
Abstract
Even after huge strides in medicine, cancer continues to be a formidable disease, which is slated to become the leading cause of death worldwide. The present study investigates the 1,3-benzodioxole and its propargyl ether derivatives as a novel histone deacetylase enzyme inhibitor in order to cure cancer, as aberrant expression of histone deacetylases (HDACs) is associated with carcinogenesis. Bioinformatics approaches were employed to carry out preclinical and pharmacological evaluations of designed benzodioxole derivatives. Furthermore, their interaction with HDAC-1 enzyme was studied through computational methods for their specific inhibitory effects and evaluated for their LD50 (oral rat acute toxicity) value. In addition to this work, three-dimensional (3D) structure of HDAC-1 enzyme was extracted and evaluated using various parameters including Ramachandran plot and molecular docking stimulation. In our study, we found that compound 7 and compound 9 have higher binding score than approved drugs (SAHA, TSA and VPA). Importantly, these compounds were found to possess good pharmacological and pharmacokinetic properties and can be considered as potent novel compound to combat the HDAC-1 enzyme to cure cancer. Compounds were also analyzed and validated with parameters like absorption, metabolism, excretion, toxicity and synthetic accessibility during the preclinical evaluation. This study paves way to search for novel and potent small chemical compounds for inhibiting HDAC-1 enzyme and in particular to combat the cancer progression by interrupting the cell cycle.
Introduction
Cancer is an uncontrolled cell growth. It has the potential to invade or spread to other parts of the body. According to the reports of American Chemical Society [Citation1], it is expected that about 1,688,780 new cancer cases will be diagnosed in 2017. In America, around 600,920 people are expected to die of cancer in 2017. In India, around 14.5 lakh new cancer cases were reported in 2016 [Citation2], and the figure is likely to reach nearly 17.3 lakh new cases in 2020. Worldwide, Cancer is the second most common cause of death, exceeding only by heart disease.
Conventionally, cancer is considered as a genetic disorder that leads to activation of oncogenes or loss of tumour suppressor genes [Citation3]. However, tumour development can also be caused by epigenetic changes like histone acetylation [Citation4]. HDAC is a class of enzymes that regulates the acetylation and deacetylation of histone, thus playing a crucial role in the modification of chromatin structure and gene expression [Citation5,Citation6]. Aberrant HDAC activity leads to tumour formation. Inactivation of histone acetyltransferase is associated with tumour formation and aberrant expression of histone deacetylase is associated with carcinogenesis [Citation7]. HDAC regulates deacetyl and acetyl group release which causes histones to be tightly twisted with DNA, thereby inhibiting transcription of tumour suppressor genes responsible for tumours to grow into cancer cells [Citation8,Citation9].
In human, at least 18 HDACs have been identified. They are grouped into four classes and comprise of two major categories: Zn2+ and NAD+ dependent [Citation10]. The classification is based on the structural, functional and phylogenetic analysis of HDACs [Citation11]. Class I comprises of HDAC-1–3 and 8 [Citation12–14] which controls many functional and regulatory mechanisms [Citation15]. Implication of class I HDACs has been monitored in haematological malignancies, resulting in differentiation and proliferation abnormalities of myeloid cells [Citation16]. Aberrant expression pattern of HDAC-1 has been observed in prostate, gastric, breast and colon [Citation17–20] cancers. Similarly, increased expression of HDAC-2 leads to onset of gastric, cervical and colorectal carcinomas [Citation21–23]. Many studies have analyzed the critical role of histone acetyl transferases (HATs) and HDACs in tumour progression, cardiac and brain disorders. [Citation24–27]. Therefore, inhibition of HDAC is a potential and effective strategy of inducing growth arrest, differentiation and finally apoptosis of cancer cells [Citation28]. Inhibition of HDAC also leads to increased sensitivity of actinotherapy and chemotherapy for cancers [Citation29] because in cancer patients, functional activity of HDAC inhibitor (HDACI) is mediated both extrinsically and intrinsically due to cell cycle arrest at G1/G2 phases and upregulation of apoptosis [Citation30,Citation31].
There are several classes of HDACIs. So far the most potent inhibitor ever discovered is trichostatin A (TSA) [Citation32], vorinostat or suberoylanilide hydroxamic acid (SAHA). SAHA belongs to the hydroxamic group and is the most widely studied for its anticancer properties [Citation9]. However, the major disadvantages associated with TSA, SAHA and other HDAC is the production, cost of production, toxicity, non-specificity and side effects [Citation32].
Another type of HDACIs is short chain carboxylic acids (aliphatic acids) like valproic acid (VPA), butyrate and sodium phenyl butyrate. VPA was the first approved HDACI for clinical treatment of bipolar disorder by the FDA [Citation33]. It is relatively a weak inhibitor. Currently, several active trials of HDACI in combination with other antitumor agents are under study for patients suffering from haematological malignancies [Citation34].
