
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A new class of cell penetrating peptides (CPPs) named peptide amphiphile was designed to improve the intracellular uptake and the antitumor activity of epirubicin (EPR). Various amphiphilic CPPs were synthesized by solid phase peptide synthesis method and were chemically conjugated to EPR. Their corresponding nanoparticles (CPPs-E4 and CPPs-E8) were prepared via non-covalent binding of the peptides and polyanions. Cytotoxicity and anti-proliferative activity were evaluated by MTT assay. Cellular uptake was examined by flow cytometry and fluorescence microscopy. The CPPs exhibited slight cytotoxicity. Binding of polyglutamate to CPPs (CPPs-E4 and CPPs-E8 nanoparticles) decreased their cytotoxicity. CPPs-E8 nanoparticles showed lower cytotoxicity than CPPs-E4 nanoparticles. Cellular uptake of K3W4K3-E8, K2W4K2-E8 and W3K4W3-E8 reached 100% with no difference between each of the mentioned CPPs and its nanoparticles at 50 µM. The anti-proliferative activity of EPR was enhanced following conjugation to peptides and nanoparticles at 25 µM. CPPs-EPR-E4 and CPPs-E8-EPR nanoparticles displayed higher anti-proliferative activity than CPPs-EPR at 25 µM. CPPs-E8-EPR nanoparticles showed higher anti-proliferative activity than CPPs-E4-EPR. K3W4K3-E8-EPR nanoparticles exhibited the highest anti-proliferative activity at 25 µM. The synthesized peptide nanoparticles are proposed as suitable carriers for improving the intracellular delivery of EPR into tumor cells with low cytotoxicity and high antitumor activity.
Graphical Abstract
Introduction
Despite vast advances in anticancer drugs during the last decade, cancer is still one of the important worries of the world [Citation1,Citation2]. Ineffective accumulation of drugs in tumors, lack of tumor specificity, heterogeneity of cancer cells, drug toxicity and drug resistance are factors that causes the lack of efficiency in cancer therapy [Citation1,Citation3,Citation4].
Epirubicin (EPR) is an anthracycline drug that has been used alone or in combination with other drugs for treatment of various cancers such as breast, ovarian, gastric and lung [Citation5,Citation6]. EPR is a stereoisomer of doxorubicin and is flavored over it. It has been shown that EPR is generally more successful than doxorubicin due to the higher tumor therapeutic efficiency and less side effects [Citation7,Citation8]. In fact, as a consequence of the reorientation of the hydroxyl group in the 4'-position of the daunosamine ring, epirubicin possess different pharmacologic properties than doxorubicin [Citation9]. First, it has a lower pKa. Accordingly, it is more lipophilic and better able to penetrate cells. Second, the glucuronidation of epirubicin and epirubicinol to inactive metabolites leads to a shorter terminal half-life for epirubicin (30 h) when compared with doxorubicin (45 h) [Citation8,Citation9]. Third, higher doses of epirubicin are required to produce the same degree of toxicity as doxorubicin. The doxorubicin-epirubicin dose ratios for similar toxicities are 1:1.2 for hematologic, 1:1.5 for nonhematologic and 1:1.8 for cardiac [Citation8,Citation10].
Although EPR exhibited activity in all phases of the cell cycle but it was most active in S and G2 phases. The antineoplastic effect of epirubicin occurs through a number of mechanisms. First, it intercalates between DNA nucleotide base pairs, resulting in the inhibition of DNA, RNA and protein synthesis. Second, the intercalation leads to topoisomerase II cleavage of DNA, which results in cytocidal activity. Third, epirubicin inhibits DNA helicase activity, which ultimately interferes with replication and transcription [Citation10,Citation11]. Various experiments have shown that EPR inhibits cell proliferation and DNA synthesis in various carcinoma cell lines. Although fewer, like chemotherapy drugs, EPR still shows side effects such as cardiac toxicity and myelosuppression [Citation7]. Development of resistance to a certain dose of anticancer drugs is another major limitation of EPR. EPR is a hydrophilic drug with high volume of distribution [Citation11].
Drug delivery systems modify the efficacy and toxicity of anticancer drugs. Moreover, drug delivery systems can avoid multi drug resistance (MDR) created through factors such as P-glycoprotein and multi drug resistance proteins (MRPs) [Citation12–18]. Chemical conjugation with a parent drug as “Prodrug” strategy is one of the drug delivery systems that has been widely used in EPR delivery [Citation12]. Several methods have been used to improve EPR delivery, which includes using nanodiamond [Citation19], SLN (solid lipid nanoparticles) [Citation20], phospholipids as multi drug resistance modulators of the epirubicin transport [Citation21], carbon nanoparticles [Citation22] and PLGA nanoparticles [Citation23].
Peptides and proteins are generally worthy in many aspects, such as their possible high potency, good selectivity and acceptable toxicity [Citation24]. Two decades ago, a new kind of peptides, commonly known as cell penetrating peptide (CPP), was discovered. These peptides have been extracted from natural proteins and have the capability to cross cellular membranes and mediate the uptake of a wide range of macromolecular cargoes [Citation1,Citation25–28]. The discovered peptides as non-invasive vectors with very limited toxicity introduced a novel field in drug delivery [Citation29]. In addition, they can be modified or designed de novo. They are typically short; usually 5–30 amino acids long [Citation28,Citation30–35]. These bioactive and biodegradable peptides are able to carry and deliver their cargos such as nucleic acids, proteins, drugs, or imaging agents, to the cytoplasm or nuclei of cells [Citation30,Citation33–35]. In the recent years, numerous natural and synthetic CPPs such as polyarginines, TAT (trans-acting activator of transcription) [Citation27] and peptide amphiphiles (PA) [Citation23] have improved the cellular uptake of various drugs such as taxol [Citation36], methotrexate [Citation16] and doxorubicin [Citation12]. Among these CPPs, peptide amphiphiles appear to be among the efficient systems in drug delivery [Citation37–39]. Universally, peptide amphiphiles consist of hydrophobic segments such as tryptophan and charged segments such as arginine or lysine [Citation37]. These peptides have a helical secondary structure with the hydrophobic and hydrophilic domains. They use the charged region for cell membrane interaction and the hydrophobic region for membrane perturbation and translocation [Citation30,Citation40]. Moreover, several studies have shown that the presence of tryptophan and backbone spacing can affect the uptake efficiency as well as its mechanism [Citation41].
In the present study several novel CPPs based on different designs containing lysine and tryptophan were synthesized. Highly active EPR (instead of doxorubicin) was utilized as a drug. These peptides were then conjugated to EPR to improve the antitumor activity of the drug in MCF-7 breast cell line. Their corresponding nano-assemblies were prepared by non-covalent binding of the peptides and polyglutamate, with two different chain lengths. Finally, cytotoxicity, intracellular uptake and antitumor activity of the synthesized peptides as well as their nanoparticles were investigated and compared.
