
Abstract
Although melatonin has been shown to exhibit a wide variety of biological functions, its effects on promotion of self-renewal in pancreatic stem cells remain unknown. In this study, we incubated murine pancreatic stem cells (PSCs) with various concentrations of melatonin (0.01, 0.1, 1, 10 or 100 μM) to screen for the optimum culture medium for increasing cell proliferation. We found that 10 μM melatonin can significantly increase proliferation and enhance expression of a stem cell marker, nestin, in PSCs via melatonin receptor 2 (MT2). Thus, we used 10 μM melatonin to study the melatonin-mediated molecular mechanisms of cell proliferation in PSCs. We applied extracellular signal-regulated kinase (ERK) pathway inhibitor SCH772984 and transforming growth factor beta (TGF-β) pathway inhibitor SB431542, along with interfering RNAs siERK1, siERK2, siSmad2, siSmad3, siSmad4 and siNestin, to melatonin-treated PSCs to research the roles of these genes in self-renewal. The results revealed a novel molecular mechanism by which melatonin promotes self-renewal of PSCs: a chain reaction in the MT2/ERK/SMAD/nestin axis promoted the aforementioned self-renewal as well as inhibited differentiation. In addition, upregulation of nestin created a positive feedback loop in the regulation of the transforming growth factor beta 1 (TGF-β1)/SMADs pathway by promoting expression of Smad4. Conversely, knockdown of nestin significantly suppressed the proliferative effect in melatonin-treated PSCs. These are all novel mechanisms through which the ERK pathway cooperatively crosstalks with the SMAD pathway to regulate nestin expression, thereby enhancing self-renewal in PSCs.
Introduction
Melatonin (N-acetyl-5-methoxytryptamine), an indolamine originally isolated from mammalian pineal tissue, takes part in many important physiological functions. It can scavenge hydroxyl and peroxyl radicals [Citation1–4]; reduce myocardial ischemia–reperfusion injury; regulate circadian rhythms; and modulate visual, reproductive, cerebrovascular, neuroendocrine and neuroimmunological activity [Citation5–7]. Nestin is an intermediate filament protein that was initially identified as a marker of neural stem and progenitor cells [Citation8]. Melatonin can modulate nestin-positive neural stem cell (NSC) functions such as proliferation, reprogramming them into NSC-derived pluripotent stem cells as found in our previous research [Citation9]. However, nestin is transiently expressed not only during development of the central nervous system but also in different cell types of embryonic and adult tissues, such as pancreatic cells, developing skeletal muscle, mesenchymal cells, cardiomyocytes and endothelial cells [Citation10]. Nestin also seems to play an important role in regeneration in response to injury in various tissues, and there are reports that nestin regulates cell proliferation in normal stem cells [Citation11] and tumor cells [Citation12]. These findings strongly suggest that nestin plays an important role in controlling cell proliferation.
In pancreatic islets, a small subset of cells express nestin. These cells, called pancreatic stem cells (PSCs), are thought to be precursors of differentiated pancreatic endocrine cells [Citation13] and play an important role in the growth and maintenance of islets [Citation14–16]. Melatonin can modulate cell function in nestin-positive neural stem cells according to previous research, which prompted us to study whether melatonin can also regulate nestin expression to influence cell fate. In addition, melatonin can combine with melatonin receptor 1 (MT1) on cell membranes to promote the progression of breast cancer by influencing nestin expression in nestin-positive breast cancer stem cells [Citation17]. In our research, we found that nestin-positive PSCs and their differentiated beta cells expressed melatonin receptors 1 (MT1) and 2 (MT2), which are G-protein-coupled receptors. These findings prompted us to study whether melatonin also affects proliferation and differentiation of primary cultured PSCs. In the current study, we measured the viability of primary cultured PSCs in vitro treated with melatonin to verify optimal PSC amplification, demonstrated that melatonin enhanced the self-renewal of the PSCs via the MT2/extracellular signal-regulated kinase (ERK)/SMAD/nestin axis, and suggested a potential molecular mechanism of melatonin that produces these effects.
Materials and methods
Ethics statement
We conducted this study in strict accordance with recommendations of the Institutional Animal Care and Use Committee of Jining Medical University, China. The protocol was approved by the Committee on the Ethics of Animal Experiments of Jining Medical University (License ID: 2017-JZ-003). All surgery was performed under pentobarbital anesthesia, and all efforts were made to minimize suffering.