According to in vitro and in vivo evaluations, variety of compounds has been synthesized and reported to have inhibitory activity against histone deacetylase (HDAC) and antiproliferation activity against human cancer cells [Citation35]. Based on pharmacophore modelling, a good HDACI should have at least three sides/regions: the attachment side of the Zn2+ cofactor/HDAC active site (Zn2+ binding group/ZBG), hydrophobic cap (CAP) and linker containing connecting unit (CU) with electronegative groups [Citation36]. In this study, we analyzed piperonal derivatives and studied their preclinical analysis in detail. The piperonal moiety along with a propargyl ether group attached to it acted as an ideal compound for HDAC inhibition. Propargyl ether derivatives have been earlier studied and validated for their cytotoxicity against cancer cells.
Recently, in many reports, benzodioxole derivatives have been assessed and validated as anticancer drugs. A number of 1,3-benzodioxoles were reported to be active in vivo against P388 lymphocytic leukaemia and other tumours like podophyllotoxin. The 1,3-benzodioxole moiety occurs commonly in many antimitotic agents such as podophyllotoxin, steganacin and combretastatin A-2 [Citation37] and acts as potent tubulin binders and antimitotic agents [Citation38,Citation39].
Moreover, it has been recently reported that a series of 1,3-benzodioxole derivatives possess a cytotoxic activity against several human tumour cell lines including human colon carcinoma cells [Citation40] and multidrug-resistant nasopharyngeal carcinoma cells [Citation41]. On basis of these studies and in order to pursue our interest in the field of drug discovery and development of new anticancer agents [Citation42,Citation43], we have focused our attention on new 1,3-benzodioxole derivatives [Citation44].
As an extension of our investigation on the structure/activity relationship of 1,3-benzodioxoles as antiproliferative agents, we have performed molecular docking and other pharmacological analysis of compounds for combating the HDAC enzyme for their anticancer activity. Molecular docking studies provide a new rational approach to drug design for the cancer, where the structure of the drug designed on basis of three-dimensional conformations of cancer receptor protein site rather than by random leads or analogy to other active structures. Pharmacology analysis was also done for molecules with better binding affinity against HDAC than standards [Citation45]. This analysis includes pharmacological properties based on Lipinski’s rule of five, bioactivity, drug likeness and drug score [Citation8]. Furthermore, in silico preclinical trials including absorption, distribution, metabolism, excretion and toxicity (ADMET) test, health effect probability and maximum recommended daily dose (MRDD) prediction were also performed in this study [Citation46,Citation47]. The synthetic accessibility and novelty of the lead compounds were also predicted.
In future, we would extend our research work on HDAC inhibiting enzymes acting as anticancer compounds. We will synthesise the anticancer compounds reported here and characterize them by IR, NMR, etc. and would perform in vitro biological assays. This research is expected to obtain novel potential inhibitors of Homo sapiens HDAC-1 which can be used as drugs cancer treatment.
Methodology
HDAC-1 structure retrieval and assessments
Three-dimensional structure of HDAC-1 enzyme (PDB ID 4BKX) of Homo sapiens origin was retrieved from the protein data bank at Research Collaboratory for Structural Bioinformatics (RCSB). All data were stored in PDB format and structural qualities were assessed. The 3D structure was first evaluated for its stereochemical properties by determining the Ramachandran plot using the Rampage server. Other 3D structure properties and its quality were analyzed using the Verify3D, ProSA and ERRAT web interface.
Rampage server (http://mordred.bioc.cam.ac.uk/∼rapper/rampage.php) was employed to study the conformations of the HDAC-1 structure. Ramachandran plot helps in visualizing the dihedral angles phi (Φ) and psi (ψ) of amino acid residues of protein structure and defines the possible allowed and disallowed conformations of protein structure [Citation48].
The ERRAT (http://services.mbi.ucla.edu/ERRAT/) interface works on the basis of the statistics of non-bonding molecular interactions among different atoms and provides the accuracy of protein structure [Citation49]. Verify3D (http://services.mbi.ucla.edu/Verify_3D/) was used to determine the compatibility of the protein structure with its primary amino acid sequences. A higher Verify3D score defines the better structural quality of the protein structure [Citation50]. ProSA server (https://prosa.services.came.sbg.ac.at/prosa.php) determines the overall quality and feasibility of the protein structure. The output result should come in the range of experimentally studied protein structures. ProSA score calculates the Z-score which indicates the overall protein structure quality and fold reliability conformations [Citation51].
Ligand designing and preparation
The known anticancer compound 1,3-benzodioxole’s benzyl propargyl ether-based derivative was synthesized and characterized by spectral and elemental analysis such as UV-IR, NMR techniques. In the current study, we are studying the preclinical and pharmacological analysis of the synthesized compounds using the bioinformatics tools and softwares. ChemDraw software was used to draw the known compounds and its derivatives. The compounds were saved in the .PDB and .mol format to dock with HDAC-1 enzyme. All compounds were optimized for molecular docking simulations for their potent molecular interaction with the HDAC-1 enzyme.