Materials and methods
Materials
Classical glass reaction vessels suitable for performing solid-phase peptide synthesis (SPPS) were made by glassblowers. All of the amino acids (lysine, tryptophan and glutamate) and Rink amid-resin were purchased from Aapptec [Citation15]. N, N, N′, N′-Tetramethyl-O-(benzotriazol-1-yl) uraniumtetrafluoroborate (TBTU) and N,Ndiisopropylethylamine (DIPEA) used as coupling reagents, dichloromethane (DCM) and N,N dimethyl formamide (DMF) used as activating reagents, phenol and triisopropylsilane (TIS) as scavengers, trifluoroacetic acid (TFA), fluorescein isothiocyanate (FITC), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were all obtained from Sigma-Aldrich [Citation15]. Potassium cyanide (KCN), ninhydrin and pyridine were from Merck (Germany). MCF-7 cells were purchased from Pasteur Institute (Iran).
Synthesis of peptides
Six linear peptides named (KW)4, (KW)5, K3W4K2, K3W4K3, W2K4W2 and W3K4W3 were synthesized using the hydrophobic (tryptophan (W)) and charged (lysine(K)) amino acids with various designs as presented in . Overall methods were carried out according to the previously reported procedure [Citation37,Citation42,Citation43]. Briefly, all peptides were synthesized manually by solid-phase peptide synthesis method, assembled on Rink-Amide AM resin by fluorenylmethyloxycarbonyl chloride (Fmoc) strategy using a fritted glass vessel. Fmoc-Rink Amide Resin (0.049 mmol, 0.3 mmol/g) was swollen in DMF for approximately 1 h under dry nitrogen. Fmoc groups at N-terminal of the linear peptide sequence of resin were removed using piperidine in DMF (20% v/v, total volume of 2 ml). Following a 30 min incubation at room temperature, Fmoc-Lys (Boc)-OH or Fmoc-Try (Boc)-OH (0.148 mmol) was coupled to the N-terminal of Rink Amide Resin in the presence of TBTU (0.148 mmol) and DIPEA (0.148 mmol) in DMF (2 ml) by mixing for 1.5 h. Completion of coupling was confirmed by Kaiser Test. The reaction solution was filtered off. Then, the obtained resin was washed with DMF (three times each with 2 ml) and DCM (three times each with 2 ml) and dried under vacuum overnight. Following the drying process, a fresh cleavage cocktail composed of TFA:phenol:water:TIS (88:5:5:2 v/v/v/v, total mixture volume of 10 ml per 100 mg peptide) was added to the resin. After shaking the mixture at room temperature for 2 h, the resin was collected and washed with another 2 ml fresh cleavage cocktail. The medium of the combined filtrates were evaporated and reduced into a minimum volume using dry nitrogen. The crude unreacted peptide was precipitated by adding excess volume (10-fold) of cold diethyl ether and centrifuged at 4000 rpm for 5 min. Finally, the product was further washed with diethyl ether (50 ml) twice and then lyophilized. The schematic peptide synthesis of K3W4K3 is illustrated in .
Figure 1. The schematic peptide synthesis of K3W4K3 (a), the schematic synthesis of FITC-labeled K3W4K3 (b) and the schematic synthesis of K3W4K3-EPR conjugate (c).
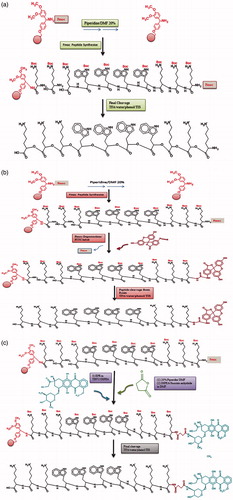
Table 1. Different cell penetrating peptide amphiphiles (CPPs) synthesized in the present study.
Table 2. The percentage of drug (EPR) loading for different peptide-EPR conjugates (CPPs-EPR).
Fluorescent labeling of the peptides on the resin
Fmoc protecting group was removed from N-terminal of all synthesized peptides using piperidine solution in DMF (20%, 2 ml, 30 min). A solution of 1/1 equivalent of FITC was provided in pyridine/DMF/DCM (12:7:5). Then FITC was attached to the peptides through adding the above solution to the resin-peptides (0.049 mmol) followed by an overnight mixing. The FITC-labeled peptides were evaluated by Kaiser test to confirm the success of labeling process [Citation43]. The schematic synthesis of FITC-labeled K3W4K3 is demonstrated in .
Preparation of nanoparticles
Peptide nanoparticles were synthesized by conjugation of the linear peptides to polyglutamate (E), with two chain lengths of E4 (EEEE) and E8 (EEEEEEEE). The synthesized linear peptides dissolved in DMSO were added to the polyglutamate E4 and E8 solution in DMSO, at different proportions of 1:1, 1:5 and 1:10 (v/v). Then, the prepared peptide solutions were sonicated so that the nanoparticles were formed through self-assembly by electrostatic attraction.
Determination of particle size
Morphology and size of the synthesized CPPs and nanoparticles were investigated by scanning electron microscopy (SEM). Each sample was dissolved in DMSO and distilled water (1:99 v/v) and was dropped onto an aluminum plate and lyophilized. The prepared slides were then visualized using a Philips XL electron microscope.
Preparation of peptide-drug conjugates
Conjugates of peptide-drug (CPP-EPR for various CPPs) were synthesized through reaction of aminegroup in peptide and hydroxyl group in EPR drug using a succinyl spacer. When succinic anhydride binds to the peptides as a linker, NH2 group of the peptide is changed to a carboxyl moiety. N-terminal of the prepared peptides was deprotected using piperidine in DMF (20%). Then the deprotected peptides were treated with succinic anhydride (1.5 equals) and DIEPA (3 equals) in DMF for 2 h. The completion of the reaction was confirmed by the Kaiser test. Following washing the resin with DMF (3 × 2 ml) and DCM (3 × 2 ml), DIEPA (0.25 ml, 1.44 mmol) was added dropwise to the mixture of succinylated peptide and TBTU (3 equals) in DMF (2 ml) under stirring for 30 min. Afterwards, EPR (1/5 equals) solution in DMF (2 ml) was added to the above mixture. The reaction mixture was stirred at room temperature for 48 h. Following drying using nitrogen gas, peptide-EPR conjugates were cleaved from the resin with cleavage cocktail composed of TFA/phenol/water/Tis (88:5:5:2 v/v/v/v, total volume of 10 ml per 100 mg peptide). Then, the corresponding nanoparticles (CPP-E4-EPR and CPP-E8-EPR each for various CPPs) were prepared similarly as mentioned in the “Preparation of nanoparticles”. The schematic synthesis of K3W4K3-EPR conjugate is shown in .
Drug loading measurement
UV spectra were obtained in order to evaluate the conjugation of EPR to various CPPs using a UV spectrophotometer. Afterwards, absorbance of the unreacted EPR for each sample and the standard solution (first EPR concentration) were measured at the wavelength of 504 nm. Loading of EPR (%) was calculated from EquationEquation (1)(1)
(1) .
(1)
(1)
Where Af and As represent the optical absorbance of the sample and the standard, respectively.