Primary cultures of murine PSCs in vitro
We isolated PSCs from the pancreases of suckling mice and cultured them in advanced RPMI 1640 medium (11.1 mM glucose) supplemented with fetal bovine serum (FBS) and cocktail factors according the method of our previous study [Citation18]. We obtained the fraction containing the majority of nestin-positive PSCs by flow cytometry (FCM) purification. We used immunofluorescence (IF) to analyze the PSCs for specific markers, including nestin, pancreas/duodenum homeobox protein 1 (PDX1), MT1 and MT2.
Differentiation of nestin-positive PSCs toward beta cells
Nicotinamide, a poly (adenosine diphosphate [ADP]–ribose) synthetase inhibitor, has been reported to prevent the development of diabetes in animal models and to promote the proliferation and differentiation of islet beta cells. In this study, we plated nestin-positive PSCs on 24-well cell culture plates. We then cultured them for 7 days in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 containing glucose (11.1 mM) and a cocktail of several growth factors, including 10 mM nicotinamide, 100 pmol/L hepatocyte growth factor (HGF) and 2 nmol/L active-A. To test whether insulin release from differentiated beta cells was glucose dependent, we used 2 glucose concentrations (5.5 mM and 20.5 mM). We measured insulin release using an Insulin ELISA Kit. We washed the beta cells with Krebs buffer and then pre-incubated them in low-glucose (5.5 mM) Krebs buffer for 2 h to remove residual insulin. We then washed them 3 times in Krebs buffer, incubated them in low-glucose Krebs buffer for 30 min and collected the supernatant. Finally, we washed the beta cells 3 times in Krebs buffer, incubated them in high-glucose (20.5 mM) Krebs buffer for 30 min and collected the supernatant. We repeated this sequence 3 additional times.
RNA interference
Small interfering RNA (siRNA) of MT1 (Gene ID: 17773), MT2 (Gene ID: 244701), ERK1 (Gene ID: 26417), ERK2 (Gene ID: 26413), Smad2 (Gene ID: 17126), Smad3 (Gene ID: 17127), Smad4 (Gene ID: 17128) and nestin (Gene ID: 18008) were designed, synthesized and cloned into lentiviral vectors. Recombinant lentiviral vectors harbored siRNAs targeting genes, as well as enhanced green fluorescence protein as a marker to detect infection efficiency, and were transfected into 293 T cells. Recombinant lentiviruses harvested from 293 T cells had titers of 3 × 106 TU/mL. We seeded and infected PSCs with recombinant lentiviruses and used positive cells for subsequent studies (siMT1, siMT2, siERK1, siERK2, siSmad2 [siS2], siSmad3 [siS3], siSmad4 [siS4] and siNestin cells). We used Western blot analysis to measure protein expression of genes targeted by siRNAs.
Proliferation assay
We analyzed PSC proliferation using FCM after Ki67 staining. Ki67 is a nuclear non-histone protein that is preferentially expressed during the late G1, S, G2 and M phases of the cell cycle; while resting, non-cycling cells (G0 phase) lack Ki67 expression. We treated PSCs in different groups and cultured them for 72 h in PSC medium. We then visualized Ki67 expression in PSCs using FCM, counting Ki67-positive stem cells from 3 random culture dishes in each of 3 independent PSC samples.
Immunofluorescent staining
We fixed the cells with 4% paraformaldehyde (PFA) in polybutylene succinate (PBS) for 1 h, then rinsed them 3 times with PBS for 5 min each time. We then permeabilized the cells with 0.01% Triton X-100 for 30 min. After rinsing them 3 times with PBS, we blocked the cells with goat serum for 30 min, then incubated them with primary antibodies in a humidified chamber at 4 °C overnight. After three 5-min washes with PBS the following day, we incubated the cells with Cy3 or fluorescein isothiocyanate (FITC)-labeled secondary antibodies at room temperature (RT) for 1 h. Cells were given a final wash with PBS 3 times for 5 min each. We recorded IF data using a confocal optical system.