Molecular docking analysis of designed compounds
In order to define potent anticancer drug targeting the HDAC enzyme, designed compounds were compared with approved compounds (SAHA, TSA and VPA) through molecular docking. Prior to molecular docking, receptor protein HDAC-1 was prepared to remove water molecules and unwanted ligands. All water molecules and small ions except zinc ion were removed using the protein preparation wizard of Whatif web interface. Zinc was not removed because it is known to work as cofactor for the protein and stabilizes the structure. Furthermore, hydrogen atoms were added to the structure to correct the tautomeric and oxidation states of amino acid residues. Thereafter, ligands were docked with HDAC-1 enzyme. Moreover, in this study, first we have docked the designed compounds using the AutoDock Vina and then with HEX 8.0, in order to validate our results with another molecular docking algorithm. AutoDock Vina was first employed for the molecular docking; it docks the flexible compound (ligand) with the protein using the gradient optimization method. This tool matches the crystalline ligand with protein molecule and this program is known to more accurate and precise than the other version of AutoDock with sophisticated algorithm [Citation52]. Thereafter, HEX 8.0 CUDA program was used for molecular docking simulations of compounds with HDAC-1 protein. HEX identifies the electrostatic potential, steric shape and the charge density of target protein structure [Citation53]. Binding residues and different bonds involved in the binding of resulting docked complexes were evaluated using the AutoDock tools interaction determining module and further docked complexes were analyzed using Chimera molecular modelling tool.
Pharmacology analysis and preclinical trials
The pharmacological properties and bioactivities of the newly designed inhibitors were determined by using various pharmacology analysis and preclinical trials. Pharmacology analysis in this study included pharmacological properties test based on Lipinski’s rule of five, bioactivity, drug likeness and drug score. The preclinical trials included ADMET test, maximum recommended daily dose and other predictions. These analyses were performed using the in silico analysis with variety of software such as SwissADME, ACD/I-Lab, FAF-Drugs2, and Molinspiration.
Synthetic accessibility and novelty of the lead compound prediction
Synthesis accessibility prediction is essential to analyze the feasibility of synthesizing a best designed drug in a wet laboratory. SwissADME web interface was used to determine the synthetic accessibility of the lead compounds. Furthermore, SciFinder was used for prediction and for predicting the novelty of the lead compounds, respectively.
Results
Structure determination and evaluation of HDAC-1
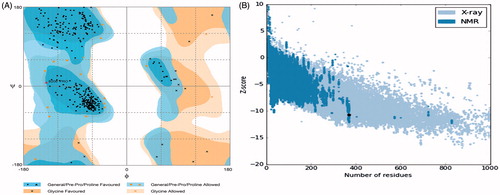
The 3D structure of human origin HDAC-1 enzyme (4BKX) was retrieved from the protein data bank. HDAC-1 structure was found in complex with metastasis-associated protein-1 and small ions including zinc ion. Also, the HDAC-1 protein structure consists of 482 amino acids. For carrying out molecular interaction studies, the structure was evaluated for its three-dimensional structural properties (). Stereochemical properties of the HDAC-1 structure were analyzed through Ramachandran plots. Rampage server was used to assess the dihedral angles and a number of amino acid residues in favoured and disfavoured regions [Citation48]. The Ramachandran plots for HDAC-1 showed 95.1% in favoured region, 4.6% in the allowed region and 0.3% in generously allowed region using the Rampage server. ERRAT interface showed that the overall quality score of the structure is 92.521, which indicates a structural resolution of ∼2.5–3 Å. Verify3D web interface also confirms the good quality of HDAC-1 protein structure with more than 80% amino acids consisting of 3D–1D profiles. ProSA score came out to be a Z-score of −10.7, as shown in the , suggesting that the HDAC-1 structure Z-score is within the range of scores of native protein structure of similar size. Structure assessments carried out with all the tools mentioned above confirmed the good quality structure of the HDAC-1 protein which could be further used for molecular studies.
Figure 1. Diagram depicting the three-dimensional structure of histone deacytlase-1 enzyme in new cartoon view.
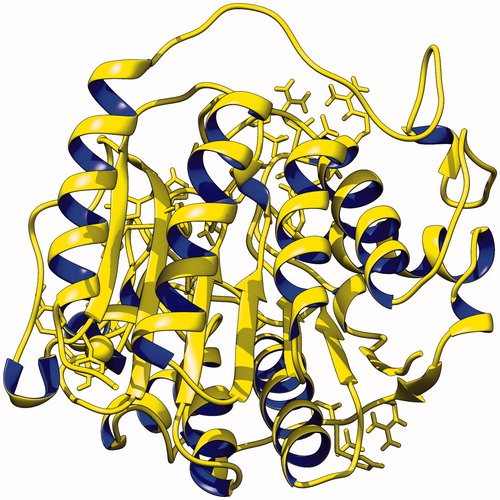
Figure 2. (A) The Ramachandran plot of HDAC-1 enzyme. Plot shows structure lies within the favoured and allowed region; (B) ProSA-web interface analysis depiction of HDAC-1 protein structure with NMR/X-ray plot of known structures.
