repeating images. A cut-off distance of 10 Å was considered to calculate the short-range electrostatics and van der Waals interactions. The SHAKE strategy [Citation12] was used to restrain all covalent bonds involving hydrogen atoms.
The domain–peptide binding free energy ΔGttl was calculated based on the structure snapshots extracted over the MD equilibrium phase using molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) method [Citation13]:
(1)
(1) where ΔEint is the nonbonded interaction energy between domain and peptide, which can be divided into electrostatic (ΔEelc) and van der Waals (ΔEvdW) potentials and was calculated with molecular mechanics (MM) approach. ΔGslv is the solvent effect associated with the interaction, which is contributed from polar (ΔGplr) and nonpolar (ΔGnplr) desolvations; the polar aspect was calculated by numerical solution of the nonlinear Poisson–Boltzmann (PB) equation, while the nonpolar facet was described using surface model (SA) as ΔGnplr = β+γ·SASA [Citation14]. The grid size for the PB calculations was set to 0.5 Å, and the interior and exterior dielectric constants were 2 and 78, respectively. –TΔS is the conformational free energy due to entropy penalty of the binding, which was computed by normal mode analysis (NMA) [Citation15].
Binding affinity assay
The peptides PPII (TSKPQTQGLA), PPIIm2 (TSKPQTPGRA), PPIIm5 (KPPTPPRA), PPIIm6 (FPPPPPRA) and PPIIm7 (YPPLPPRA) were synthesized via commercial approach. GST-tagged human c-Src SH3 domain (residues 85–140) was expressed as a fusion protein in Escherichia coli BL21. Briefly, bacteria were lysed in buffer 5 mM DTT, pH 7.3, 100 mM NaCl, 3 mM KCl and 10 mM Na2HPO4. The bacterial lysate was incubated with GST Bind Resin for 2 h at 4 °C. The domain proteins were eluted in buffer 5 mM DTT, pH 8.0, 40 mM Tris–HCl and 20 mM reduced glutathione.
Fluorescence analysis of peptide binding to c-Src SH3 domain was carried out at room temperature using a protocol modified from our previous work [Citation6]. Briefly, the changes in fluorescence emission spectra of domain protein upon peptide binding were monitored using a Perkin-Elmer fluorescence spectrophotometer. The excitation wavelength was 280 nm (4 nm slit), and the emission wavelength was 350 nm. To measure the dissociation constant (Kd), peptides were added to protein solution (1 μM) until no significant dependent changes in fluorescence intensity and maximum wavelength were observed. Binding constants were calculated from the 1:1 stoichiometry by nonlinear fitting of titration curves [Citation16].
Results and discussion
Protein context plays a crucial role in native SH3–PPII binding
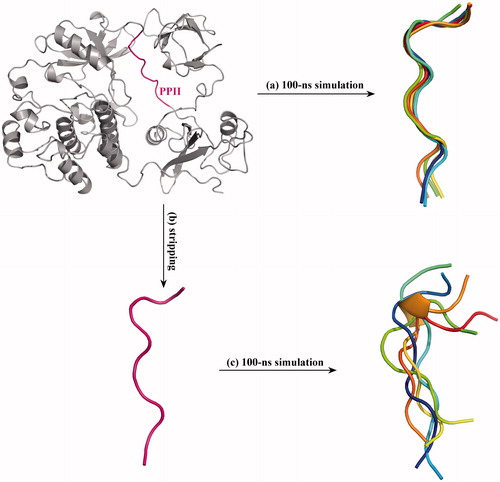
As can be seen in , the PPII is located between the SH2 and CD domains of c-Src kinase, which is tightly packed in the kinase protein context. It is worth noting that the term protein context indicates the whole protein system, which does not only mean the residues that covalently connect with the PPII in primary sequence, but also contains the spatial effect of intramolecular interactions with other regions of the protein. By surveying the crystal structure data of peptide-mediated protein interactions, Stein and Aloy [Citation17] found that the context plays a crucial role in determining interaction stability and specificity by either improving the affinity with the native partner or impeding non-native interactions. In addition, it is known that the c-Src PPII (247TSKPQTQGLA256) is a non-typical SH3 ligand that possesses only one proline (P250) residue but does not contain the consensus SH3 recognition motif PxxP [Citation18]. Thus, it is suggested that the protein context may help SH3–PPII binding. To verify this notion, full-length c-Src kinase as well as the PPII peptide stripped from the kinase protein was separately subjected to 100-ns MD simulations. Over the simulations, five structural snapshots of PPII (either in full-length kinase protein or in stripped state) were saved at 0, 25, 50, 75 and 100 ns, and superposed onto each other. As compared in , the PPII segment in protein context exhibits low disorder and thermal motion [root-mean-square deviation (rmsd) (Cα) = 0.35 Å over the simulations], which can be well structured as a typical left-handed helix of poly-l-proline II that is suited for interacting with SH3 domain (). In contrast, the stripped PPII peptide is highly flexible [rmsd (Cα) = 1.86 Å over the simulations] that cannot hold in the poly-l-proline II helical structure and thus cannot be recognized by SH3 domain directly ().