Western blotting
We lysed the cells with Protein Extraction Reagent (Pierce Biotechnology [Thermo Fisher Scientific], Waltham, MA) supplemented with a protease inhibitor (phenylmethylsulfonyl fluoride). We measured protein concentrations within extracts using a bicinchoninic acid (BCA) assay (Pierce Biotechnology) and equalized them with extraction reagent. We loaded equal amounts of extracts and subjected them to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer onto nitrocellulose membranes. Primary antibodies (nestin, 1:300; MT1, 1:500; MT2, 1:500; ERK1/2, 1:500; P-ERK1/2, 1:800; Smad3, 1:500; Smad2, 1:500; Smad4, 1:800; and glyceraldehyde 3-phosphate dehydrogenase [GAPDH], 1:10,000) and horseradish peroxidase-coupled secondary antibodies (1:2000) were purchased from Abcam (Cambridge, UK). We probed the membranes using an ultra-enhanced chemiluminescence Western blot reagent. We used GAPDH as an internal control, and we measured protein concentration with Image J software (http://imagej.nih.gov/ij/).
Immunohistochemistry (IHC)
We dissected pancreas samples from suckling mice, fixed them in a 4% PFA/PBS solution overnight, and embedded them in paraffin. We then cut 5-μm longitudinal sections from the paraffin blocks, rehydrated them with xylene followed by decreasing concentrations of ethanol, microwaved them in 0.01 M sodium citrate (pH 6.0) for 20 min, and permeabilized them with 1% Triton X-100 in PBS prior to incubation with primary antibodies at 4 °C overnight. We used primary antibodies against nestin, MT1 and MT2 at dilutions of 1:100, 1:50 and 1:100, respectively. We then incubated the sections with GARB (1:400) and GAMB (1:100) for 3 h at RT and then with FITC/Cy5-conjugated streptavidin (1:50) for 3 h at RT. After counterstaining the sections with 4′,6-diamidino-2-phenylindole (DAPI), we obtained images by confocal microscopy.
Polymerase chain reaction (PCR)
We isolated total RNA from PSCs using TRIzol Reagent (Invitrogen, Carlsbad, CA). We performed cDNA synthesis using the High Capacity cDNA Synthesis Kit (Applied Biosystems, Foster City, CA) with 2 ng total RNA as the template. Sequence-specific reverse transcription (RT)–PCR primers for nestin, MT1, MT2, and the GAPDH endogenous control were synthesized by Sangon Biotech (Shanghai, China).
Statistical analysis
We performed statistical analyses of our data with a 1-way analysis of variance [ANOVA] followed by the Tukey–Kramer honest significant difference (HSD) test for the 3 sets of results (p < .05 was considered statistically significant). We assessed our statistical analyses with JMP Statistical Discovery software (SAS Institute, Cary, NC).
Experimental design
We isolated PSCs from the pancreases of suckling mice and cultured them in advanced RPMI 1640 medium, then differentiated beta cells from these PSCs using methods as previously reported [Citation18]. To test co-expression of melatonin receptors and nestin in PSCs and beta cells, we used IF, IHC and PCR to analyze the expression of MT1, MT2 and nestin in these cells and in murine tissue. To study the effect of melatonin on PSC proliferation, we supplemented melatonin in culture medium at 0.01, 0.1, 1, 10 or 100 μM. We then treated PSCs with melatonin and measured cell proliferation using FCM after Ki67 staining. We selected the optimal melatonin treatment for PSC proliferation for supplementation in differentiation medium of beta cells, and we measured proinsulin expression using FCM. We tested potential mechanisms of melatonin-mediated cell proliferation using ERK pathway inhibitor (SCH772984), TGF-β pathway inhibitor (SB431542) and RNA interference (RNAi). For all experiments, we dissolved melatonin in Dulbecco’s Modified Eagle’s Medium (DMEM) overnight, and we repeated all experiments independently at least 3 times.