Binding interaction studies using molecular docking
Molecular interaction studies of designed compounds with target protein HDAC-1 were performed. In order to define potent anticancer drug targeting the HDAC enzyme, designed compounds were compared with approved compounds (SAHA, TSA and VPA) using molecular docking. Prepared HDAC-1 protein structure was docked with designed compounds and then with approved drugs using two molecular docking algorithms, AutoDock Vina and HEX 8.0.
To begin with, AutoDock Vina molecular docking program was used; input files were converted into PDBQT files. AutoDock tool was used to remove the water molecules and to add the polar hydrogen atoms to the HDAC-1 protein structure. For molecular docking, PDBQT files were uploaded with the grid spacing of 0.375 Å and also grid centre coordinates were set at X, Y and Z axes at 60 × 60 × 60. The grid conformation box was placed on the interacting sites of the HDAC-1 enzyme, which provides the space for compound rotation and translations. Resulting docked complexes of protein and compounds were analyzed using AutoDock tools. Among all resulting docked compounds with HDAC-1, compound 7 and compound 9 both resulted in −10.6 kJ/mol docking score, better than that for approved drugs SAHA, TSA and VPA with docking scores of −9.2, −10.3 and −5.9 kJ/mol, respectively, through the AutoDock Vina () (). Both compounds were found to interact potently to the binding groove of HDAC-1 enzyme, as shown in .
Figure 3. Depiction of molecular docking complexes. (A) Interacting complex of HDAC-1 enzyme with the first lead compound 7. New cartoon ribbon view of the HDAC-1 enzyme with interacting compound-7, at its interior. Also shown the enlarged view of compound-7 at right and (B) interacting complex of the HDAC-1 enzyme with compound 9. New cartoon ribbon view of the HDAC-1 enzyme with interacting compound-9, at its interior binding groove. Also shown the enlarged view of compound-9 at right.
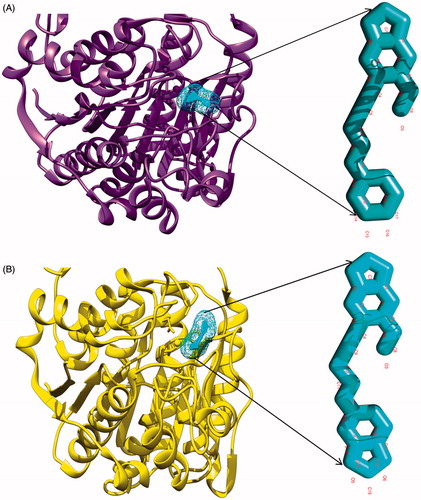
Figure 4. (A) Surface view of interaction of compound 7 to the binding groove of HDAC-1 enzyme and (B) surface view of interaction of compound 9 into the binding groove of HDAC-1enzyme.
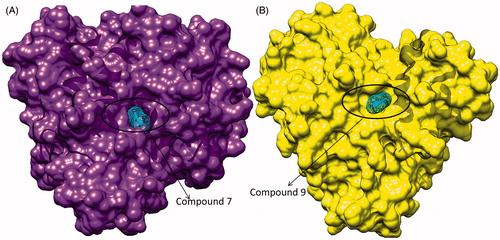
Table 1. Molecular binding free energy scores of designed compounds with the receptor HDAC-1 enzyme, using the AutoDock Vina and HEX 8.0 molecular docking software.
Further molecular docking analysis was accomplished using HEX 8.0 to obtain the best native conformation of protein–ligand docked complex. Input parameters for docking were the HDAC-1 structure, PDB coordinate files as receptor protein and designed propargyl ether derivatives of benzodioxole as ligand files with set parameters. On the basis of shape and electrostatic parameters, the total energy of interacting complexes were determined and resulting final search was set to 25 (N = 25) conformations of the complexes. The range of molecular interaction angular search was confined by selecting interface amino acid residues of HDAC-1 with an angle range of 45°. Other interaction parameters of interaction were set to default values as per algorithm. The best model of docking complex which showed the largest binding affinity was subjected to further molecular interaction studies. Notably, we found more similar results with HEX 8.0 like AutoDock Vina. HEX 8.0 showed that the compound 7 and compound 9 have a binding score of −195.61 and −205.92 kJ/mol, respectively, higher than the SAHA, TSA and VPA with docking score of −172.02, −184.54 and −140.19 kJ/mol, respectively. These molecular docking simulation studies suggest that two compounds 7 and 9 can be considered as potent HDAC-1 enzyme inhibitor candidate to be used as an anticancerous drug. Furthermore, these two compounds were evaluated through the preclinical tool and pharmacological analysis to be defined as a potent drug candidate.