Cell culture
Cellular studies were performed using MCF-7 (human breast adenocarcinoma) cell line. The cells were cultured on cell culture flasks in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) and 1% antibiotic-antimycotic (Penicillin-Streptomycin; Gibco, Grand Island, NY) in an incubator (5% CO2) at 37 °C.
Cytotoxicity and anti-proliferative assay
The MTT assay was carried out to evaluate and compare the cytotoxicity and anti-proliferative activity of various samples (different CPPs, CPPs-E4 and CPPs-E8 nanoparticles for cytotoxicity; different CPPs, CPPs-EPR, CPPs-E4-EPR, CPPs-E8-EPR and EPR alone for anti-proliferative activity) each at different concentrations. MCF-7 cell suspension at a density of 2 × 104 cells/well was seeded into 96-well microplates. Following incubation at 37 °C and 5% CO2 for 24 h and medium aspiration, the cells were treated with the 1/10 diluted samples in serum-free culture medium each at different final concentrations of 5, 10 and 25 μM for cytotoxicity and 1, 5, 10 and 25 μM for anti-proliferative assay. The cells were incubated for 24 or 48 h and then the medium was aspirated and replaced with 50 µl of MTT solution (2 mg/ml). After 4 h incubation, the medium was aspirated again and insoluble formazan crystals in each well were dissolved in mixture of dimethyl sulfoxide (DMSO, 200 µl) and Sorensen’s phosphate buffer (25 µl). The absorbance was measured by a microplate reader (Bio-Tek, Winooski, VT) at a wavelength (λ) of 570 nm with reference to 650 nm. All of the results were blank corrected. The experiment was run for six replicates. Anti-proliferative activity was calculated according to EquationEquation (2)(2)
(2) .
(2)
(2)
Where AS570 and AS650 correspond to the absorbance of treated samples at the respective λ = 570 and 650 nm; AC570 and AC650 are the corresponding absorbance of untreated control cells.
Fluorescence microscopy
The cellular uptake of CPPs and the corresponding nanoparticles were visualized using fluorescence microscopy. MCF-7 cells were seeded on the coverslips placed inside the six well plates at a density of 1 × 105 cells per well and incubated at 37 °C and 5% CO2 for 24 h. Then, the cells were treated with the FITC-CPPs and FITC-CPPs-E8 nanoparticles in serum-free culture medium each at the concentration of 10, 25 and 50 μM and incubated for 2 h. Subsequently, the medium of each well was aspirated and the cells were washed three times with cold phosphate buffered saline (PBS). For fixing the cells, 200 µl of 2% formaldehyde solution in PBS was added to each well and incubated at 37 °C for 15 min followed by washing three times with PBS. Then, the samples were visualized by a fluorescence microscope (Olympus IX81, Olympus Optical Co. Ltd., Japan).
Cell uptake quantification by flow cytometry
For cellular uptake quantification, flow cytometry assay was performed. MCF-7 cells were seeded in 6-well plates at a density of 2 × 105 cells/well and incubated at 37 °C and 5% CO2 for 24 h. Following culture medium aspiration and washing with PBS, the cells were treated with FITC-CPPs and FITC-CPPs-E8 nanoparticles in serum-free culture medium each at concentrations of 10, 25, and 50 μM. After 2 h incubation at 37 °C, the cells were washed three times with PBS and detached from the well by 3 min incubation at 37 °C with 0.53 mM of 0.25% trypsin/EDTA. Then, the cells were harvested, centrifuged, resuspended in 0.5 ml cold PBS and homogenized by pipetting several times. The cells were kept on ice until measurement of the treated cell-associated fluorescence intensity of FITC at the emission wavelength of 519 nm through FL1 channel for total of 10,000 events (cells/sample) using a FACSCalibur flow cytometer (Becton Dickinson, Holdrege, NE). Cell Quest software (Becton Dickinson, Holdrege, NE) was used for data acquisition. The analysis of fluorescence intensities was performed subsequently via the above mentioned software for the gated singlet cells.
Statistical analysis
The data were expressed as mean ± standard deviation of 3–6 determinations. Statistical analysis was performed using Graphpad Prism Software Inc. version 5.04 (Inc., La Jolla, CA). Different samples were compared using two-way analysis of variance (ANOVA). The differences were quoted statistically significant where p < .05. The significance level for pair wise comparisons was adjusted by Bonferroni method using Student t-test.
Results
Different synthesized CPPs and the corresponding nanoparticles were characterized by scanning electron microscopy (supplementary data, Figure S1), followed by cytotoxicity, cellular uptake and anti-proliferative activity.
Cytotoxicity
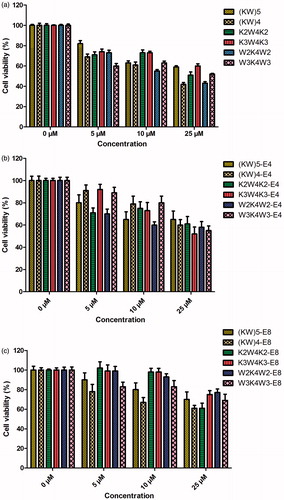
As an efficient delivery vector of anti-cancer drugs, blank CPPs and nanoparticles must have low levels of toxicity against cells. Therefore, the cytotoxicity of all synthesized CPPs and the corresponding nanoparticles (E4 and E8) were evaluated in MCF-7 cancer cells as a function of concentrations (5, 10 and 25 µM) after 24 and 48 h incubation time. For blank peptides, i.e. CPPs, cytotoxicity was concentration dependent (p < .001). The toxicity results for all of the nanoparticles showed that an increase in the concentration of peptides resulted in an increase in the toxicity after incubation for 24 (supplementary data, Figure S2) and 48 h (). The increase in the toxicity of nanoparticles found to be statistically significant (p < .0001). Following 24 h of incubation of the peptides with MCF-7 cells, all of the CPPs showed low cytotoxicity (<20%) at the concentration of 5 µM. (KW)5, K2W4K2 and W3K4W3 showed the lowest cytotoxicity (16, 21 and 22%, respectively) at the concentration of 10 µM (p < .001). In addition, at the highest concentration (25 µM), cytotoxicity of W3K4W3 was the lowest (22%) followed by (KW)5 and K3W4K3 peptides (32 and 35%, p < .00001). Incubation time also affected the cytotoxicity of the peptides. Increasing the incubation time from 24 (supplementary data, Figure S2) to 48 h () resulted in higher cytotoxicities (p < .0001). As illustrated in , (KW)5 showed the lowest cytotoxicity at concentration of 5 µM (15%, p < .01). The lowest cytotoxicity was observed for K2W4K2 and K3W4K3 (25 and 26%, p < .001) when the concentration was 10 µM. In the cases of (KW)5 and K3W4K3 (42and 42%, p < .01) the lowest toxicity was obtained at 25 µM. Binding of E4 to peptides led to a significant reduction in the cytotoxicity at different concentrations for both 24 (p < .0001) and 48 h (p < .001) incubation times (). As demonstrated in , binding of E8 to peptides also resulted in a significant decrease in the cytotoxicity at different concentrations for all nanoparticles at the both incubation times, 24 and 48 h (p < .00001 and .0001, respectively). Interestingly, CPPs-E8 nanoparticles showed lower cytotoxicity than CPPs-E4 nanoparticles (p < .001). Therefore, CPPs-E8 nanoparticles possessed the lowest cytotoxicity among all nanoparticles. All of CPPs-E8 nanoparticles showed very low cytotoxicity (<20%) even at the highest concentration (25 µM) except (KW)4-E8 (at 10 and 25 µM) and K2W4K2-E8 (just at 25 µM).
