
Abstract
Since physiological and pathological processes occur at nano-environments, nanotechnology has considered as an efficient tool for designing of next generation specific biomolecules with enhanced pharmacodynamic and pharmacodynamic properties. In the current investigation, by control of the size and hydrodynamic volume at the nanoscale, for the first time, physicochemical and pharmacokinetic properties of an anti-VEGFA nanobody was remarkably improved by attachment of a Proline-Alanine-Serine (PAS) rich sequence. The results elucidated unexpected impressive effects of PAS sequence on physicochemical properties especially on size, hydrodynamics radius, and even solubility of nanobody. CD analysis revealed an increment in random coil structure of the PASylated protein in comparison to native one without any change in charge state or binding kinetic parameters of nanobody assessed by isoelectric focusing and surface plasmon resonance measurements, respectively. In vitro biological activities of nanobody were not affected by coupling of the PAS sequence. In contrast, the terminal half-life was significantly increased by a factor of 14 for the nanobody-PAS after single dose IV injection to the mice. Our study demonstrated that the control of size in the design of small therapeutic proteins has a promising effect on the stability and solubility, in addition to their physiochemical and pharmacokinetic properties. The designed new anti-VEGFA nanobody could promise a better therapeutic agent with a long administration intervals and lower dose, which in turn leads to a better patient compliance.
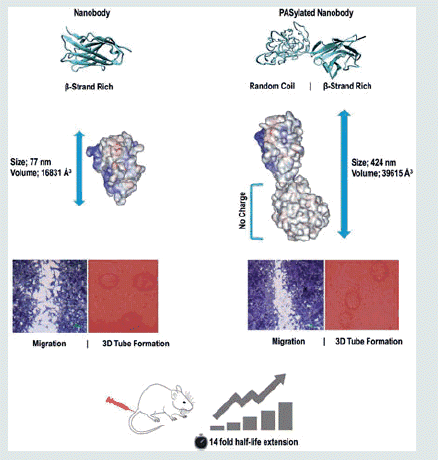
Size adjustment of an anti-VEGF nanobody at the nanoscale by the attachment of a natural PAS polymer remarkably improves physicochemical properties, as well as a pharmacokinetic profile without any change in biological activity of the miniaturized antibody.
Graphical Abstract
Introduction
All natural biomolecules exert their vital roles in the body at the nanoscale in the physiological conditions. Similarly, this nano-environment is also very important in pathological situations. For example in the angiogenesis process, VEGFA has a pivotal role in the regulation of vascularization in both physiological and pathological conditions. Many drugs have developed for inhibition of VEGFA, but among them, conventional monoclonal antibodies like bevacizumab have pharmaceutically considered because of their specific and effective functions [Citation1]. Despite long plasma half-life of the whole antibodies that increase the contact duration with the target antigen, the poor penetration rate into solid tissues as well as asymmetric distribution results in inappropriate targeting of the tumours clinically [Citation2].
Recently, miniaturized antibodies, known as nanobodies, have considered as efficacious therapeutics agents in the medicine due to nanoscale dimension [Citation3]. They have good distribution as well as high penetration rate to locations which could not be accessible by large molecules [Citation4]. Nanobodies, which naturally occur in the serum of Camelidae, are derived from heavy chain variable antibody fragments and are the known smallest antibody fragments that bind specifically to the target molecules [Citation5]. Recent studies have shown that antibody fragments have sufficient penetration rate to the solid tumours due to the small size and can be more effective than large-sized whole commercially available antibodies [Citation3,Citation6]. In addition, nanobodies can recognize hidden antigenic sites that normally are not detectable by whole antibodies [Citation7]. They are more stable and soluble compared to the whole antibodies. They can also be produced in bacterial systems that make their production process easier in terms of cost and time at industrial scale [Citation8].
The main disadvantage of nanobodies is their rapid excretion from the body after administration, which in turn needs to be injected at repeated dose and limits the practical clinical use [Citation9]. The reason based on this fact that the mean glomerular pore size is approximately 6–8 nm [Citation10] while the nanobodies have prolate shape with a dimension of about 2.5 × 4 nm. Because of too small sizes, these miniaturized antibodies likely to be eliminated rapidly from glomerular filtration [Citation8]. Several technologies have been developed for solving this issue including Fc fusion, albumin conjugation, glycoengineering [Citation11], and PEGylation [Citation10,Citation12–15]. However, they suffer from some limitations like undesirable and/or unpredictable effects on the structure, physicochemical, and biological properties of the native protein, the requirement of additional purification steps, use of animal cells for production, high immunogenicity and polydispersity, and the high cost of finished product [Citation11,Citation12,Citation16–18].
Recently, the use of PEG mimetic peptides was expanded to improve the pharmacokinetic properties of recombinant proteins for medicinal purposes. The special one, introduced by XL-protein GmbH, is called PAS sequence (proline, alanine, and serine) which usually between 100 and 600 amino acids genetically added to the N- or C-terminus of the target protein. It has unique properties such as compatible with the biological environment, high water solubility, no negative or positive charges or polydispersity, and mainly random coil structures similar to PEG polymer. These criteria lead to increase in hydrodynamic radius as well as the higher solubility of the target protein without any chemical coupling reactions [Citation19]. Other advantages of the PAS sequence include; lack of immunogenicity, lack of significant changes in pI of fused protein, lack of accumulation in the kidney, resistance to plasma proteases, and manufacture of active proteins in E. coli expression systems without long and difficult downstream processes [Citation20].
In the current investigation, for the first time, a novel nano-size adjusted miniaturized antibody against VEGFA was designed by the attachment of a size controlled PAS sequence. The physicochemical properties, antiproliferative, cell migration, and angiogenesis effects, as well as pharmacokinetic parameters of the developed miniaturized antibody, were compared to the native protein. Moreover, the effect of PAS#1(200) sequence on structural conformation of fused protein was also computationally investigated.
Materials and methods
Reagents and media
Anti-VEGFA nanobody (Nb) gene in pHEN6 expression plasmid was a gift by Dr. Behdani (Department of Biotechnology, Pasteur Institute of Iran) and recombinant human VEGFA165 were obtained from R&D Systems (Minneapolis, MN). Bevacizumab and HRP-conjugated anti-His antibody were purchased from Roche (Basel, Switzerland). DNA ligase, restriction enzymes, and DNA ladder were obtained from Fermentas (Thermo Fisher Scientific Inc., Waltham, MA). All other chemicals were procured from Merck (Darmstadt, Germany). Primary human umbilical vein endothelial cells (HUVEC) were purchased from Lonza Biologics (Walkersville, MD) and cultured in endothelial cell basal medium (EBM) (Lonza Biologics) supplemented with 2% FBS in a 37 °C incubator with 5% CO2. HEK293 cell line was obtained from Pasteur Institute of Iran and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Lonza Biologics) supplemented with 2% FBS in a 37 °C incubator with 5% CO2. Female BALB/c mice were purchased from Pasteur Institute of Iran and animal study was approved by Animal Ethics Committee of Pasteur Institute of Iran (Ethical Code: 94/0201/10563).