Figure 2. Superposition of MD-derived PPII structural snapshots. (a) The crystal structure of full-length c-Src protein was subjected to 100-ns MD simulations, and the structural snapshots of PPII segment in the full-length protein were saved at 0, 25, 50, 75 and 100 ns and superposed onto each other. (b) The PPII peptide was stripped from the c-Src crystal structure, (c) which was then subjected to 100-ns MD simulations, and its structural snapshots were saved at 0, 25, 50, 75 and 100 ns and superposed onto each other.
According to our previous investigation, protein–peptide recognition is co-dominated by direct readout of the intermolecular interaction between protein receptor and peptide ligand, and by indirectly readout of entropy penalty upon the interaction [Citation19]. In this respect, it is suggested that protein context can largely improve the structural rigidity of PPII segment in c-Src kinase and effectively minimize the entropy penalty involved PPII binding to SH3 domain. Here, the binding energetics of PPII segment (in full-length c-Src) and PPII peptide (in stripped state) to SH3 domain were calculated and decomposed to interaction energy (ΔEint), solvent effect (ΔGslv) and entropy penalty (–TΔS) [Citation20]. As listed in , the total binding free energies (ΔGttl) of PPII segment and PPII peptide to SH3 domain are −7.4 and −5.2 kcal/mol, respectively, indicating that the former can bind tightly to the domain as compared to the latter, albeit both share the same sequence. As can be seen, although the stripped PPII peptide has higher interaction energy than PPII segment (ΔEint = –103.4 versus –98.2 kcal/mol), the solvent effect and, in particular, entropy penalty of stripped peptide are considerably unfavourable as compared to intact segment (ΔGslv = 58.4 versus 68.3 kcal/mol and –TΔS = 43.8 versus 22.5 kcal/mol). The large entropy penalty of free peptide binding to SH3 domain is expected since the binding should reduce a considerable degree of freedom of the highly flexible peptide. Here, the binding affinity of stripped PPII peptide to SH3 domain was determined as Kd = 160 μM using fluorescence-based assay, which is quite moderate as compared to that of some other known c-Src SH3 binders, such as PLR1, RLP2, Sos and mSosl. These peptides were obtained from the native partner proteins of c-Src kinase but have been measured to have a higher SH3 affinity than the PPII peptide (Kd = 59, 8, 67 and 5.7 μM, respectively, [Citation21] (). By examining peptide sequence patterns, it is found that the four peptides possess the standard PxxP motif and contain three or more proline residues. It is known that the proline is an important conformational restrictor for protein and peptide owing to its closed ring side chain. Thus, the proline-rich peptides PLR1, RLP2, Sos and mSosl should have low flexibility and intrinsic disorder relative to PPII peptide. This can be substantiated by rmsd of backbone Cα fluctuation during 100-ns MD simulations of these isolated peptides in solution. It is evident in that the four peptides have smaller rmsd values (0.68, 0.55, 0.97 and 0.84 Å, respectively) than PPII peptide (rmsd = 1.86 Å); this could be attributed to the conformational constraint addressed by proline residues involved in these proline-rich peptides. As might be expected, the four proline-rich peptides also show lower entropy penalty (–TΔS < 35.4 kcal/mol) and higher binding energy (ΔGttl< −5.6 kcal/mol) as compared to PPII peptide (–TΔS = 43.8 kcal/mol and ΔGttl = −5.2 kcal/mol). Overall, it is suggested that the PPII segment does not contain the standard binding motif PxxP of SH3 domain and should be restricted into a structured poly-l-proline II helical conformation only within the full-length c-Src protein context; stripping PPII segment from protein context would largely increase the PPII’s flexibility and thus incur considerable entropy penalty upon binding to SH3 domain.