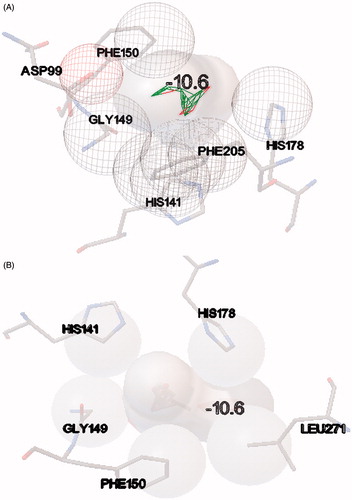
The resulting docked complexes were further analyzed with PDBsum tool to identify the involved hydrophobic interactions between designed compounds and HDAC-1 protein. The results showed that compound 7 has strong hydrophobic interactions with HDAC-1 through amino acids aspartate 99, histidine 141, glycine 149, phenylalanine 150, histidine 178 and phenylalanine 205. Compound 9 was also found to have strong hydrophobic interactions at similar position as compound 7 to HDAC-1, through residues histidine 141, glycine 149, phenylalanine 150, histidine 178 and leucine 271 (). Higher binding molecular docking scores and strong molecular interactions suggested that these two compounds could be a potent candidate to inhibit the HDAC-1 enzyme in cancer. The compounds were further evaluated for preclinical and pharmacological properties.
Figure 5. (A) Depiction of hydrophobic interactions between the compound 7 and HDAC-1 enzyme, derived through molecular docking and (B) depiction of hydrophobic interactions between the compound 9 and HDAC-1 enzyme, using PDBsum web interface.
Pharmacology analysis
In this study, the pharmacology analysis included testing of pharmacological properties based on Lipinski’s rule of five. Lipinski’s rule of five is based on the approach that the human body contains about 90% of water. Drug molecules should be hydrophobic enough to last longer in the body and not easily excreted, as this will enhance its bioactivity efficiency in the body. It is quite a good approach for drug lead design, especially for hydrophobicity and oral bioavailability prediction. This rule is modified Lipinski’s rule of five and consists of the following: molecular weight (MW) is from 160 to 500 Da, octanol–water partition coefficient (logP) is between −0.4 and +5.6, molar refractivity is from 40 to 130 cm3, hydrogen donor is less than 5 atoms, hydrogen acceptor is less than 10 atoms, and topological polar surface area (TPSA) is not more than 140 A˚2. Molinspiration software was used to study the pharmacological properties of designed propargyl ether derivatives.
It was seen that both compounds 7 and 9 resulted in no violations with Lipinski rule of five and followed all the requisite properties needed for a potent drug which was same as the pharmacological properties of standard drugs SAHA, TSA and VPA (). These results also indicate that compounds are hydrophobic as they have a lower solubility in water than the three standards. With their good hydrophobicity, it can be concluded that compounds will be able to circulate longer and are not easily excreted out from the body, which enhances their activities and interactions with the target protein HDAC-1.
Table 2. Pharmacological properties of standard compounds and lead compounds.
The other pharmacology parameter analyzed in this study is drug likeness. The drug likeness is a pharmacological parameter that indicates fragments and bioactivity similarity between designed ligands and approved drugs [Citation54].
The drug likeness score also includes the combined score of several bioactivity parameters such as GPCR (G-protein coupled receptor) ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitor properties [Citation55]. We found that both compounds 7 and 9 possess high drug likeness using Lipinski rule of five and other algorithms, with standard drugs using SwissADME interface. These results concluded that compound 7 and compound 9 have high fragments and bioactivity. They also possess similarity with the existing drugs and could be developed as novel drugs for cancer treatment.
In silico preclinical trial
In the earliest in silico preclinical trial of the designed inhibitor compounds 7 and 9, pharmacokinetic properties and toxicity prediction were studied. pkCSM and FAF-Drugs2 interfaces were used to predict the pharmacokinetic properties. pkCSM can further predict the maximum recommended daily dose of the designed ligands if they were to be developed as cancer drugs.
Based on the hydrophobicity of the designed compounds, the oral bioavailability can also be calculated using FAF-Drugs2 and pkCSM software. FAF-Drugs2 prediction shows that both compounds have a good oral bioavailability based on Veber’s and Egan’s rules.
By using pkCSM, maximum recommended tolerated dose was calculated for both compounds. Compound 7 was found to have high tolerance with recommended tolerated dose (0.433 log mg/kg/day) and compound 9 (0.289 log mg/kg/day). The results were based on the maximum recommended starting dose in phase-1 clinical trials, based on the 1222 experimental data of human clinical trials.