3D structure modelling and energy minimizations
In order to investigate whether or not PAS#1(200) polypeptide has any deleterious effect on molecular characteristics of nanobody, 3D-structures of nanobody and PAS#1(200) polypeptide were built by the I-TASSER server version 2.1 [Citation21]. Then, N-terminus of modelled PAS#1(200) was linked to the C-terminus of nanobody via a peptide bond using Chimera program version 1.9 [Citation22] and finally the generated structures were energy minimized by NAMD package version 2.11 [Citation23] using the CHARMM 27 force field [Citation24]. Structural energy minimizations were performed for 50 ns using conjugate gradients method and VMD 1.9.2 was used to take snapshots and implement pre- and post-processing of energy minimization steps [Citation25]. A detailed structural conformation assessment was performed on fused protein in terms of molecular volume, whole RMSD, and surface area accessibility.
Expression plasmids
The anti-VEGFA nanobody gene was previously generated by phage display technique and cloned into the pHEN6 expression plasmid which contained His-tag and an ampicillin resistance marker [Citation26]. PAS#1 sequence [ASPAAPAPASPAAPAPS-APA] [Citation20] with a length of 200 amino acids was genetically fused to the C-terminus of nanobody gene. The His-tag sequence was added to the end of construct for detection and purification purposes. The construct was then synthesized by Biomatik (Cambridge, Canada) and subsequently cloned into pET-26b(+) expression vector using NdeI and EcoRI restriction sites.
Expression and purification
Nanobody-PAS#1(200) expression plasmid was transformed to BL21 (DE3) E. coli and cultured in Terrific broth media with 50 mg/ml kanamycin. Recombinant gene expression was induced using 1 mm isopropyl-β-D-thiogalactoside when the OD600 nm was 0.5 for 16–20 h at 30 °C. Immediately thereafter, the cells were collected by centrifugation at 8000 g for 10 min and periplasmic extraction was performed in the presence of 500 mm sucrose, 1 mm EDTA and 100 mm Tris/HCl adjusted to pH 8.0. The PASylated nanobody was purified from the extract by Ni+-NTA column chromatography (Qiagen, Hilden, Germany) according to the manufacture’s instruction. The purified protein was buffer exchanged against PBS and concentrated by ultrafiltration using Amicon Ultra centrifugal filter units (3000 MWCO; 50 ml; Millipore, Billerica, MA). Finally, the samples were stored at −70 °C for further steps. A similar procedure was employed for the expression and purification of the native protein. Expression of PASylated nanobody and the native protein were studied using 15% SDS-PAGE and the gels were stained with Coomassie brilliant blue R-250 (Sigma-Aldrich, St. Louis, MO). After blotting, the membranes were blocked by buffer containing skim milk 2.5%, glycerol 2.5% in TTBS containing 20 mm Tris, 500 mm NaCl, pH 7.4, 0.05% Tween-20 for 16 h at 4 °C. The membranes (Bio-Rad, Hercules, CA) were then washed three times with TBST buffer and incubated with anti-His HRP conjugated antibody [(1:10,000 dilution) (Roche)] for detection of the native protein and the PASylated nanobody. Finally, the membranes were stained with the Amersham ECL Prime Western Blotting Detection reagent (GE Healthcare Life Sciences, Chalfont, UK) according to manufacturer’s instructions (GE) and the chemiluminescence was detected by a Kodak Image Station 4000 mm pro digital imaging system (Kodak, Rochester, NY). The protein concentrations were determined by Bradford method [Citation27].
Circular dichroism spectroscopy (CD)
The secondary structures of anti-VGEFA nanobody and its PASylated form were investigated by a J-810 spectropolarimeter (JASCO Corporation, Tokyo, Japan). Spectra were recorded in the wavelength range of 190–250 nm at room temperature (bandwidth 1 nm, scan speed 200 nm/min, response 1 s) using 500 μg/ml protein solutions in water for injection. Spectra were monitored with the instrument software and the molar ellipticity ΘM was calculated according to the following equation:
Where Qobs is the measured ellipticity, c is the protein concentration [mol/l], and d is the path length of the quartz cuvette [cm] [Citation28].
Charge characterization
Isoelectric focusing (IEF) analysis of anti-VGEFA nanobody and its PASylated form were performed using the manufacturer’s instruction (Bio-Rad). Fifty micrograms of each purified protein were individually dissolved in rehydration buffer (8 M urea, 4% CHAPS, 0.2% 100 × Biolyte and 50 mm DTT), loaded onto IPG strips (7 cm, non-linear pH 3–10) and kept at room temperature for 16 h. IEF was performed using three steps of increasing voltage from 250 V to 14 kV. Finally, IPG strips were stained by Bio-Rad special dye (Bio-Rad) for 45 min and destained by 40% methanol – 10% glacial acetic acid solution.
Dynamic light scattering (DLS)
Size distributions of anti-VGEFA nanobody and its PASylated form were measured using Brookhaven Zeta Plus Instruments Corporation (Zeta PALS, Holtsville, NY) at room temperature with a wavelength of 658 nm, and the results were analyzed with Nano Brook 90Plus Particle Size Analyzer version 5.2 (Brookhaven Instruments, Holtsville, NY). The purified proteins were prepared by centrifugation at 13,000 g for 10 min and 0.5 mg/ml of each sample was filtered through 0.22 μm polytetrafluoroethylene filters (Millipore).
Size exclusion chromatography (SEC)
Molecular mass comparison of anti-VGEFA nanobody and its PASylated form were performed by size exclusion chromatography (SEC) (KNAUER AZURA Analytical HPLC system, Germany). Twenty micrograms of each of the purified protein was applied to a G2000 SEC column [(300 × 7.8 mm) (GE-Healthcare, Milwaukee, WI)] with 0.05 M Na2HPO4, 0.05 M NaH2PO4, and 0.15 M NaCl (pH 6.8) as running buffer (flow rate: 0.8 ml/min and chart speed: 1 cm/min). Samples were centrifuged at 14,000 g for 15 min prior to injection. Standard proteins including thyroglobulin (bovine, 670 kDa), γ-globulin (bovine, 158 kDa), ovalbumin (chicken, 44 kDa), myoglobin (horse, 17 kDa), and vitamin B12 (1.35 kDa) were employed as a marker. The detection was carried out at a wavelength of 280 nm.
Stability assay
To estimate the impact of the PAS#1(200) sequence instability of nanobody at the freeze-thaw condition and time storage, the purified samples were frozen at −70 °C for 3 months and the size distribution were measured after thawing by DLS as previously mentioned.