Table 1. The binding energetics and affinity of peptide ligands to c-Src SH3 domain.
Optimization of the sequence pattern of c-Src SH3 peptide binders
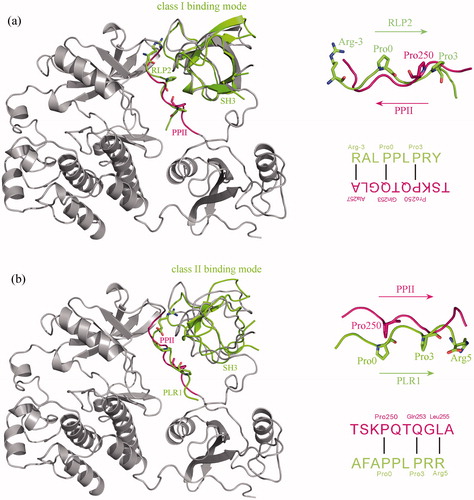
Above structural, energetic and dynamics investigations revealed that the nonstandard PPII peptide cannot bind tightly to c-Src SH3 domain in stripped state, which can only interact effectively with the domain when it is integrated into the full-length protein context; proline-rich peptides containing the standard PxxP motif seem to be good candidates of c-Src SH3 binders. Considering that peptide ligands can bind to c-Src SH3 domain in two opposite orientations [Citation21] and both class I (+xxPxxP) and class II (PxxPx+) proline-rich peptides are recognized by the domain [Citation22], we herein examined the binding mode of PPII segment (247TSKP250QTQGLA256) to SH3 domain in c-Src kinase. Previously, Feng et al. reported the crystal structures of c-Src SH3 domain in complex with class I peptide ligand RLP2: R−3ALP0PLP3RY (PDB: 1PRL) and class II peptide ligand RLP1: AFAP0PLP3RR5 (PDB: 1RLP), which were separately superposed onto the SH3–PPII adduct of c-Src kinase crystal structure (PDB: 2SRC). It is evident in that the binding orientation of PPII segment in domain pocket is consistent with the PLR1 peptide, where the PPII residues Pro250, Gln253 and Leu255 match well with the three key motif residues Pro0, Pro3 and Arg5 of PLR1 peptide, respectively (see ), suggesting that the native binding mode of PPII segment to SH3 domain is class II, albeit the PPII does not contain a typical class II motif.
Figure 3. Superposition of class I c-Src SH3–PLR2 complex crystal structure (PDB: 1PRL) (a) and class II c-Src SH3–PLR1 complex crystal structure (PDB: 1RLP) (b) onto the SH3–PPII adduct of c-Src kinase crystal structure (PDB: 2SRC). From the superposition the binding mode of PPII to SH3 domain is revealed as class II, where the three key residues Pro0, Pro3 and Arg5 of class II motif P0xxP3x+5 in PLR1 peptide can well match the residues Pro250, Gln253 and Leu255 of PPII, respectively.