Figure 2. MTT-based cytotoxicity of different CPPs (a), CPPs-E4 nanoparticles (b) and CPPs-E8 nanoparticles (c) at various concentrations (5, 10 and 25 µM) after 48 h incubation in MCF-7 cell line at 37 °C. Data represent Mean ± SD for six replicates.
Cellular uptake by fluorescence microscopy
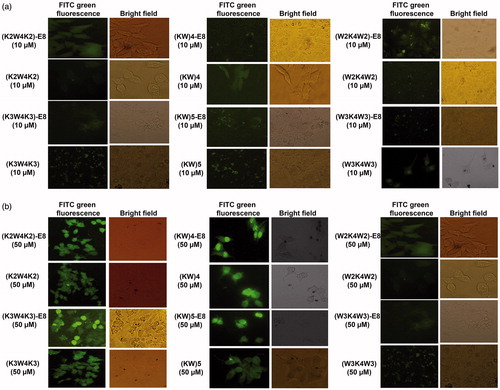
The cellular uptake (intracellular localization) was first visualized by fluorescence microscopy. MCF-7 cells were incubated with various FITC-CPPs and FITC-CPPs-E8 at concentrations of 10, 25 and 50 µM, respectively for 2 h. As illustrated in , cell treatments with high level concentrations of the samples (25 μM, supplementary data Figure S3 and 50 μM, ) led to increase the cellular uptake in comparison with lower concentration (10 μM, ) for both CPPs and nanoparticles. Besides, it was indicated that CPPs and CPPs-E8 nanoparticles of K3W4K3, W3K4W3, (KW)5 and (KW)4 accumulated in the nucleus of the cells, while K2W4K2 peptide and its corresponding nanoparticle localized in the cytoplasm. It was demonstrated that W2K4W2 (both peptide and its nanoparticles) had the lowest cellular uptake.
Figure 3. Fluorescence microscopy images of MCF-7 cells incubated with FITC-labeled CPPs and their corresponding nanoparticles (CPPs-E8) at concentration of 10 (a) and 50 µM (b), respectively after 2 h incubation at 37 °C.
Cellular uptake by flow cytometry
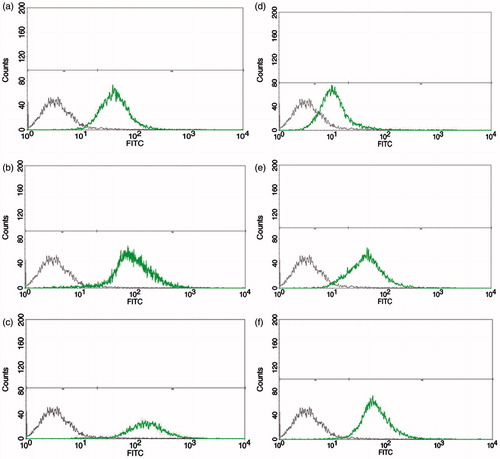
Flow cytometry was performed in order to evaluate and compare the cellular uptake ability of different peptides and their nanoparticles (FITC-CPPs and FITC-CPPs-E8 nanoparticles). Just like fluorescence microscopy, MCF-7 cells were incubated with different concentrations of the samples for 2 h at 37 °C. As shown in , the uptake was concentration dependent for all of the CPPs and nanoparticles. In other words the percentage of cellular fluorescence increased with increasing the concentration up to 25 µM (p < .0001). At this concentration, the cell fluorescence reached the maximum percentage (about 100%) for all of the CPPs except W2K4W2 () which was very low (about 20%). By increasing the concentration from 25 to 50 µM, the cell fluorescence remained stable (at plateau) for K3W4K3 (), W3K4W3 () and K2W4K2 (). However, in the case of (KW)4 () and (KW)5 () the cell fluorescence decreased to about 75%. Among all of the CPPs, W2K4W2 () exhibited the lowest uptake (p < .00001). For all of the nanoparticles (), cellular uptake was lower compared to their corresponding CPPs at the concentration of 10 (p < .0001) and 25 µM (p < .001), respectively. The fluorescent signal of K3W4K3-E8 (), K2W4K2-E8 () and W3K4W3-E8 () reached the same percentage with the corresponding CPPs, i.e. the maximum percentage of approximately 100% at the concentration of 50 µM and there was no significant difference between each of the mentioned CPPs and its nanoparticle (p > .05). Cellular uptake of (KW)4-E8 () and (KW)5-E8 () was significantly higher in comparison with their corresponding CPPs at the concentration of 50 µM (p < .0001 for both). Although, the cell uptake of W2K4W2-E8 () was significantly higher than its corresponding CPP (W2K4W2) at the concentrations of 25 ( < .0001) and 50 μM (p < .0001), respectively it still exhibited the lowest percentage of cellular uptake compared to all other CPPs-E8 nanoparticles (p < .00001). Flow cytometry histograms of K3W4K3 and K3W4K3-E8 each at various concentrations of 10, 25 and 50 µM are presented in .
Figure 4. Cellular uptake (%) of FITC-CPPs and the corresponding nanoparticles (FITC-CPPs-E8) at different concentration of 10, 25 and 50 µM, respectively after 2 h incubation in MCF-7 cell at 37 °C. Data represent Mean ± SD for three replicates. FITC-CPPs and FITC-CPPs and FITC-CPPs-E8 nanoparticles are presented by solid lines with the symbol of ▪ and dashed lines with the symbol of •, respectively.
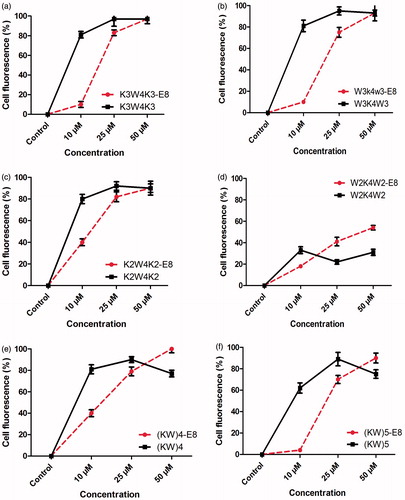
Figure 5. Flow cytometry histograms of MCF-7 cells treated for 2 h with K3W4K3 (left) and K3W4K3-E8 (right) at various concentrations of 10 (a and d), 25 (b and e) and 50 (c and f) µM, respectively. First peak corresponds to untreated control cells.