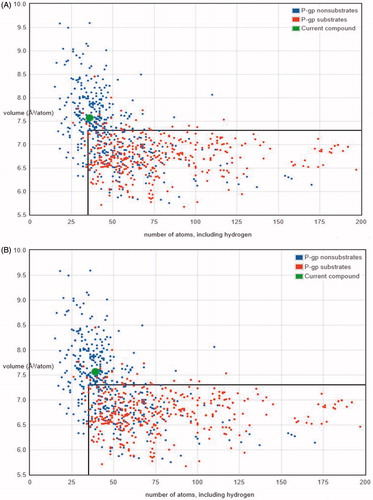
Another important consideration during preclinical analysis trial was to analyze the P-glycoprotein (P-gp) non-substrate candidature. P-gp works as efflux transporter which pumps drugs and other compounds and its substrate out of the cell, which was found to be one of explanation for its resistance to various chemotherapeutics for cancer. It has been found that P-gp is a critical transporter of drug which consequently results in resistance to anticancer drugs like imatinib, lonafarnib and taxanes [Citation56,Citation57]. Therefore, we analyzed the lead compounds 7 and 9 for P-gp candidature or not using the Biozyne web interface. We found both compounds 7 and 9 are not substrates of P-gp protein with very high probability, also shown in the . This suggests that the lead compounds have very less chance to efflux out of the cell, thus resulting in maximum effect.
Figure 6. Graphs between the numbers of atoms versus the volume of compounds. (A) Compound 7 was found to be under the P-gp non-substrate region (out of box region in graph), whereas P-gp substrates compounds comes under the box region and (B) compound 9 was also found to be under P-gp non-substrate region (out of the box region in graph), whereas P-gp substrates comes under the region inside the box in graph.
In passive absorption of drugs, drugs are absorbed through the intestinal mucosa or epithelial cells that do not use energy or ATP contribution along the process. Intestinal absorption for compound 7 and compound 9 was found to be 97.497% and 97.457%, respectively, during passive absorption. It was notable to see that both do not result in skin sensation ().
Table 3. Preclinical trial of lead compounds (compounds 7 and 9) using pkCSM and FAF-Drugs2.
Another stage of in silico preclinical trial was to assess the inhibition of cytochrome p450 by lead compounds. Since compounds should combat the cytochrome p450 enzyme in order to maintain a substantial concentration of compounds in the body for prolonged effect, we found out that both compounds combat the cytochrome p450.
Thereafter, we determined that our compounds remain unbound during circulation, since mostly drugs exist in equilibrium, i.e. either unbound or bound to the serum proteins in plasma. The efficacy of drug might get affected due to binding of blood proteins to the drug present in blood. The drug’s potency to transverse through cell membrane will decrease as binding of the drug to the drug protein increases. Further, the compounds were tested for renal clearance. They were analyzed for their transportation through organic cation transporter 2 (a renal transporter). We found that both drugs were likely to be organic cation transporter 2 substrate for transportation for renal clearance.
During preclinical studies, it is important to assess the toxic potency of compounds. To determine the relative toxicity of compounds by a standard measurement of acute toxicity, lethal dosage values (LD50) were determined. LD50 is the concentration of compound that causes the death of 50% testing animals. LD50 estimation for compound 7 and compound 9 was estimated to be 2.027 and 2.273 mol/kg, respectively, from a model built from over 10,000 chemical compounds tested on rats.
Oral rat chronic toxicity of compounds was also analyzed. This parameter determines the lowest dose of compounds that results in adverse effects. We found that large concentration of compound 9 (2.052 log mg/day) and compound 7 (1.805 log mg/day) would not result in adverse effects on rats. We may conclude from the clinical study above, carried out with bioinformatics tool that both compound 7 and compound 9 have drug requisite pharmacokinetic properties and could be used for further in vitro and other biological assays to be developed as potent anticancer drug.
Synthetic accessibility and novelty of the lead compound prediction
The next step in drug discovery after computer-aided drug discovery and development is to determine the in vitro and in vivo bioactivity for synthesis of these drugs. For this reason, synthetic accessibility and lead likeliness of in silico designed ligands have to be assessed to evaluate the synthetic probability and feasibility of the ligands. Screening and informational analysis of the contribution of particular fragments and the complexity of the structures derived from a database containing millions of already synthesised compounds form the basis of this prediction. So from this method, the synthetic accessibility of drug lead compounds could be estimated from de novo synthesis approach.
SwissADME software was employed to predict the synthetic accessibility and lead likeliness of compounds as a potent anticancer drug. These tools can estimate the percentage of synthetic accessibility based on the existence of starting materials and structural or residual complexity [Citation58]. Synthetic accessibility predictions of the standard compounds and compound 7 and compound 9 are presented in . It can be seen that compounds 7 and 9 have higher synthetic accessibility values than the standard compounds SAHA, TSA and VPA. We also found that compounds 7 and 9 possess higher lead likeliness values; on the other hand, known drugs such as SAHA and VPA do not have lead drug likeliness.
Table 4. Synthetic accessibility analyses of standard compounds and lead compounds using SwissADME.