Next, the SH3–PPII adduct portion was separated from the crystal structure of full-length c-Src kinase, which was then subjected to 100-ns MD simulations for structural relaxing and dynamics equilibrium. The equilibrium conformation of SH3–PPII complex is shown in . It is seen that the PPII peptide can bind to SH3 domain in a standard poly-l-proline II helical conformation, where the 10 peptide residues seem to play different role in the domain–peptide binding. As described above, the Pro250, Gln253 and Leu255 correspond to the three key motif residues and their side-chains do not point to the domain. However, as can be seen in , all the three residues can form hydrogen bonds with SH3 domain through their backbone, suggesting that mutation of these residues would not influence the backbone interaction substantially [Citation23]. In this consideration, the residue Gln253 was mutated to Pro253, resulting in peptide PPIIm1 (TSKPQTPGLA), and then the residue Leu255 was mutated separately to Arg255 and Lys255, resulting in peptides PPIIm2 (TSKPQTPGRA) and PPIIm3 (TSKPQTPGKA), respectively, that meet the consensus class II binding motif PxxPx+. The binding energetics of PPIIm1, PPIIm2 and PPIIm3 to SH3 domain was calculated and decomposed using MM/PBSA/NMA (). As might be expected, the PPIIm1 has lower flexibility (rmsd = 1.42 Å) and entropy penalty (–TΔS = 39.2 kcal/mol) as compared to the wild-type PPII peptide (rmsd = 1.86 Å and –TΔS = 45.8 kcal/mol), although the mutation seems to impair the domain–peptide interaction stability slightly (ΔEint changes from –103.4 to –99.6 kcal/mol). The further mutated versions PPIIm2 and PPIIm3 have also improved binding capability relative to the PPIIm1, with ΔGttl increase from −4.5 to −5.6 and −5.2 kcal/mol, respectively, albeit the increase is moderate. This is anticipated if considering that the positively charged Arg and Lys can interact with the domain via long-range electrostatic forces.
Figure 4. The intermolecular interaction between c-Src SH3 domain and PPII peptide. (a) The equilibrium conformation of c-Src SH3 domain in complex with PPII peptide. The residue positions that play different roles in the domain–peptide binding are highlighted in different colours. Magenta (Pro250, Gln253 and Leu255): key residues in the PxxPx + motif. Indigo (Lys249 and Thr252): the residues pointing to the domain pocket. Yellow (Gln251 and Gly254): the residues pointing out of the domain pocket. Green (Thr247 and Ser248): the two N-terminal residues that are far away from the domain. Hoar (Ala256): the C-terminal residue that interacts slightly with the domain. (b) Hydrogen bonds and hydrophobic interactions across the complex interface of c-Src SH3 domain with PPII peptide (produced using LigPlot program, Citation23]. (c) Computational alanine scanning of PPII peptide binding to c-Src SH3 domain. Positive and negative ΔΔGAla values indicate favourable and unfavourable residues in the domain–peptide binding, respectively.
![Figure 4. The intermolecular interaction between c-Src SH3 domain and PPII peptide. (a) The equilibrium conformation of c-Src SH3 domain in complex with PPII peptide. The residue positions that play different roles in the domain–peptide binding are highlighted in different colours. Magenta (Pro250, Gln253 and Leu255): key residues in the PxxPx + motif. Indigo (Lys249 and Thr252): the residues pointing to the domain pocket. Yellow (Gln251 and Gly254): the residues pointing out of the domain pocket. Green (Thr247 and Ser248): the two N-terminal residues that are far away from the domain. Hoar (Ala256): the C-terminal residue that interacts slightly with the domain. (b) Hydrogen bonds and hydrophobic interactions across the complex interface of c-Src SH3 domain with PPII peptide (produced using LigPlot program, Citation23]. (c) Computational alanine scanning of PPII peptide binding to c-Src SH3 domain. Positive and negative ΔΔGAla values indicate favourable and unfavourable residues in the domain–peptide binding, respectively.](/cms/asset/941e1ea4-2936-449a-8115-380a6403bce8/ianb_a_1360327_f0004_c.jpg)
Table 2. The binding energetics and affinity of PPII-derived peptides to c-Src SH3 domain.
It is seen from that the two N-terminal residues Thr247 and Ser248 are far away from the domain, suggesting that they may only play a marginal role in the domain–peptide binding. Therefore, we just deleted the two residues to generate a truncated version of PPIIm2 peptide, namely PPIIm4 (KPQTPGRA) and recalculated its binding energetics to the domain. As seen in , the binding potency of the truncated PPIIm4 has only a very modest decrease relative to PPIIm2, with ΔGttl change from –5.6 to –5.3 kcal/mol, but the shorter PPIIm4 exhibits lower flexibility and smaller entropy penalty than the longer PPIIm2 (–TΔS = 32.6 versus 42.0 kcal/mol).