Drug loading measurement
Following the conjugation of EPR to different CPPs, conjugation was confirmed using UV spectrophotometry (supplementary data, Figure S4). The percentages of EPR loading calculated are presented in . The loading percentages were found to be in the range of 50–77%.
Antitumor activity of drug loaded CPPs
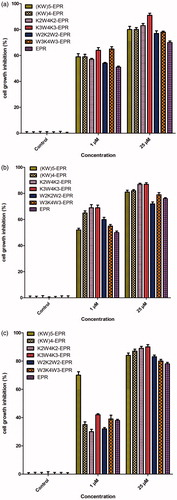
To determine whether the synthesized peptides and nanoparticles can be utilized for delivery of biologically relevant doses of EPR to cells, EPR was conjugated to them and the antitumor (anti-proliferative) activity of EPR was then examined and compared in MCF-7 cells at various concentrations. As illustrated in Figure S5(a) (24 h, supplementary data) and (48 h), even the low concentration of 1 µM resulted in a significant increase in antitumor activity. As demonstrated in (48 h), anti-proliferative activity was concentration dependent which increased by increasing the concentration (p < .001). Moreover, incubation time also led to higher anti-proliferative activity (p < .01, supplementary data, Figure S5 for 24 h; for 48 h). Following the conjugation of EPR to the CPPs, CPPs-E4 and CPPs-E8 nanoparticles, the anti-proliferative activity of the drug increased significantly (p < .001) at the highest concentration (25 µM). Free drug (EPR) showed 63% anti-proliferative activity (37% cell viability) at the concentrations of 25 µM. The anti-proliferative activity increased to 64–79% and 77–91%, respectively () for all CPPs-EPR conjugates at 25 µM at the respective incubation times of 24 (p < .05, supplementary data, Figure S5(a)) and 48 h (p < .01, ). Among the six CPP-EPR conjugates at 25 µM concentration, K3W4K3-EPR exhibited the highest anti-proliferative activity (80%) and W2K4W2-EPR as well as (KW)4-EPR possessed the lowest anti-proliferative activity (62%) after 24 h incubation (supplementary data, Figure S5(a)). Following the increase of incubation time to 48 h at 25 µM concentration, K3W4K3-EPR and W2K4W2-EPR showed the highest (90%) and the lowest (74%) anti-proliferative activity, respectively, as demonstrated in . CPPs-EPR-nanoparticles displayed higher anti-proliferative activity compared to CPPs-EPR conjugates (p < .05) at both incubation times especially at higher concentrations. Among all CPPs-EPR-nanoparticles, CPPs-E8-EPR (Figure S5(c) ,supplementary data and ) showed higher anti-proliferative activity than CPPs-E4-EPR (Figure S5(b), supplementary data and ) at both incubation times (p < .05 and p < .01). As illustrated in K3W4K3-E8-EPR nanoparticles exhibited the highest anti-proliferative activity (92%) for all CPPs-E8-EPR at 25 µM following incubation for 48 h (p < .05).
Figure 6. MTT-based anti-proliferative activity of different CPPs-EPR (a), CPPs-E4-EPR (b) and CPPs-E8-EPR (c) in comparison with free drug (EPR) at various concentrations (1, 5, 10 and 25 µM) after 48 h incubation in MCF-7 cells at 37° C. Data represent Mean ± SD for six replicates.
Discussion
Coupling of EPR to CPPs has been employed as one of the specific methods to deliver the drug molecules into various cell lines. In the present study, different peptide sequence designs were chosen for peptide synthesis to examine how the presence of tryptophan and its position along with lysine and then peptide conjugation with two different chain lengths of polyglutamate may influence the cytotoxicity, the cellular uptake and the anti-proliferative activity of the drug.
The cytotoxicity of the synthesized CPPs and the corresponding nanoparticles (-E4 and -E8) was investigated as a function of concentrations at two incubation times of 24 (supplementary data, Figure S2) and 48 h (). The cytotoxicity was concentration dependent for CPPs. All of the CPPs showed low cytotoxicity at concentration of 5 µM (Figure S2(a), supplementary data and ). As also reported before, the CPPs shows either nothing or low cytotoxicity at low concentrations while their cytotoxicity may increase in higher doses [Citation44]. At the highest concentration (25 µM), for 24 h incubation the cytotoxicity of W3K4W3 was the lowest followed by (KW)5 and K3W4K3 (Figure S2(a), supplementary data) and for 48 h incubation the lowest cytotoxicity belonged to (KW)5 and K3W4K3 (). Taken all data together, it was shown that increasing the number of amino acids might result in decreased cytotoxicity so that the peptides with 10 amino acids (higher number of amino acids) exhibited lower toxicity than peptides composed of eight amino acids (lower number of amino acids). Displacement of lysine and tryptophan had impact on CPPs cytotoxicity since they showed different cytoxicities. However, it cannot be concluded that the alternative design is better than the block design or vice versa. Binding of polyglutamate, E4 () and E8 (), to the peptides led to a significant decrease in the cytotoxicity. It was in agreement with another study which previously reported that masking a positively charged CPP with a negatively charged polyglutamate, can reduce the toxicity of peptide in vivo [Citation44]. Interestingly, CPPs-E8 nanoparticles showed even lower cytotoxicity than CPPs-E4 nanoparticles. It indicates that polyglutamate chain length can be considered as an important factor in cytotoxicity and increasing the chain length (in range of study, from four to eight) leads to a significant decrease in cytotoxicity.
Cellular uptake of the synthesized CPPs labeled with FITC (FITC-CPPs) and the corresponding nanoparticles (FITC-CPPs-E8 nanoparticles) was first investigated by fluorescence microscopy. The green fluorescence of FITC existed inside the cells even at the lowest concentration (10 μM). It is indicated that the peptides and nanoparticles possess a good ability for entering the cells even at low concentrations [Citation45–48]. Increasing the concentration from 10 μM () to 25 μM (supplementary data, Figure S3) or the highest concentration of 50 μM () led to a clear increase in cellular uptake for both CPPs and nanoparticles which is an indicative of the significant role of concentration [Citation48–50]. Besides, it was shown that the CPPs and CPPs-E8 nanoparticles of K3W4K3, W3K4W3, (KW)5 and (KW)4 accumulated in the nucleus of the cells that is an indicative of their fast ability to pass other barriers inside the cells and reaching the nucleus, while K2W4K2 peptide and its corresponding nanoparticle localized in the cytoplasm. There are different uptake pathways for penetrating of the CPPs to the cells [Citation45,Citation46,Citation48]. K2W4K2 might be translocated by a different internalization mechanism from the other peptides. The probable mechanism for this peptide might be the caveolin pathway [Citation51]. It was reported that accumulation of the caveolins in the endoplasmic reticulum leads to an apparent concentration of the proteins in lipid droplets [Citation52]. Hence, it is possible that K2W4K2 may be associated with lipid droplets or other cytoplasmic particles and this peptide might need longer time to enter to the nucleus [Citation51]. Both peptide and-E8 nanoparticle of W2K4W2 displayed the lowest fluorescence inside the cells which is possibly due to its low cellular uptake, which means its weak internalization into the cells.