Binding potency
The binding of the native protein and the PASylated nanobody to human rhVEGFA165 was determined by an enzyme-linked immunosorbent assay (ELISA). One hundred microliters of the recombinant human VEGFA165 [(rhVEGFA165; R&D systems)] was coated in 96-well microtitre plate for overnight at a concentration of 50 ng/ml in 50 mm NaHCO3 adjusted to pH 9.6 at 4 °C. The wells were then blocked with 200 µl skim milk 3% (w/v) in PBS for 1 h at room temperature and washed three times with PBS/T. Different concentrations of the native protein, PASylated nanobody, and bevacizumab (control) (10−4–10−16 M) were added to the wells and further incubated for 1 h at room temperature. The wells were then washed three times with PBS/T and incubated with anti-His HRP conjugated antibody [(Roche) (1:2000 dilution)] for 1 h at room temperature. Finally, 100 μl 3, 3', 5, 5'-tetramethylbenzidine [(TMB, ready to use) (PishtazTeb, Tehran, Iran)] was added as a substrate to the wells and the absorbance was measured at 450 nm by a microplate reader spectrophotometer (EPOCH, BioTeK).
Surface plasmon resonance-based kinetics measurement
Surface plasmon resonance (SPR) experiments were accomplished by Biacore X100 (GE Healthcare) at 25 °C using HBS-EP running buffer (10 mm HEPES pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.005% (v/v) detergent p20, pH 7.4) on CM5 (CM5 carboxymethyl dextran) sensor chip (XanTecBioanalytics GmbH) that had been coated with anti-His antibodies (Biacore His-capture kit, GE Healthcare) and amine coupling kit (GE Healthcare). The native protein and PASylated nanobody were injected in an appropriate concentration at a flow rate of 30 µl/min for 180 s. with a surface density of about 800 resonance units (RU). The rhVEGFA165 (R&D Systems) was injected at different concentrations (0.312, 0.625, 1.25, 2.5, 5, and 10 μg/ml) HBS-EP buffer at a flow rate of 30 µl/min for 180 s. Chip regeneration was achieved with regeneration buffer (10 mm glycine-HCl at pH 1.5) to eliminate all the binding proteins. Kinetic analyses were performed by Biacore X100 Evaluation software version 2 and the ka (association rate constant), kd (dissociation rate constant) and KD (equilibrium binding constants) parameters were finally determined.
VEGF neutralization assay
In order to evaluate the antiproliferative effect of the PASylated nanobody, 1 × 104 cells (HUVECs, Lonza) were cultured in EBM (Lonza) supplemented with 2% FBS in 96-well culture plate (triplicate) at a 37 °C incubator with 5% CO2. HEK293 cells were used as a VEGFR2 negative cell line. Different concentrations (0.25–20 µg/ml) of the native protein, PASylated nanobody and bevacizumab (control) were mixed with a constant amount of 50 ng/ml VEGFA in a separate 96-well microplate and incubated at 37 °C for 4 h. The mixtures were then transferred to the culture wells and incubated for 72 h. Thirty microliters of MTT solution (5 mg/ml) Merck was added to the wells and incubated for 4 h at 37 °C. The dye was finally solubilized by 100 µl of dimethyl sulphoxide (Sigma-Aldrich) and the absorbance was measured at a wavelength of 570 nm using a microplate reader spectrophotometer (EPOCH, BioTeK).
In vitro cell migration assay
In vitro cell migration assay was done by Scratch Wound Healing Assay. Briefly, HUVEC cells (1 × 104 cells) were cultured in a 24-well culture plate as described previously to reach ∼70–80% cell confluence. The monolayer cells were gently wounded with a sterile pipette tip across the centre. After washing with PBS, different concentrations (2–15 µg/ml) of the native protein, PASylated nanobody and bevacizumab (control) were mixed with a constant amount of 50 ng/ml VEGFA and incubated at 37 °C for 4 h. The mixtures were transferred to the cultured cells and incubated for 48 h. Then, the cells were washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 10 min. Finally, the cells were stained with Giemsa’s stain and the cell migration was inspected by an inverted microscope (TS100; Nikon, Tokyo, Japan) equipped with a digital camera (Nikon, ELWD 0.3/OD75).
3D capillary tube formation assay
3D capillary tube formation assay was performed using standard method [Citation29]. In summary, HUVEC cells were mixed with sterilized cytodex-3 microcarrier beads (Amersham, Pharmacia Biotech, Piscataway, NJ) at a ratio of 30 cells per bead in 1 ml of DMEM medium with 10% FBS and shaken slowly every 20 min for 4 h. After that, the mixtures were transferred to a 24-well tissue culture plate and incubated for 12–16 h in a 37 °C incubator with 5% CO2. For collagen gel formation, seven volumes of cold collagen solution with one volume of 10× minimal essential medium (MEM) and two volumes of sodium bicarbonate (11.76 mg/ml) were mixed and kept on ice to avoid immediate gelation. Then, beads with cells were re-suspended in type I collagen gel. One hundred microliters of collagen/bead mixture were added to each well of a 96-well tissue culture plate and permitted to clot for 20 min at 37 °C. Then, 200 µl of DMEM was added to each well. For studying the anti-angiogenic effect, different concentrations (0.25–20 µg/ml) of the native protein, PASylated nanobody and bevacizumab (control) were mixed with a constant amount of 50 ng/ml VEGFA on a separate plate and incubated at 37 °C for 4 h. Subsequently, the mixtures were added to the cells and incubated for 3–5 d. Finally, the anti-tubulogenesis effects were inspected by an inverted microscope (TS100; Nikon) equipped with a digital camera (Nikon, ELWD 0.3/OD75).
In vivo pharmacokinetic study
Forty-five female BALB/c mice (18–20 g) (Pasteur Institute of Iran) were randomly divided into two groups for pharmacokinetic study (PK). All mice had received an i.v. the dose of 5 mg protein/kg b.w. of the native protein and the PASylated nanobody. Blood samples were taken after 15 min, 30 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 24 h, and 48 h of injection and the plasma was obtained by centrifugation at 14,000 g for 20 min at4 °C and stored at −20° C. The determination of protein concentration was performed by a homemade ELISA of plasma half-life calculations. Briefly, 96-well microtitre plates were coated with 100 µl of the recombinant human VEGFA165 [(rhVEGFA165; R&D systems)] at a concentration of 1 µg/ml for the native protein and 50 ng/ml for PASylated nanobody, in 50 mm NaHCO3 buffer (pH 9.6) and incubated at 4 °C overnight. After that, the wells were blocked with 200 µl 3% (w/v) skim milk in PBS for 1 h at room temperature and washed three times with PBS/T. The plasma samples were prepared in dilution series in PBS/T supplemented with up to 0.5% (v/v) untreated mouse plasma and incubated for 1 h. Then, the wells were washed three times with PBS/T and incubated with anti-His HRP conjugated antibody (1:2000 dilution) (Roche) for 1 h at room temperature. Finally, the substrate (TMB, ready to use) (PishtazTeb) was added and the absorbance was measured at 450 nm by a microplate spectrophotometer (EPOCH, BioTeK). The terminal half-life was determined using a log-linear trapezoidal method and the mean area under the curve (AUC0–∞), clearance (CL), volume of distribution (Vd) values was also calculated.