We also performed novelty evaluation by searching ligand chemical structures on SciFinder that identifies similar structures, tautomers, structural and geometrical isomers. It was important to note that the designed compound 7 and compound 9 were not found on SciFinder database. According to this result, it could be predicted that the designed inhibitors were novel, i.e. they have never been synthesized before and could be considered as potential lead for anticancer drug designing.
Discussion
Present study focuses on the anticancer drug design and preclinical evaluation of drugs by targeting the HDAC, an important cell cycle regulator in cancer. HDACIs represent the important class of anticancer agents, which can arrest the cancerous cell differentiation and induce apoptosis. Molecular docking and preclinical studies of series of designed drugs were performed. Docking studies assist in evaluating the efficacy of the drug, with the most detailed possible view of drug–receptor interaction, and give the structural insights to mechanistic interaction of drug and receptor. Molecular docking defines the binding scoring function between the anticancer drugs and the target receptor proteins, on the basis of free energy value, hydrophobic interactions and hydrogen bond formations. Importantly, docking scoring function along with pharmacokinetic and pharmacodynamic profile of drug has been correlated with the experimental and clinically studied protein–drug interactions, which paves way to design the potent and safe anticancer drug using molecular docking approach. The combination of in silico drug design (based on de novo lead molecule design approach), molecular docking, pharmacology analysis, virtual screening and manual interpretation is more efficient than synthesis methods to discover new effective inhibitors and drug leads that may have a great impact for future experimental studies. Computational or in silico methods play an important role in drug discovery and development by improving the efficiency of novel HDACIs. Unlike conventional methods, this method limits the use of chemicals for synthesis and biological testing, thus greatly reducing resource requirements. It is expected that this study will produce potent HDACIs as drugs for cancer treatment.
In this study, novel potential inhibitors for cancer targeting Homo sapiens histone deacetylase-1 enzyme were designed by in silico approach and evaluated using preclinical studies. According to pharmacophore model, HDACIs consist of three main parts: zinc binding group (ZBG), linker and hydrophobic cap (CAP). Using this 1,3-benzodioxole compound series based on propargyl ether moiety has been designed and modified. To test the pharmacological properties and bioactivities of designed ligands, in silico studies have been performed. The higher free energy binding score through molecular interaction of compounds with HDAC-1 enzyme active site was the basis of best ligand selection molecular docking simulation. The selection yielded two compounds (compounds 7 and 9) which have better binding affinity (lower free energy of binding) than SAHA, TSA and VPA, standards drugs for cancer. Molecular interaction analysis of HDAC-1 enzyme compound complex showed that compounds have strong hydrophobic interactions. Fragment propargyl ether derivative of benzodioxole acts as a zinc binding group of HDAC-1 enzyme with the Zn2+ cofactor.
Pharmacology analysis was then performed to assess bioactivity and pharmacological properties. Both compounds meet Lipinski’s rule of five and show drug likeliness. They also possess lower solubility in water than standards. Hence, these are the potential compounds as anticancer candidates that would enhance the bioactivity due to delayed excretion.
In silico preclinical trial was conducted to evaluate the pharmacokinetics (ADMET) properties of the lead compounds including toxicological properties, passive absorption and oral bioavailability prediction. Based on our results, they also have a high passive absorption and good oral bioavailability based on Veber’s and Egan’s rules and do not possess the toxicity to human. Moreover, we also found that compounds have high tolerance to human cells and high LD50 value for rats. The compound 7 and compound 9 also have a good synthetic accessibility. It could be inferred through these assessments that all designed ligands are potential to be novel HDACIs and as potential cancer drug leads. These compounds could be developed as novel potent drugs for cancer targeting HDAC-1 enzyme.
Acknowledgements
We are grateful to the Council of Scientific and Industrial Research (CSIR) for providing financial assistance and necessary funds to Prof. Ramesh Chandra. We are also grateful to the University of Delhi for providing support and necessary facilities to carry out research work. Neeraj Kumar particularly thanks Department of Biotechnology (DBT-JRF) for providing the fellowship for ongoing research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Cancer facts and figures 2017(US). Atlanta (GA): American Cancer Society(US); 2017.
- A report, Over 17 lakh new cancer cases in India by 2020: ICMR; 2016. Available from: http://icmr.nic.in/icmrsql/archive/2016/7.pdf
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784.
- Khan O, Thangue L. NB HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2011;90:85–94.
- Marks P, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202.
- Jacobson S, Pillus L. Modifying chromatin and concepts of cancer. Curr Opin Genet Dev. 1999;9:175–184.
- Tambunan USF, Parikesit AA. HPV bioinformatics: in silico detection, drug design and prevention agent development. In: Topics on cervical cancer with an advocacy for prevention. Croatia: Intech Publishing; 2012.
- Singh VK, Kumar N, Kalsan M, et al. Mechanism of induction: induced pluripotent stem cells (iPSCs). J Stem Cells. 2015;10:43.
- De Ruijter A, Van Gennip A, Caron H, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749.
- Gregoretti I, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31.
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51.