It was reported that the addition of tryptophan amino acid could improve cellular uptake efficiency and peptides with sequences containing tryptophan in the middle or along the peptide sequence resulted in high uptake [Citation41]. Interestingly, it was observed that the cytotoxicity decreased markedly and the cellular uptake increased partly following the coupling of E8 and E4 to CPPs and synthesizing the nanoparticles. CPPs-E8 showed less cytotoxicity and more improved delivery of EPR compared to CPPs-E4. The various CPPs sequences may lead to different modes of cellular uptake and therefore different levels of uptake. CPPs can traverse membranes in order to enter cells via different uptake mechanisms. Despite of many studies, the mechanism by which they enter cells is still a subject of dispute. For a long time, it was believed that CPPs would most likely enter cells by a passive process, which is temperature and receptor independent [Citation53]. Endocytosis, being the most common process used by cells to absorb materials from their environment, can also be used as one of the translocation pathway of CPPs into the cells [Citation53,Citation54]. Endocytosis is a generic term for several different processes, such as phagocytosis for large particles and pinocytosis for smaller ones, as well as receptor-mediated endocytosis in which clathrin or caveolin pits are involved [Citation25,Citation45,Citation53]. Several receptors were uncovered to be involved in internalization of CPPs, such as chemokine receptors, syndecans [Citation55,Citation56], neuropilins [Citation57,Citation58], the family of integrins [Citation59], homing sequences and positively-charged scavenger receptors [Citation60,Citation61]. Micropinocytosis appears to be another pathway for some of CPPs that is mediated by positively charged residues interacting with phosphoinositides. A non-endocytic, receptor-free, energy-independent cellular process is another mechanism of translocation across biologic membranes used by the CPPs, including formation of inverted micelles, direct translocation through the lipid bilayer, and pore formation on the membrane [Citation53,Citation54,Citation62,Citation63]. CPPs with high content in cationic residues are first absorbed at the cell surface to the numerous anionic moieties, such as sialic or phospholipidic acid or heparan sulfate proteoglycans [Citation53,Citation64]. Which of these mechanisms a CPP will use is dependent on several parameters such as its concentration, size (with cargo), temperature, modifications of CPPs or their cargo and cell type [Citation45,Citation53,Citation62]. The cell membrane is a heterogeneous double layer where some regions are denser and some others have more fluidity due to the presence of different lipid compositions. These properties in turn along with CPPs properties can result in a variety of signaling pathways and different levels and modes of uptake [Citation65–69]. Since the synthesized amphipathic peptides commonly have positive charge and cancer cell membranes typically carry a net negative charge [Citation70], the elevated expression of anionic molecules such as proteoglycans [Citation71] and the electrostatic attraction between them is believed that can play a role as one of the mechanisms in the cellular uptake of these peptides [Citation31].
To determine the antitumor activity of the synthesized peptides and the nanoparticles, EPR was conjugated to these synthesized peptides using succinyl hydrolysable spacer. It is well known that succinyl spacer can be hydrolyzed by the esterase enzymes which are present in the cells and biological fluids [Citation72–74] and consequently may allow the drug to release after entering the cells. As demonstrated in , anti-proliferative activity was concentration dependent which increased by an increase in the concentration of CPP-EPR that is in agreement with another study [Citation75]. Following the conjugation of EPR to the CPPs, CPPs-E4 and CPPs-E8 nanoparticles, the anti-proliferative activity of the drug increased significantly at the highest concentration (25 µM). Free drug (EPR) showed 63% anti-proliferative activity. The antitumor activity increased to 63–79% and 87–91% ( and S3(a)) for all CPPs-EPR conjugates at 25 µM at the respective incubation time of 24 and 48 h, respectively. It can be concluded that at the highest concentration, peptides improves the antitumor activity of EPR in comparison to free EPR. The enhanced antitumor activity of CPPs-EPR conjugates in comparison with free EPR can be related to their high cellular uptake tendency. Among all CPP-EPR conjugates at 25 µM, K3W4K3-EPR exhibited the highest anti-proliferative activity and W2K4W2-EPR as well as (KW)4-EPR possessed the lowest anti-proliferative activity after 24 h incubation. Following the increase in incubation time to 48 h, K3W4K3-EPR and W2K4W2-EPR showed the highest and the lowest anti-proliferative activity, respectively (). The lowest antitumor activity of W2K4W2-EPR can be due to its lowest cellular uptake demonstrated by both fluorescence microscopy and flow cytometry. CPPs-EPR-nanoparticles displayed higher anti-proliferative activity compared to free EPR and also CPPs-EPR conjugates at both incubation times especially at higher concentrations while overall they showed very low cytotoxicity even at the highest concentration. Therefore, their enhanced anti-proliferative activity is possibly related to their high cellular uptake tendency. Generally, CPPs () and CPPs-E4 () or CPPs-E8 () nanoparticles displayed improved anti-proliferative activity compared to free EPR (at higher concentrations). This enhanced antitumor activity could be attributed to the high cellular uptake tendency of the synthesized peptides or nanoparticles [Citation76]. Nanoparticles showed greater intracellular uptake and less cytotoxicity. Moreover, the effect of EPR was better for nanoparticles compared to CPPs at higher concentration levels. The reason for this may be due to the changes in conformation and the charge of peptides of nanoparticles and diverse uptake pathways following the interaction with cell membrane. It was reported that at low concentration, endocytosis of peptides could occur which may result in endosomal entrapped peptides and possible metabolic degradation [Citation77,Citation78]. However numerous evidences suggest that direct penetration does occur at high concentrations (above 10 µm) [Citation77,Citation78]. Pathways of the uptake depended concentration can explain variation of cytotoxicity and uptake of peptides. As shown, the uptake of peptides and following antitumor activity of EPR-CPPs and EPR-CPPs-E8 increased at high concentrations and this can possibly due to direct translocation at high concentration.
Conclusion
In summary, different linear peptides were successfully synthesized separately or conjugated to EPR. Polyglutamate (E4 or E8) was also conjugated to the peptides and peptides-EPR to prepare nanoparticles. The effect of various CPPs and their nanoparticles was then evaluated against a breast cancer cell line (MCF-7). Results showed that cytotoxicity was concentration and time dependent. Binding of polyglutamate to CPPs resulted in a significant decrease in cytotoxicity. Interestingly, CPPs-E8 nanoparticles showed lower cytotoxicity than CPPs-E4. Uptake was concentration dependent for all of the CPPs and nanoparticles up to 25 µM. Among all of the CPPs, W2K4W2 exhibited the lowest uptake. K3W4K3-E8, K2W4K2-E8 and W3K4W3-E8 reached the highest uptake at the concentration of 50 µM and there was no significant difference between each of them and their nanoparticles. Following the conjugation of EPR to the CPPs and nanoparticles, the anti-proliferative activity of EPR increased significantly at the highest concentration (25 µM). CPPs-EPR-nanoparticles displayed higher anti-proliferative activity compared to CPPs-EPR conjugates especially at 25 µM. Interestingly, CPPs–E8-EPR nanoparticles showed higher anti-proliferative activity than CPPs–E4-EPR. K3W4K3-E8-EPR nanoparticles exhibited the lowest cytotoxicity, high cellular uptake and the highest anti-proliferative activity at 25 µM following 48 h incubation. Taking together the aforementioned advantages, the peptide nanoparticles are proposed as more potential nanosystems for cellular delivery of drugs at high concentration levels compared to CPPs, but they should also be tested in-vivo.