Statistical analysis and curve plotting
Graph plotting and statistical analysis were done using Prism 6 software package (GraphPad Software, Inc., La Jolla, CA) and a p value less than .05 was regarded as statistically significant in all assays. For in vitro assays, the statistical analyses were performed using two-way analysis of variance (ANOVA).
Results
3D modelling and energy minimizations
Top generated models by I-TASSER were subjected to secondary structure analysis. Nanobody revealed a beta-strand structure expected for the spatial conformation of nanobodies deposited in PDB database (see 4I13, 5E0Q, and 1I3V PDB files). However, an unstructured and mainly random coil was obtained for PAS sequence. Selected structures were subjected to energy minimization runs to obtain the more equilibrated structures. Snapshots at 0, 25, and 50 ns of energy minimizations are shown in . The nanobody region of energy minimized structures was superposed () and TM-score was calculated [Citation25,Citation30]. TM-scores indicated 94% similarity in nanobody region of two energy minimized structures. Additionally, backbone RMSD calculations realized no detrimental effect of PAS sequence on structure stability of nanobody (). Data indicated that PASylated nanobody has more than twice the volume than unfused form. Moreover, as shown in , the surface accessible surface area (SASA) of whole proteins and CDR3 loops did not elucidate any spatial restriction of PAS sequence on native protein.
Figure 1. Snapshots before and after energy minimizations step runs for (a) nanobody and (b) PASylated nanobody. As depicted, no steric hindrance was observed for CDR3 region (red colour) in both proteins. (c) Superimposition of nanobody fragment revealed just a small spatial distortion in the three-dimensional structure of nanobody.
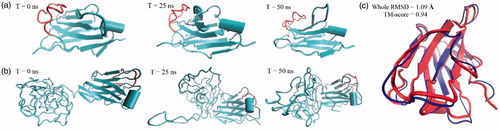
Table 1. Calculated conformational parameters for equilibrated structures.
Table 2. Calculated kinetic and affinity parameters for PASylated nanobody and native protein [ka: association rate constant; kd: dissociation rate constant and KD: equilibrium binding constant].
Electrophoretic mobility assessment
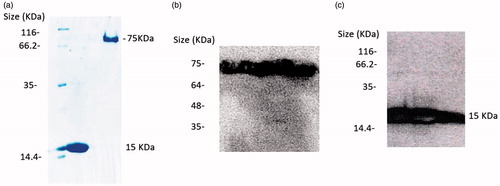
PASylated and native proteins were expressed in the periplasm of E. coli in a soluble form. Polyacrylamide gel electrophoresis remarkably revealed that the PASylated nanobody showed a significantly reduced electrophoretic mobility with a migration at around ∼75 kDa (). The bonds were confirmed with western blot using chemiluminescence ().
Figure 2. (a) Electrophoretic mobility of anti-VGEFA nanobody and the PASylated form on 15% SDS-polyacrylamide gel. Lane 1: Marker; Lane 2: Nanobody; Lane 3: Nb-PAS#1(200). SDS-PAGE investigation shows Ni-NTA-purified nanobody and PASylated nanobody have molecular weights of 15 and 75 kDa, respectively. Western blot analysis using ECL method identified specified bonds for the proteins (b and c).
Hydrodynamic radius distribution and molecular weight analysis
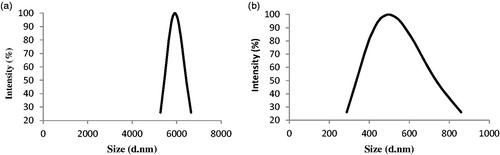
The effect of PAS#1(200) sequence on the hydrodynamic radius and molecular weight of nanobody were investigated using DLS and SEC techniques, respectively. A 5.5-fold increase in hydrodynamics radius was observed due to attachment of PAS#1(200) sequences compared to native protein (). However, in SEC method, a single peak corresponded to the PASylated nanobody was observed in the chromatogram with an increase in size in comparison to the calculated molecular mass of 32 kDa (). Mass spectroscopy confirmed the expected molecular mass (28591.3867 Da) as well as monodisperse composition (just one peak) of the PASylated protein, without any premature or truncated products ().
Figure 3. Hydrodynamic radius and molecular weight characterizations. (a) SEC of native protein and PASylated nanobody was performed using molecular weight markers [(1) 670 kDa (Thyroglobulin, bovine), (2) 158 kDa (γ-globulin, bovine), (3) 44 kDa (Ovalbumin, chicken), (4) 17 kDa (Myoglobin, horse) and (5) 1.35 kDa (Vitamin B12)]. As depicted, a remarkable increase has seen in molecular weight of PASylated nanobody. Size distribution analysis of (b) native and (c) PASylated nanobody revealed main peaks at 77.16 and 424 nm for native and PASylated form, respectively. (d) Mass spectrometric analysis by MALDI-TOF/TOF. A single peak with a molecular weight of 28591.3867 Da was observed for Nb-PAS#1(200).
![Figure 3. Hydrodynamic radius and molecular weight characterizations. (a) SEC of native protein and PASylated nanobody was performed using molecular weight markers [(1) 670 kDa (Thyroglobulin, bovine), (2) 158 kDa (γ-globulin, bovine), (3) 44 kDa (Ovalbumin, chicken), (4) 17 kDa (Myoglobin, horse) and (5) 1.35 kDa (Vitamin B12)]. As depicted, a remarkable increase has seen in molecular weight of PASylated nanobody. Size distribution analysis of (b) native and (c) PASylated nanobody revealed main peaks at 77.16 and 424 nm for native and PASylated form, respectively. (d) Mass spectrometric analysis by MALDI-TOF/TOF. A single peak with a molecular weight of 28591.3867 Da was observed for Nb-PAS#1(200).](/cms/asset/078dd510-442a-460b-ab26-c710a2c78314/ianb_a_1369426_f0003_c.jpg)
Protein aggregation assay
To determine the effect of the PAS#1(200) sequence on the solubility of native protein, DLS was performed on the samples again after 3 months incubation at −70 °C. The results revealed that the solubility of PASylated protein was considerably affected by attachment of PAS#1(200) sequence which about 77-fold increase in size was observed for native protein while no change was seen in PASylated nanobody at the same conditions ().
Figure 4. Light scattering for assessing protein aggregates. A peak shift to micron size range was observed for native protein after 3 months incubation (77.16–5919.9 nm), while no significant change was seen for PASylated form at the same conditions (424–496.5 nm).