The peptide residues Gln251 and Gly254 point out of the domain pocket and should contribute limitedly to the domain–peptide binding (). Thus, we herein considered mutating them to Pro for improving the peptide rigidity, resulting in PPIIm5 peptide (KPPTPPRA), which, as expected, has a decreased flexibility and small entropy penalty as compared to PPIIm4 (–TΔS changes from 32.6 to 25.9 kcal/mol). In addition, the mutation can also moderately reduce the unfavourable solvent effect involved in peptide binding (ΔGslv changes from 64.8 to 62.4 kcal/mol), as proline is a nonpolar amino acid that can be compatible well with the hydrophobic pocket of SH3 domain. Consequently, the total binding energy of PPIIm5 is improved substantially as compared to PPIIm4 (ΔGttl increases from −5.3 to −6.7 kcal/mol).
In order to substantiate the computational findings, two rationally designed mutants PPIIm2 and PPIIm5 were selected, and their binding affinities towards c-Src SH3 domain were determined as Kd = 72 and 34 μM, respectively, using fluorescence-based assays. As described above, the PPIIm2 contains a complete class II binding motif (PxxPx+) and the PPIIm5 is a further mutation to restrict the peptide conformation. Consistently, the two peptide mutants exhibit moderately or considerably increased potency as compared to the wild-type PPII (Kd = 160 μM), and PPIIm5 can bind more tightly than PPIIm2, suggesting that the theoretical prediction is well in line with experimental analysis, which can be used to guide the rational peptide design and optimization.
Structure-based redesign of anchor residues Lys249 and Thr252
It is shown in that the side-chains of charged/polar residues Lys249 and Thr252 point directly to the peptide-binding pocket of SH3 domain; nonbonded analysis also found that the two residues can separately form a salt bridge and a hydrogen bond with the domain residues Asp91 and Arg95, respectively (). As can be seen in , both the two residues locate in an amphipathic microenvironment that are surrounded by a number of nonpolar/aromatic domain residues (Tyr90, Tyr92, Pro133, Tyr136 and Trp118) and polar/charged domain residues (Asp91, Arg95 and Asn135). In this respect, the peptide Lys249 and Thr252 are suggested as anchor residues that should interact effectively with the domain. This can be further solidified by computational alanine scanning. As shown in , most of native PPII residues (Ser248, Lys249, Pro250, Thr252, Gln253 and Leu255) play a favourable role (ΔΔGAla>0) in the domain–peptide binding, where the Lys249 and Thr252 are most important as mutation of them to neutral Ala residue would cause >1 kcal/mol binding free energy loss for the peptide ligand. In addition, there are also few residues (Thr247, Gln251 and Gly254) that contribute unfavourably (ΔΔGAla<0) to the peptide binding, albeit the unfavourable effect is very modest.
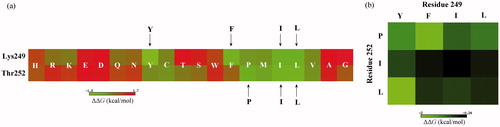
We systematically mutated the two anchor residues to other 19 amino acid types and calculated the change in MM/PBSA-derived peptide binding free energy upon mutation. The mutation was performed on the PPIIm5 peptide in complex with c-Src SH3 domain using the rotamer-based SCWRL4 program [Citation24], followed by a force field minimization to relax the mutated complex systems. Consequently, a (2 × 19) matrix representing the systematic single-point mutation profile for Lys249 and Thr252 was obtained, where element m(i,j) of the matrix indicates the domain–peptide binding free energy change ΔΔG upon mutation of peptide ith anchor residue (Lys249 or Thr252) to other amino acid type j (j = 1, 2, 3 ··· 19). In this way, we can obtain the favourable (ΔΔG < 0) and unfavourable (ΔΔG > 0) mutations at the two anchor residues (). It is evident that most mutations are unfavourable to the peptide binding; this is expected because parent peptide PPIIm5 has already been optimized to possess a moderately high potency (Kd = 34 μM) and, hence, mutations generally impair the potency. However, there are also few mutations that can substantially improve the peptide binding, including the amino acids Y, F, I and L at Lys249 residue and the amino acids P, I and L at Thr252 residue. As can be seen, these favourable substitutions are all hydrophobic and/or aromatic amino acid types; their physicochemical property differs significantly from that of the native charged Lys249 and polar Thr252.