Samaneh_et_al._Supplementary_material.doc
Download MS Word (6.1 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Dissanayake S, Denny WA, Gamage S, et al. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J Control Release. 2017;250:62–76.
- Wang H, Sun M, Li D, et al. Redox sensitive PEG controlled octa arginine and targeting peptide co-modified nanostructured lipid carriers for enhanced tumour penetrating and targeting in vitro and in vivo. Artif Cells Nanomed Biotechnol. Forthcoming. [cited 2017 Mar 31]. doi:10.1080/21691401.2017.1307214
- Han C, Li Y, Sun M, et al. Small peptide-modified nanostructured lipid carriers distribution and targeting to EGFR-overexpressing tumor in vivo. Artif Cells Nanomed Biotechnol. 2014;42:161–166.
- Yu M, Han S, Kou Z, et al. Lipid nanoparticle-based co-delivery of epirubicin and BCL-2 siRNA for enhanced intracellular drug release and reversing multidrug resistance. Artif Cells Nanomed Biotechnol. Forthcoming. [cited 2017 Apr 10]. doi:10.1080/21691401.2017.1307215
- Plosker GL, Faulds D. Epirubicin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cancer chemotherapy. Drugs. 1993;45:788–856.
- Robert J. Clinical pharmacokinetics of epirubicin. Clin Pharmacokinet. 1994;26:428–438.
- Charak S, Jangir DK, Tyagi G, et al. Interaction studies of epirubicin with DNA using spectroscopic techniques. J Mol Struc. 2011;1000:150–154.
- Launchbury AP, Habboubi N. Epirubicin and doxorubicin: a comparison of their characteristics, therapeutic activity and toxicity. Cancer Treat Rev. 1993;19:197–228.
- Beslija S. The role of anthracyclines/anthraquinones in metastatic breast cancer. Breast Cancer Res Treat. 2003;81:25–32.
- Ormrod D, Holm K, Goa K, et al. Epirubicin: a review of its efficacy as adjuvant therapy and in the treatment of metastatic disease in breast cancer. Drugs Aging. 1999;15:389–416.
- Coukell AJ, Faulds D. Epirubicin. An updated review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of breast cancer. Drugs. 1997;53:453–482.
- Nasrolahi Shirazi A, Tiwari R, Chhikara BS, et al. Design and biological evaluation of cell-penetrating peptide–doxorubicin conjugates as prodrugs. Mol Pharmaceutics. 2013;10:488–499.
- Yang X, Deng W, Fu L, et al. Folate‐functionalized polymeric micelles for tumor targeted delivery of a potent multidrug‐resistance modulator FG020326. J Biomed Mater Res. 2008;86:48–60.
- Chavanpatil MD, Khdair A, Gerard B, et al. Surfactant–polymer nanoparticles overcome P-glycoprotein-mediated drug efflux. Mol Pharmaceutics. 2007;4:730–738.
- Sadava D, Coleman A, Kane SE. Liposomal daunorubicin overcomes drug resistance in human breast, ovarian and lung carcinoma cells. J Liposome Res. 2002;12:301–309.
- Lindgren M, Rosenthal-Aizman K, Saar K, et al. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem Pharmacol. 2006;71:416–425.
- Farvadi F, Tamaddon A, Sobhani Z, et al. Polyionic complex of single-walled carbon nanotubes and PEG-grafted-hyperbranched polyethyleneimine (PEG-PEI-SWNT) for an improved doxorubicin loading and delivery: development and in vitro characterization. Artif Cells Nanomed Biotechnol. 2017;45:855–863.
- Dhankhar R, Vyas SP, Jain AK, et al. Advances in novel drug delivery strategies for breast cancer therapy. Artif Cells Nanomed Biotechnol. 2010;38:230–249.
- Wang X, Low XC, Hou W, et al. Epirubicin-adsorbed nanodiamonds kill chemoresistant hepatic cancer stem cells. ACS Nano. 2014;8:12151–12166.
- Hu L, Jia Y. Preparation and characterization of solid lipid nanoparticles loaded with epirubicin for pulmonary delivery. Pharmazie. 2010;65:585–587.
- Lo YL. Phospholipids as multidrug resistance modulators of the transport of epirubicin in human intestinal epithelial Caco-2 cell layers and everted gut sacs of rats. Biochem Pharmacol. 2000;60:1381–1390.
- Yang Q, Wang XD, Chen J, et al. A clinical study on regional lymphatic chemotherapy using an activated carbon nanoparticle–epirubicin in patients with breast cancer. Tumor Biol. 2012;33:2341–2348.
- Birnbaum DT, Brannon-Peppas L. Molecular weight distribution changes during degradation and release of PLGA nanoparticles containing epirubicin HCl. J Biomater Sci Polym Ed. 2003;14:87–102.
- Sahu KK, Minz S, Kaurav M, et al. Proteins and peptides: the need to improve them as promising therapeutics for ulcerative colitis. Artif Cells Nanomed Biotechnol. 2016;44:642–653.
- Farkhani SM, Valizadeh A, Karami H, et al. Cell penetrating peptides: efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides. 2014;57:78–94.
- Langel U. Handbook of cell-penetrating peptides. Boca Raton (FL): CRC press; 2006.
- Kristensen M, Nielsen HM. Cell-penetrating peptides as tools to enhance non-injectable delivery of biopharmaceuticals. Tissue Barriers. 2016;4:e1178369.
- Jin C, Bai L, Lin L, et al. Paclitaxel-loaded nanoparticles decorated with bivalent fragment HAb18 F (ab’) 2 and cell penetrating peptide for improved therapeutic effect on hepatocellular carcinoma. Artif Cells Nanomed Biotechnol. Forthcoming. [cited 2017 Aug 4]. doi:10.1080/21691401.2017.1360325
- Clavier S, Du X, Sagan S, et al. An integrated cross-linking-MS approach to investigate cell penetrating peptides interacting partners. EuPA Open Proteom. 2014;3:229–238.
- Figueiredo IR, Freire JM, Flores L, et al. Cell‐penetrating peptides: a tool for effective delivery in gene‐targeted therapies. IUBMB Life. 2014;66:182–194.
- Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17:850–860.
- Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206.
- Najafi-Hajivar S, Zakeri-Milani P, Mohammadi H, et al. Overview on experimental models of interactions between nanoparticles and the immune system. Biomed Pharmacother. 2016;83:1365–1378.
- Niazi M, Zakeri-Milani P, Najafi Hajivar S, et al. Nano-based strategies to overcome p-glycoprotein-mediated drug resistance. Expert Opin Drug Metab Toxicol. 2016;12:1021–1033.