Secondary structure analysis
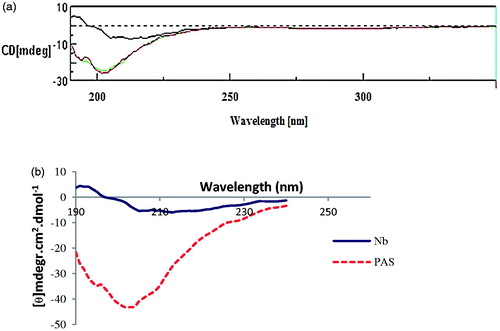
To gain information about conformational effects of the PAS#1(200) sequence circular dichroism (CD) spectroscopy was performed for both proteins (). Molar differences in CD spectra were then calculated with regarding native protein. The CD spectrum of the PASylated fusion protein revealed a major negative minimum shift around 204 nm () corresponding to an increase in random coil structure of the protein.
Figure 5. Secondary structure analysis by Far-UV CD spectroscopy. (a) CD spectra of nanobody (upper line) and PASylated nanobody (lower lines). (b) Molar ellipticity calculations demonstrate negative shift around 204 nm in the curve of PASylated nanobody compared to native protein.
Charge characterization
The charging behaviour of the fusion protein was investigated using the IEF technique. Data revealed PAS#1(200) sequence had a negligible effect on the isoelectric point (pI) of native protein with pI about of 6.98 ().
Figure 6. Isoelectric focusing of PASylated nanobody and native protein revealed both proteins have a similar net charge with a pI around 7 [Lane 1; Marker; Lane2; Nanobody, Lane 3; Nanobody-PAS#1(200)].
![Figure 6. Isoelectric focusing of PASylated nanobody and native protein revealed both proteins have a similar net charge with a pI around 7 [Lane 1; Marker; Lane2; Nanobody, Lane 3; Nanobody-PAS#1(200)].](/cms/asset/ef99b086-cd08-4ae7-906e-3bc512d6ef8a/ianb_a_1369426_f0006_b.jpg)
Binding potency and SPR-based kinetics measurements
Binding potencies of PASylated nanobody and native protein to rhVEGFA165 were determined by an ELISA. The EC50s were obtained as 1.1 × 10−7, 1.8 × 10−5 and 7.1 × 10−14 M for PASylated nanobody, nanobody, and bevacizumab as control, respectively, using non-linear curve fitting. The results indicated PAS#1(200) sequence did not affect antigen binding of native protein ().
Figure 7. Binding assessment of nanobody and nanobody-PAS#1(200) by ELISA. Bevacizumab was used as positive control and data are represented as mean ± SD. The results showed that the PASylated nanobody is reached to saturation state at lower concentrations than the native protein.
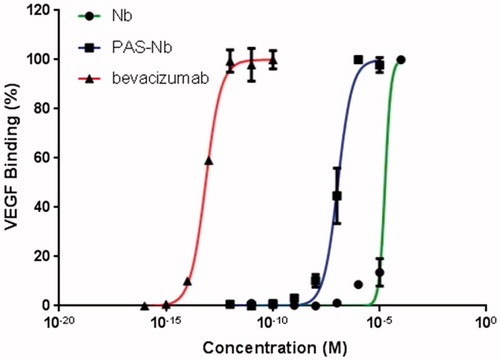
Figure 8. Biacore sensorgrams obtained by binding kinetics assay. (a) Nanobody with rhVEGFA165. (b) PASylated nanobody with rhVEGFA165. Curves from top to bottom corresponded to the solutions of 0.312, 0.625, 1.25, 2.5, 5, and 10 μg/ml of VEGFA165.
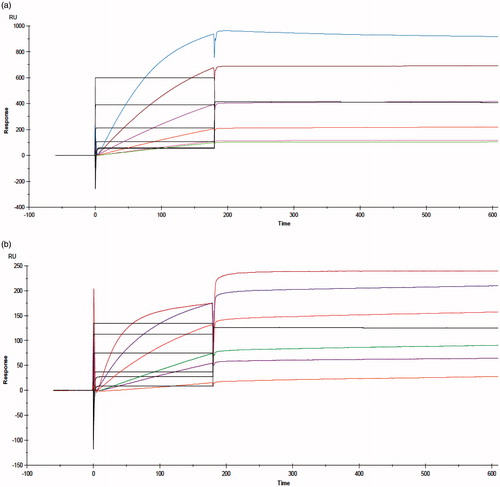
More investigation on the thermodynamic affinities and binding kinetics against rhVEGFA165 were performed by SPR spectroscopy on CM5 chip (SPR, Biacore X100) (). demonstrates the results of kinetic and affinity parameters. The binding constant (KD) of PASylated Nanobody was so close to the native one. These data were confirmed that PAS#1(200) polypeptide do not have undesirable effects on the binding kinetics of native protein.
Antiproliferation assay
Antiproliferative activity of PASylated nanobody, nanobody and bevacizumab were evaluated by a dose-response MTT assay using HUVEC cells. HEK293 were used as non-expressing VEGFR2 cells for the control and the data were analyzed using two-way ANOVA statistical model. In all groups, significant differences (p < .05) were observed in comparison to VEGFA group while it was not for others (PASylated nanobody, nanobody, and bevacizumab) (). In addition, no significant antiproliferative activities were observed for groups on HEK293 as VEGFR2 negative cells ().
Figure 9. Antiproliferative activity of PASylated nanobody, nanobody and bevacizumab on (a) HUVECs and (b) HEK293. Data are represented as mean ± SD and asterisks show the statistical level of significance between cell proliferation of the samples and VEGFA group (***p<.001).
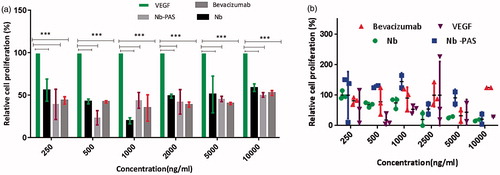
Figure 10. (a) Anti-migration activity of PASylated nanobody in comparison to the native protein on HUVECs. Data are represented as mean ± SD and bevacizumab was used as a control. (b) Representative snapshots, captured from migration assay on HUVECs (1) Control (tfinal), (2) scratched cells (tinitial) (3) nanobody (2000 ng/ml) and (4) Nb-PAS#1(200) (2000 ng/ml). As depicted in the figure, both Nb-PAS#1(200) and nanobody strongly inhibit the migration of HUVECs at a concentration of 2000 ng/ml.