Figure 5. The mutation energy profile of c-Src SH3–peptide interaction. The mutation energies (ΔΔG) are coloured by binding energy change upon PPIIm5 peptide residue mutation. (a) Systematic single-point mutation profile of the anchor residues Lys249 and Thr252 of PPIIm5 peptide, where each residue was mutated to all other 19 amino acid types. (b) Favourable double-point mutation profile of the anchor residues Lys249 and Thr252 of PPIIm5 peptide, where the mutation only consider the combination of four favourable amino acids Y, F, I and L at residue 249 and three favourable amino acids P, I and L at residue 252.
Theoretically, these favourable amino acids at the two anchor residue positions could work together to effectively promote peptide binding, albeit they were analysed in an independent binding manner. This is reasonable if considering that protein–peptide recognition commonly meets the independent binding of side-chains (IBS) hypothesis [Citation25], which is expected to be also applicable for the current case since the peptide ligands adopt a linearly extended conformation to interact with SH3 domain. Therefore, a (4 × 3) matrix representing the systematic combination of four amino acids (Y, F, I and L) at Lys249 and the amino acids (P, I and L) at Thr252 was generated, where element m(i,j) of the matrix is the domain–peptide binding free energy change ΔΔG upon mutation of two peptide anchor residues Lys249 and Thr252 to amino acid types i (i= Y, F, I and L) and j (j= P, I and L). It is seen from that all the 12 double-point mutations can modestly or moderately improve peptide binding, with ΔΔG range between –0.24 and –2 kcal/mol. In particular, the double-point mutations (Lys249Phe/Thr252Pro) and (Lys249Tyr/Thr252Leu) were predicted to largely increase the peptide binding capability (ΔΔG=–1.95 and –2.01 kcal/mol, respectively). Here, the two promising mutants PPIIm6 (FPPPPPRA) and PPIIm7 (YPPLPPRA) were synthesized and purified, and their binding potency was assayed against the recombinant protein of human c-Src SH3 domain. Consistently, the two designed peptides exhibit a moderately and considerably increased affinity (Kd = 15 and 5.7 μM, respectively) relative to PPIIm5 (Kd = 34 μM). Previously, the 12-mer peptide VSLARRPLPPLP and its analogue APPLPPRNRPRL have been discovered by Feng et al. [Citation26] as potent binders of c-Src SH3 domain, which exhibited SH3-binding potency at submicromolar level (Kd = 0.45 and 1.2 μM, respectively). Although the two previously discovered peptides have higher affinity than the peptide mutants designed in the current work, they are much longer and not derived from the cognate PPII sequence of c-Src SH3 domain, and thus cannot be regarded as self-inhibitory peptides. Here, we attempt to investigate the structural basis, energetic property and dynamics behaviour of SH3–SBP recognition and interaction and, based on the harvested knowledge, to optimize and refine the SBP peptide with increased affinity that can competitively target the SH3–SBP interaction. In this respect, the designed peptides of this study may not have the highest binding affinity for c-Src SH3 domain in all discovered SH3 binders. As pointed out, the complex conformations of c-Src SH3 domain with the wild-type PPII and the designed high-activity mutant PPIIm6 were equilibrated using MD simulations. Based on equilibrated conformations, the nonbonded interaction patterns across the two complex interfaces were identified using PLIP server [Citation27] and shown in . Evidently, the two N-terminal residues Thr247 and Ser248 of PPII peptide do not interact effectively with SH3 domain and the C-terminal residue Ala256 of the two peptides (PPII and PPIIm6) can only form a weak hydrophobic contact with the domain Trp118 residue. Thus, the two termini should contribute limitedly to peptide binding. There are a similar nonbonded profile between the residue positions 250, 251, 253 and 255 of the two peptides, suggesting that the mutations would not influence the established domain–peptide interactions essentially. However, it is worth noting that the mutations of two peptide anchor residues 249 and 252 can reshape their nonbonded interaction patterns with the domain. The PPII Lys249 can form a salt bridge with domain Asp91, which was replaced by a hydrogen bond with domain Asp91 as well as an additional hydrophobic force and a π···π stacking with domain Tyr92 when mutating the peptide Lys249 to Tyr249. The PPII Thr252 can form a specific hydrogen bond with domain Arg95, which is substituted by three nonspecific hydrophobic forces with domain Tyr92, Pro133 and Tyr136 upon the peptide Thr252Leu mutation. In addition, the peptide mutation Gly254Pro can define a new hydrophobic contact with domain Trp118, which is also considered to improve peptide rigidity and to minimize entropy penalty for domain–peptide binding.