- Mussa Farkhani S, Asoudeh Fard A, Zakeri-Milani P, et al. Enhancing antitumor activity of silver nanoparticles by modification with cell-penetrating peptides. Artif Cells Nanomed Biotechnol. 2017;45:1029–1035.
- Dubikovskaya EA, Thorne SH, Pillow TH, et al. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octa arginine transporters. Proc Natl Acad Sci U S A. 2008;105:12128–12133.
- Mohammadi S, Mojarrad JS, Zakeri-Milani P, et al. Synthesis and in vitro evaluation of amphiphilic peptides and their nanostructured conjugates. Adv Pharm Bull. 2015;5:41.
- Kim JK, Anderson J, Jun HW, et al. Self-assembling peptide amphiphile-based nanofiber gel for bioresponsive cisplatin delivery. Mol Pharmaceutics. 2009;6:978–985.
- Nasrolahi Shirazi A, Oh D, Tiwari RK, et al. Peptide amphiphile containing arginine and fatty acyl chains as molecular transporters. Mol Pharmaceutics. 2013;10:4717–4727.
- Gautam A, Singh H, Tyagi A, et al. CPP site: a curated database of cell penetrating peptides. Database. 2012;2012:bas015.
- Rydberg HA, Matson M, Åmand HL, et al. Effects of tryptophan content and backbone spacing on the uptake efficiency of cell-penetrating peptides. Biochemistry. 2012;51:5531–5539.
- Farkhani SM, Johari-ahar M, Zakeri-Milani P, et al. Enhanced cellular internalization of CdTe quantum dots mediated by arginine-and tryptophan-rich cell-penetrating peptides as efficient carriers. Artif Cells Nanomed Biotechnol. 2016;44:1424–1428.
- Zakeri-Milani P, Farkhani SM, Shirani A, et al. Cellular uptake and anti-tumor activity of gemcitabine conjugated with new amphiphilic cell penetrating peptides. EXCLI J. 2017;16:650–662.
- Aguilera TA, Olson ES, Timmers MM, et al. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr Biol. 2009;1:371–381.
- Madani F, Lindberg S, Langel Ü, et al. Mechanisms of cellular uptake of cell-penetrating peptides. Biophys J. 2011;2011:414729.
- Lorents A, Kodavali PK, Oskolkov N, et al. Cell-penetrating peptides split into two groups based on modulation of intracellular calcium concentration. J Biol Chem. 2012;287:16880–16889.
- Myrberg H, Zhang L, Mäe M, et al. Design of a tumor-homing cell-penetrating peptide. Bioconjug Chem. 2008;19:70–75.
- Ziegler A, Seelig J. Contributions of glycosaminoglycan binding and clustering to the biological uptake of the nonamphipathic cell-penetrating peptide WR9. Biochemistry. 2011;50:4650–4664.
- Hällbrink M, Oehlke J, Papsdorf G, et al. Uptake of cell-penetrating peptides is dependent on peptide-to-cell ratio rather than on peptide concentration. Biochim Biophys Acta. 2004;1667:222–228.
- Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60:580–597.
- Zaro JL, Vekich JE, Tran T, et al. Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol Pharmaceutics. 2009;6:337–344.
- Ostermeyer AG, Paci JM, Zeng Y, et al. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol. 2001;152:1071–1078.
- Durzyńska J, Przysiecka Ł, Nawrot R, et al. Viral and other cell-penetrating peptides as vectors of therapeutic agents in medicine. J Pharmacol Exp Ther. 2015;354:32–42.
- Choi YS, David AE. Cell penetrating peptides and the mechanisms for intracellular entry. Curr Pharm Biotechnol. 2014;15:192–199.
- Letoha T, Keller-Pintér A, Kusz E, et al. Cell-penetrating peptide exploited syndecans. Biochim Biophys Acta. 2010;1798:2258–2265.
- Kawaguchi Y, Takeuchi T, Kuwata K, et al. Syndecan-4 is a receptor for clathrin-mediated endocytosis of arginine-rich cell-penetrating peptides. Bioconjugate Chem. 2016;27:1119–1130.
- Prud'homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012;3:921.
- Pang HB, Braun GB, Ruoslahti E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci Adv. 2015;1:e1500821.
- Mokhtarieh AA, Kim S, Lee Y, et al. Novel cell penetrating peptides with multiple motifs composed of RGD and its analogs. Biochem Biophys Res Commun. 2013;432:359–364.
- Reissmann S. Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci. 2014;20:760–784.
- Juks C, Lorents A, Arukuusk P, et al. Cell-penetrating peptides recruit type a scavenger receptors to the plasma membrane for cellular delivery of nucleic acids. FASEB J. 2017;31:975–988.
- Lindgren M, Langel Ü. Classes and prediction of cell-penetrating peptides. In: Langel Ü., editor. Cell-penetrating peptides: methods and protocols. New York: Humana Press; 2011. p. 3–19.
- Thorén PE, Persson D, Lincoln P, et al. Membrane destabilizing properties of cell-penetrating peptides. Biophys Chem. 2005;114:169–179.
- Pujals S, Fernández-Carneado J, López-Iglesias C, et al. Mechanistic aspects of CPP-mediated intracellular drug delivery: relevance of CPP self-assembly. Biochim Biophys Acta. 2006;1758:264–279.
- Jacobson K, Mouritsen OG, Anderson RGW. Lipid rafts: at a crossroad between cell biology and physics. Nat Cell Biol. 2007;9:7–14.
- Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3:a004697.
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603.
- McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596.
- van Meer G, de Kroon AIPM. Lipid map of the mammalian cell. J Cell Sci. 2011;124:5–8.
- Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur J Pharmacol. 2009;625:190–194.
- Ziegler A, Seelig J. Binding and clustering of glycosaminoglycans: a common property of mono- and multivalent cell-penetrating compounds. Biophys J. 2008;94:2142–2149.
- Cavallaro G, Mariano L, Salmaso S, et al. Folate-mediated targeting of polymeric conjugates of gemcitabine. Int J Pharm. 2006;307:258–269.
- Gao Y, Katsuraya K, Kaneko Y, et al. Synthesis, enzymatic hydrolysis, and anti-HIV activity of AZT − spacer − curdlan sulfates. Macromolecules. 1999;32:8319–8324.
- Williams FM. Clinical significance of esterases in man. Clin Pharmacokinet. 1985;10:392–403.
- Gopal R, Na H, Seo CH, et al. Antifungal activity of (KW) n or (RW) n peptide against Fusarium solani and Fusarium oxysporum. Int J Mol Sci. 2012;13:15042–15053.
- Aroui S, Brahim S, Waard MD, et al. Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: a comparative study. Biochem Biophys Res Commun. 2010;391:419–425.
- Brock R. The uptake of arginine-rich cell-penetrating peptides: putting the puzzle together. Bioconjugate Chem. 2014;25:863–868.
- Mellert K, Lamla M, Scheffzek K, et al. Enhancing endosomal escape of transduced proteins by photochemical internalisation. PLoS One. 2012;7:e52473.