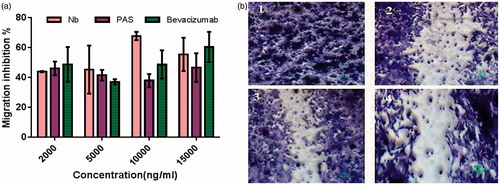
In vitro migration assay
Anti-migration activity was performed by scratch wound healing assay on HUVECs. Quantitative data were obtained by distance measurements using Image J software. Statistical analysis indicated that the PAS#1(200) did not impose significant negative effect (p < .05) on the migration activity of the native protein (). Moreover, no significant difference was found to be between bevacizumab and other groups.
3D capillary tube formation assay
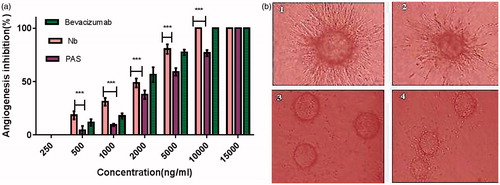
Since the endothelial cells (ECs) are able to form tubular structures on the cytodex matrix, this method was used for assessment of angiogenesis to define the effect of PAS#1(200) sequence on both morphological differentiation of ECs into capillary-like structures and inhibitory effect on the tube formation. Untreated wells exhibited a branching pattern of capillary tube structures while a dose-dependent anti-tubulogenesis behaviour was observed for the both PASylated and native proteins (). However, a significant decrease in inhibitory effect was observed for the PASylated nanobody in comparison to native protein (p < .001, ). Moreover, a significant inhibitory effect (p < .05) was observed for native protein at a concentration of 1000 ng/ml in compare with bevacizumab.
Figure 11. (a) Anti-angiogenesis effects of PASylated nanobody assessed by in vitro tube formation method. Data are represented as mean ± SD and asterisks show the significant level of capillary branch-inhibitory difference between native protein and PASylated nanobody (***p < .001). (b) Representative snapshots captured from the tube formation assay on HUVECs (1) Control (no drug), (2) nanobody (2000 ng/ml), (3) nanobody (10,000 ng/ml) and (4) Nb-PAS#1(200) (10,000 ng/ml).
Pharmacokinetic assessments
Pharmacokinetic profiles of the proteins were studied in female BALB/c mice after i.v. injection of 5 mg/kg of each protein. Plasma concentrations of each sample were determined using an indirect ELISA at defined time intervals. The curves were fitted with a two-decay regression model and pharmacokinetic parameters were calculated using a log-linear trapezoidal method (). Data revealed that the terminal half-life (T1/2) for the PASylated nanobody was significantly increased, by a factor of 14, relative to native protein. Furthermore, the volume of distribution (Vd), and clearance (CL) of PASylated nanobody were also remarkably improved (). The AUC for the PASylated nanobody was also significantly increased, with a factor of 294, compared to native protein ().
Figure 12. Pharmacokinetic profiles of PASylated nanobody and native protein at an intravenous dose of 5 mg/kg in BALB/c mice. As depicted in the figure, native protein and the PASylated nanobody are not detectable in the plasma after 6 and 54 h, respectively.
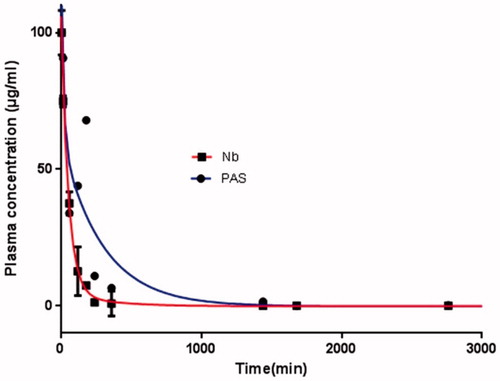
Table 3. Calculated pharmacokinetic parameters using a log-linear trapezoidal method for PASylated nanobody and native protein.
Discussion
In order to have concurrently advantageous of the nanosize dimension of small molecules and target specificity of conventional antibodies, we developed a prolonged acting of an anti-VEGFA miniaturized antibody for angiogenesis inhibition by attachment of a natural amino acid polymer having a controllable size at the nanoscale. Miniaturized antibodies have potential applications in both diagnosis and treatment programs, but in vivo performance are often hampered by a very short serum half-life. PASylation technology has recently provided an easy way to design new biopharmaceuticals with improved pharmacokinetic properties [Citation31]. It acts via genetic fusion of long unstructured amino acid sequences to the target protein which leads to a remarkable increase in the nanosize of the protein and a subsequent reduced glomerular filtration rate (GFR). Moreover, it allows practically efficient expression in microbial systems along with eukaryotic hosts, because of special biochemical features including lack of charge, and hydrophilic nature of the sequence [Citation20].
In this study, for the first time, PASylation was employed for size adjustment of a miniaturized antibody to improve its physicochemical properties, stability, and solubility as well as pharmacokinetic profile. For this purpose, a previously designed anti-VEGFA nanobody was fused to 10 repeats of PAS#1sequence, expressed in E. coli and changes in electrophoretic mobility, mass, charge, secondary structure, hydrodynamic volume, as well as binding potency and kinetic affinity to VEGFA were evaluated. In addition, in vitro antiproliferative, cell migration, and angiogenesis, as well as pharmacokinetic parameters of the PASylated anti-VEGFA nanobody were compared to the native anti-VEGFA nanobody.
Surprisingly, PAS#1(200) sequence could considerably affect the physical properties of anti-VEGFA nanobody. Likewise, an unexpected two-fold decrement in the electrophoretic mobility of protein under study could be observed. This un-contemplated mobility shift can be clearly explained by the hydrophilic nature and spatial conformation of PAS sequence. Because the real increase in mass upon coupling of PAS sequence must be about 17 kDa with an apparent increment of mass to 28.595 kDa. In the other hand, the IEF results confirmed the sequence has not affected the charge of macromolecule as previously shown by Schlapschy in a similar study [Citation20]. Therefore, the hydrophilic intrinsic of sequence followed by a more condensed water molecules network around the sequence cause a lower binding capacity to the SDS, which normally binds to proteins via its hydrophobic part and provides main driving force for the mobility through negatively charged sulphate head groups in an electric field [Citation20,Citation28]. In addition, secondary structure investigation elucidated an increase in random coil structure of the macromolecule, which in turn, donates flexibility to the sequence and increases occupied a volume of the macromolecule in the solvent much more than usual [Citation20,Citation28,Citation32].
The hydrophilicity and unstructured nature of PAS sequence were more tangible when long-term storage of PASylated nanobody at −70 °C did not reveal any aggregation by DLS. It must be mentioned that in this study, the nanobody was very prone to aggregation even at short time storage or in downstream processing (data not shown). As seen in the SEC chromatogram, a minor peak related to aggregate forms of nanobody remained even after long centrifugation times. Therefore, in addition to enhanced stability at harsh conditions, it seems the PAS sequence significantly increases the solubility of the protein. The reason could be explained by the exposure of polar groups of unstructured chain that are in contact with solvent molecules [Citation20]. This property is unique among PEG mimetic peptides. Even in some cases, it has been reported the fused sequence severely decreases the solubility of the sequence in glycine-rich amino acid sequence [Citation33].