- Hubbard T, Barker D, Birney E, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41.
- Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151.
- Millard CJ, Watson PJ, Celardo I, et al. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67.
- Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62:18–34.
- Halkidou K, Gaughan L, Cook S, et al. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189.
- Choi JH, Kwon HJ, Yoon BI, et al. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res. 2001;92:1300–1304.
- Zhang Z, Yamashita H, Toyama T, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res Treat. 2005;94:11–16.
- Wilson AJ, Byun D-S, Popova N, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006; 281:13548–13558.
- Song J, Noh JH, Lee JH, et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS. 2005;113:264–268.
- Huang B, Laban M, Leung CH, et al. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404.
- Zhu P, Martin E, Mengwasser J, et al. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–463.
- Baradari V, Huether A, Höpfner M, et al. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr-Relat Cancer. 2006;13:1237–1250.
- Hahn CK, Ross KN, Warrington IM, et al. Expression-based screening identifies the combination of histone deacetylase inhibitors and retinoids for neuroblastoma differentiation. Proc Natl Acad Sci USA. 2008;105:9751–9756.
- McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–319.
- Thomas EA. Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol Neurobiol. 2009;40:33–45.
- Rajak H, Kumar P, Parmar P, et al. Appraisal of GABA and PABA as linker: design and synthesis of novel benzamide based histone deacetylase inhibitors. Eur J Med Chem. 2012;53:390–397.
- Geng L, Cuneo KC, Fu A, et al. Histone Deacetylase (HDAC) inhibitor LBH589 Increases Duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66:11298–11304.
- Mai A, Massa S, Rotili D, et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev. 2005;25:261–309.
- Kim YB, Ki SW, Yosnida M, et al. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. J Antibiot. 2000;53:1191–1200.
- Nair SB, Teli MK, Pradeep H, et al. Computational identification of novel histone deacetylase inhibitors by docking based QSAR. Comput Biol Med. 2012;42:697–705.
- Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252.
- Tan J, Cang S, Ma Y, et al. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:5–17.
- Fournel M, Bonfils C, Hou Y, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768.
- Mohan R, Sharma AK, Gupta S, et al. Design, synthesis, and biological evaluation of novel 2,4-thiazolidinedione derivatives as histone deacetylase inhibitors targeting liver cancer cell line. Med. Chem Res. 2011;21:1156–1165.
- Hamel E. Antimitotic natural products and their interactions with tubulin. Med Res Rev. 1996;16:207–231.
- Batra JK, Jurd L, Hamel E. Structure/function studies with derivatives of 6-benzyl-1,3-benzodioxole, a new class of synthetic compounds which inhibit tubulin polymerization and mitosis. Mol Pharmacol. 1985;27:94–102.
- Jurd L, Narayanan VL, Paull KD. In vivo antitumor activity of 6-benzyl-1,3-benzodioxole derivatives against P388, L1210, B16, and M5076 murine models. J Med Chem. 1987; 30:1752–1756.
- Yokota S, Kitahara M, Nagata K. Benzylidene lactam compound KNK347, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 2000;60:2942–2948.
- Xia Y, Yang Z, Xia P, et al. Antitumor agents. Part 202: novel 2?-amino chalcones. Design, synthesis, and biological evaluation. Bioorg Med Chem Lett. 2000;10:699–701.
- Chimirri A, Grasso S, Monforte AM, et al. Synthesis and antitumor activity evaluation of D2 -1,2,4-oxadiazolines. Pharmaco. 1996;51:125–129.
- Grasso S, Micale N, Monforte AM, et al. Synthesis and in vitro antitumour activity evaluation of 1-aryl-1H,3H-thiazolo[4,3-b]quinazolines. Eur J Med Chem. 2000;35:1115–1119.
- Micale N, Zappala M, Grasso S. Synthesis and in vitro antitumor activity of 1-3-benzodioxole derivatives. Farmaco 2002;57:854–859.
- Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8:1555–1572.
- Moroy G, Martiny VY, Vayer P, et al. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today. 2012;17:44–55.
- Van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2:192–204.
- Lovell SC, Davis IW, Arendall WB III, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450.
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519.
- Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;5:83–85.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:407–410.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comp Chem. 2010;3:455–461.
- Ghoorah AW, Smail-Tabbone M, Devignes MD, Ritchie DW. Protein docking using case-based reasoning. Proteins. 2013;81:2150–2158.
- Hartenfeller M, Zettl H, Walter M, et al. DOGS: reaction-driven de novo design of bioactive compounds. PLoS Comput Biol. 2012;8:e1002380.
- Kubinyi H. Success stories of computer-aided design. In: Ekins S, editor. Computer applications in pharmaceutical research and development. Hoboken (NJ): John Wiley & Sons Inc; 2006.
- Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502.
- Tsuji A. P-glycoprotein-mediated efflux transport of anticancer drugs at the blood-brain barrier. Ther Drug Monit. 1998;20:588–590.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:4.