Figure 6. Comparison between the nonbonded interaction patterns across the complex interfaces of c-Src SH3 domain with PPII and PPIIm7 peptide ligands. The nonbonded interactions were identified using PLIP server [Citation27].
![Figure 6. Comparison between the nonbonded interaction patterns across the complex interfaces of c-Src SH3 domain with PPII and PPIIm7 peptide ligands. The nonbonded interactions were identified using PLIP server [Citation27].](/cms/asset/31ab0508-aab1-449e-a768-91263c273481/ianb_a_1360327_f0006_c.jpg)
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 31671361 and 31200993), the Science and Technology Project of Sichuan Province (No. 2015JY0252) and the Fundamental Research Funds for the Central Universities of China (Nos. ZYGX2016J186 and ZYGX2015Z006).
Disclosure statement
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Additional information
Funding
References
- Makarov AA, Monaselidze DR, Esipova NG. Intermolecular interactions of globular proteins in the crystal state. Int J Quantum Chem. 1979;16:437–444.
- Underwood DJ. Protein structures from domain packing-a game of twenty questions? Biophys J. 1995;69:2183–2184.
- Petsalaki E, Russell RB. Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol. 2008;19:344–350.
- Yang C, Zhang S, He P, et al. Self-binding peptides: folding or binding? J Chem Inf Model. 2015;55:329–342.
- Yang C, Zhang S, Bai Z, et al. A two-step binding mechanism for the self-binding peptide recognition of target domains. Mol Biosyst. 2016;12:1201–1213.
- Bai Z, Hou S, Zhang S, et al. Targeting self-binding peptides as a novel strategy to regulate protein activity and function: a case study on the proto-oncogene tyrosine protein kinase c-Src. J Chem Inf Model. 2017;57:835–845.
- Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667–678.
- Hubbard SR. Src autoinhibition: let us count the ways. Nat Struct Biol. 1999;6:711–714.
- Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012.
- Case DA, Cheatham TE, 3rd, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) Method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092.
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341.
- Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson–Boltzmann surface area method. Mol Inform. 2012;31:114–122.
- Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897.
- Case D. Normal mode analysis of protein dynamics. Curr Opin Struct Biol. 1994;4:285–290.
- Schweimer K, Hoffmann S, Bauer F, et al. Structural investigation of the binding of a herpesviral protein to the SH3 domain of tyrosine kinase Lck. Biochemistry. 2002;41:5120–5130.
- Stein A, Aloy P. Contextual specificity in peptide-mediated protein interactions. PLoS One. 2008;3:e2524.
- Saksela K, Permi P. SH3 domain ligand binding: what's the consensus and where's the specificity? FEBS Lett. 2012;586:2609–2614.
- Yu H, Zhou P, Deng M, et al. Indirect readout in protein–peptide recognition: a different story from classical biomolecular recognition. J Chem Inf Model. 2014;54:2022–2032.
- Li N, Wei M. Conversion of MIG6 peptide from the nonbinder to binder of lung cancer-related EGFR by phosphorylation and cyclization. Artif Cells Nanomed Biotechnol. 2017;45:1023–1028.
- Feng S, Chen JK, Yu H, et al. Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3–ligand interactions. Science. 1994;266:1241–1247.
- Kami K, Takeya R, Sumimoto H, et al. Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67(phox), Grb2 and Pex13p. EMBO J. 2002;21:4268–4276.
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein–ligand interactions. Protein Eng. 1995;8:127–134.
- Krivov GG, Shapovalov MV, Dunbrack RL. Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795.
- Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–175.
- Feng S, Kasahara C, Rickles RJ, et al. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc Natl Acad Sci USA. 1995;92:12408–12415.
- Salentin S, Schreiber S, Haupt VJ, et al. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447.