As it is discussed above, the net charge of macromolecule did not affect after PASylation. Therefore, there is no need to consumedly modify or add more steps in the downstream processes of proteins after PASylation. In contrast, this is often occurring in PEGylation technology that often suffers from extra labouring procedures in purification steps, which in turn, leads to larger costs of the finished pharmaceutical product. Moreover, it is shown that attachment of negatively or positively charged fusions cause the improper distribution of the drug in tissues and also alter the affinity of the drug to the receptor. This phenomenon is observed for XTENylation technology due to the coupling of negatively charged glutamic acid-rich sequences [Citation20].
Successful and sufficient penetration of the drug into the target tissue is one of the most important steps in the efficacy of treatment in solid tumours. Although monoclonal antibodies have been approved for treatment of cancerous diseases since 1997 [Citation34], however, due to a larger size and rigid structure of a macromolecule, the treatment efficacy of drugs in such situations are hampered [Citation2]. The small size of miniaturized antibodies and on the other hand, as showed by CD spectroscopy in the current study, the elasticity, as well as the controllable size of PAS sequence provides a better permeability of the PASylated drug into the target.
The computational study realized that the PAS sequence forms a random coil structure which also confirmed by CD. The unstructured shape of PAS sequence does not conformationally interrupt or alter the key structural segments required for receptor binding by adding PAS sequence to the C-terminus of the protein. The computational study also elucidated no change in secondary structure of nanobody was observed after attachment to the PAS#1(200) sequence. Binding kinetics investigation also indicated no change in binding constant (KD) was observed after coupling of the sequence. This is surprising because no linker for the sequence attachment was employed. It must be mentioned that for half-life extension by any other related technologies, even the usage of a linker cannot prevent a reduction in the receptor affinity. Since CDR3 loop have been suggested as key determinant antigen recognition and important nanobodies active sites [Citation35], we more focused on CDR3 region in in silico study. SASA calculation of CDR3 loops point out that PASylation chain does not create spatial prevention and the CDR3 loop is free enough accessible to interact or bind to the other biomolecules. The same results were also observed in the other PASylation studies [Citation2,Citation36], however, in one study, it was reported that the association rate constant (kon) of PASylated leptin was reduced little as compared to native protein [Citation32]. The smaller affinity parameters for either nanobody or PASylated form in comparison to the commercially available monoclonal antibody (Bevacizumab, 58 pM) is described by the availability of more antigen binding sites for VEGFA binding [Citation37].
Despite remarkable changes in physicochemical properties of fused protein, in vitro biological activity was not altered after coupling of the sequence when studied by antiproliferative, migration and angiogenesis assays. Proliferation assay showed PAS fused protein and un-fused form inhibited equally proliferation of ECs, however, none of them could prevent the proliferation of HEK293 cells because of lacking VEGFR2 receptor. The scratch wound healing assay was used to study cell migration in vitro [Citation38]. Nanobody and PASylated form, as well as bevacizumab similarly, inhibited the migration of ECs at a dose range of 2–15 µg/ml. Additionally, an EC branching morphogenesis assay was done to investigate the anti-vascularizing effect of the PAS sequence. Quantification of the number of capillary-like structures in microscopic images well clarified the inhibition of tube formation in ECs on a three-dimensional basement membrane matrix. Although this inhibition phenomenon was observed in a dose-depended manner for all proteins, nevertheless the inhibitory effect was significantly more in the case of nanobody as compared to fused protein. This reduction could be explained by the smaller size of nanobody, which allows more penetration of the molecule into its site of action because this decreased inhibitory effect could be compensated by adding higher concentrations of the PASylated protein to obtain the same inhibitory effect. In addition, as depicted in the images, the length of such branched structures was also apparently decreased after treatment with all proteins.
Despite advantages of miniaturized antibodies for medical uses, as mentioned, the poor pharmacokinetic properties usually restrict their clinical applications. Therefore, many investigations are undergoing to overcome the obstacle by various group of investigators. Ablynx recently attempted to compensate the poor pharmacokinetic of nanobodies by dimerization of two distinct nanobodies which one of them targets serum albumin for in vivo half-life extension. This approach acts via two different but simultaneous pathways (i) change in the hydrodynamic radius and (ii) FcRn-mediated recycling. In one study, the binding of a nanobody to anti-albumin nanobody decreased clearance by a factor of 376 as the terminal half-life extended to 4.9 d in monkeys compared to unfused form [Citation39]. In the second study, binding of anti-albumin nanobody to high-affinity anti-Il-6 R nanobody (Alx-0061) increased the half-life of nanobody to 6.6 d in Cynomolgus monkeys after a single dose intravenous administration [Citation40]. Another approach, used by Pfizer, is increasing the hydrodynamic radius of a macromolecule by the binding the different size of poly(ethylene glycol) polymers to the protein which extensively increased plasma half-life of PEGylated anti-TNF nanobody [Citation41]. Nonetheless, the arbitrary control of plasma retention time, unexpected effect of polymer- or structurally defined protein on the biological property as well as the laborious and complex downstream processes, limited such approaches to become a simple and general method for creating long-acting biopharmaceuticals so far.
PEG mimetic peptides technology, however, shows a promising prospect in this field. As it is shown in our study, due to the coupling of the PAS sequence, the fused protein exhibited a lower bi-exponential decay with a single dose intravenous administration. The terminal half-life (t1/2) was significantly increased by a factor of 14 for the Nanobody-PAS#1(200). These results confirm a strong influence of the PAS#1(200) sequence on in vivo pharmacokinetic profile of nanobody. Computational analysis showed an increase in hydrodynamics volume of PASylated protein, which is one of the most critical parameters for half-life extension with a factor of more than 2. Such enhanced improvement in pharmacokinetic profile was also reported in our previous study. A 15.6-fold half-life extension of erythropoietin was observed by the attachment of the same length of PAS sequence without any change in biological activities in vitro and in vivo [Citation42]. As it is shown in our study, enhanced pharmacokinetic parameters of nanobody could be explained by changes in physicochemical properties of the protein. According to the data obtained from CD and SEC experiments, increased hydrodynamic volume and a random chain behaviour of PAS polypeptides led to an impressive reduction in clearance through GFR, similar with coupling by PEG [Citation20]. The long-term retention of the protein in the circulation, as well as enhanced penetration into the target tissue, provides more availability of the drug in the tumour nano-environment.
Conclusions
Our study demonstrated that nanosized controllable natural polymers, like PAS sequence, could efficiently compensate poor intrinsic characteristics of small therapeutic proteins. The simplicity and applicability of this technology with superior effects on stability, solubility, and pharmacokinetic without any deleterious impact on the potency, will make it a magic technology for developing of bio better therapeutics.
Acknowledgements
The authors wish to express their deep gratitude to Ms. Azarian, Mr. Moazzami, Mr. Chiani, Dr. Khajeh and Dr. Dabirmanesh for technical supports. We also thank Dr. Namvar Asl and Dr. Dizaji for in vivo experiments. The computational resource for in silico study was provided by Sharif University of Technology. This project was financially supported by the Pasteur Institute of Iran.
Disclosure statement
The authors report no conflicts of interest in this work.
References
- El‐Kenawi AE, El-Remessy AB. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol. 2013;170:712–729.
- Mendler CT, Friedrich L, Laitinen I, et al. High contrast tumor imaging with radio-labeled antibody Fab fragments tailored for optimized pharmacokinetics via PASylation. MAbs. 2015;7:96–109.
- Herrington-Symes AP, Farys M, Khalili H, et al. Antibody fragments: prolonging circulation half-life special issue-antibody research. Adv Biosci Biotechnol. 2013;4:689–698.
- Wu D, Gao Y, Qi Y, et al. Peptide-based cancer therapy: opportunity and challenge. Cancer Lett. 2014;351:13–22.
- Kong Q, Yao Y, Chen R, et al. [Progress in nanobody and its application in diagnosis]. Sheng Wu Gong Cheng Xue Bao. 2014;30:1351–1361.
- Nelson AL. Antibody fragments: hope and hype. MAbs. 2010;2:77–83.
- Harmsen M, De Haard H. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77:13–22.
- Kijanka M, Dorresteijn B, Oliveira S, et al. Nanobody-based cancer therapy of solid tumors. Nanomedicine (Lond). 2015;10:161–174.
- Dingermann T. Book review: therapeutic proteins: strategies to modulate their plasma half‐lives. Biotechnology J. 2013;8:163–164.
- Kontermann RE. Half?life modulating strategies?an introduction. Therapeutic proteins: strategies to modulate their plasma half-lives. 2012.
- Samoudi M, Tabandeh F, Minuchehr Z, et al. Rational design of hyper-glycosylated interferon beta analogs: a computational strategy for glycoengineering. J Mol Graph Model. 2015;56:31–42.
- Maleki A, Madadkar-Sobhani A, Roohvand F, et al. Design, modeling, and expression of erythropoietin cysteine analogs in Pichia pastoris: improvement of mean residence times and in vivo activities through cysteine-specific PEGylation. Eur J Pharm Biopharm. 2012;80:499–507.
- Ahangari Cohan R, Madadkar-Sobhani A, Khanahmad H, et al. Design, modeling, expression, and chemoselective PEGylation of a new nanosize cysteine analog of erythropoietin. Int J Nanomedicine. 2011;6:1217–1227.
- Hamidi M, Rafiei P, Azadi A. Designing PEGylated therapeutic molecules: advantages in ADMET properties. Expert Opin Drug Discov. 2008;3:1293–1307.
- Sleep D. Albumin and its application in drug delivery. Expert Opin Drug Deliv. 2015;12:793–812.
- Schmidt SR. Fusion proteins: applications and challenges. Fusion protein technologies for biopharmaceuticals. John Wiley & Sons, Inc.; 2013.
- Turecek PL, Bossard MJ, Schoetens F, et al. PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs. J Pharm Sci. 2016;105:460–475.
- Garay RP, El-Gewely R, Armstrong JK, et al. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin Drug Deliv. 2012;9:1319–1323.
- Harari D, Kuhn N, Abramovich R, et al. Enhanced in vivo efficacy of a type I interferon superagonist with extended plasma half-life in a mouse model of multiple sclerosis. J Biol Chem. 2014;289:29014–29029.
- Schlapschy M, Binder U, Börger C, et al. PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng Des Sel. 2013;26:489–501.
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802.
- Mackerell AD Jr, Feig M, Brooks CL III. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–28.
- Kazemi-Lomedasht F, Behdani M, Bagheri KP, et al. Inhibition of angiogenesis in human endothelial cell using VEGF specific nanobody. Mol Immunol. 2015;65:58–67.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254.
- Zvonova EA, Ershov AV, Ershova OA, et al. PASylation technology improves recombinant interferon-β1b solubility, stability, and biological activity. Appl Microbiol Biotechnol. 2017;101:1975–1987.
- Ghavamipour F, Shahangian SS, Sajedi RH, et al. Development of a highly-potent anti-angiogenic VEGF8-109 heterodimer by directed blocking of its VEGFR-2 binding site. FEBS J. 2014;281:4479–4494.
- Xu J, Zhang Y. How significant is a protein structure similarity with TM-score =0.5? Bioinformatics. 2010;26:889–895.
- Skerra A, Theobald I, Schlapschy M, inventors; Technische Universitat Munchen, Munich (DE), assignee. Biological active proteins having increased in vivo and/or in vitro stability. US Patent 9260494 B2. 2013.
- Morath V, Bolze F, Schlapschy M, et al. PASylation of murine leptin leads to extended plasma half-life and enhanced in vivo efficacy. Mol Pharm. 2015;12:1431–1442.
- Schlapschy M, Theobald I, Mack H, et al. Fusion of a recombinant antibody fragment with a homo-amino-acid polymer: effects on biophysical properties and prolonged plasma half-life. Protein Eng Des Sel. 2007;20:273–284.
- Oldham RK, Dillman RO. Monoclonal antibodies in cancer therapy: 25 years of progress. J Clin Oncol. 2008;26:1774–1777.
- Kromann-Hansen T, Oldenburg E, Yung KW, et al. A Camelid-derived antibody fragment targeting the active site of a serine protease balances between inhibitor and substrate behavior. J Biol Chem. 2016;291:15156–15168.
- Harari D, Kallweit N, Abramovich R, et al. 73: Enhanced in vivo efficacy using a novel long-life type I interferon variant in a mouse model of multiple sclerosis. Cytokine. 2014;70:45.
- Papadopoulos N, Martin J, Ruan Q, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–185.
- Liang C-C, Park AY, Guan J-L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333.
- Hoefman S, Ottevaere I, Baumeister J, et al. Pre-clinical intravenous serum pharmacokinetics of albumin binding and non-half-life extended Nanobodies®. Antibodies. 2015;4:141–156.
- Van Roy M, Ververken C, Beirnaert E, et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther. 2015;17:135.
- Vugmeyster Y, Entrican CA, Joyce AP, et al. Pharmacokinetic, biodistribution, and biophysical profiles of TNF nanobodies conjugated to linear or branched poly (ethylene glycol). Bioconjug Chem. 2012;23:1452–1462.
- Hedayati MH, Norouzian D, Aminian M, et al. Molecular design, expression and evaluation of PASylated human recombinant erythropoietin with enhanced functional properties. Protein J. 2017;36:36–48.