Results
Co-expression of melatonin receptor and nestin in murine PSCs
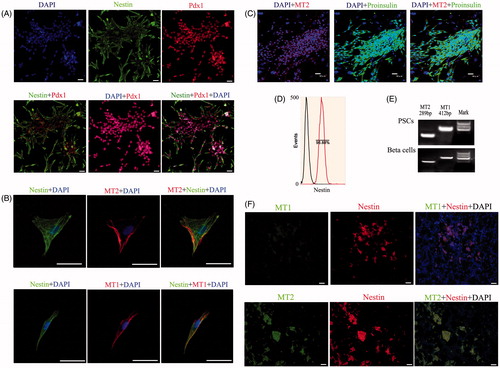
We isolated PSCs from the pancreases of suckling mice, primarily cultured them in advanced RPMI 1640 medium supplemented with cocktail factors, and purified them using nestin antibody and FCM. Pdx1, a transcription factor, is recognized as a marker of pancreatic stem cells in pancreatic development, so we determined the phenotype of pancreatic precursors in vivo. shows nestin and Pdx1 expression using IF; cells in clusters were positive for these proteins. The plasticity of stem cells is their main characteristic. Thus, we used a cocktail of growth factors to induce the formation of beta cells from PSCs, then detected the expression of proinsulin in islet-like structures using IF staining (). Melatonin receptors in mammals include 2 high-affinity receptors, MT1 and MT2, which we tested using IF and RT-PCR. shows that MT1, MT2 and nestin were co-expressed in PSCs. In addition, our IHC results demonstrated that all 3 were also co-expressed in pancreatic islet structures (). Taken together, our results demonstrated that nestin, MT1 and MT2 were co-expressed in PSCs both in vitro and in vivo.
Figure 1. Expression of specific genes in PSCs in vitro and in vivo. (A) Detection of PSC markers by IF staining. Nestin staining was located in the cytoplasm, while Pdx1 staining was located in the nucleus. The nestin, Pdx1 and DAPI signals appear as garland-like structures in merged images. Pdx1, a transcription factor, is recognized as a marker of PSCs in pancreas development (bar = 50 μm). (B) Detection of melatonin receptors by IF staining in PSCs in vitro. Melatonin receptors include MT1 and MT2, which were co-expressed in PSCs (bar = 200 μm). (C) IF analysis of sections in differentiated beta cell clusters stained for proinsulin and MT2 (bar = 50 μm). (D) Purification of PSCs by FCM using nestin antibody. (E) RT-PCR analysis of sections from differentiated beta cells and PSCs tested for MT1 and MT2. (F) Co-expression of nestin, MT1 and MT2 in pancreatic tissue using IHC. The results demonstrated that these genes were co-expressed in pancreatic islet structures (bar = 50 μm).
Melatonin influences nestin expression and cell proliferation in murine PSCs
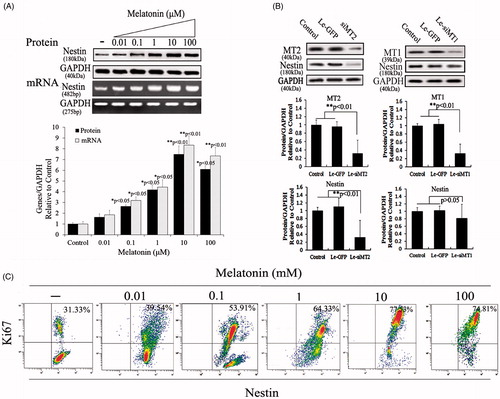
To determine whether the increased nestin expression in PSCs was due to melatonin treatment, we added various melatonin concentrations (0.01, 0.1, 1, 10 or 100 μM) to PSC cultures to test for changes in nestin expression. Results indicated that nestin expression levels increased in a concentration-dependent manner in melatonin-treated PSCs. PSCs incubated with 10 μM melatonin had significantly increased nestin expression levels compared with the other groups (p < .01); however, no significant change in expression level occurred between 10 μM and 100 μM (p > .05) (). To detect the role of melatonin receptors in nestin expression, siMT1 and siMT2 were overexpressed in melatonin-induced PSCs to determine which melatonin receptors play a role. The results indicated that MT2 is an important melatonin receptor for the induction of nestin expression in PSCs (). After treatment with various melatonin concentrations for 72 h, we measured cell viability data via Ki67 assay; the number of Ki67-positive cells indicated that proliferation had increased and that melatonin treatment had improved PSC viability in a dose-dependent manner compared with untreated cells. However, PSCs treated with 10 μM melatonin showed no significant change in cell proliferation compared with PSCs treated with 100 μM melatonin (). Therefore, we selected 10 μM melatonin as the dose to promote cell proliferation of PSCs.
Figure 2. Melatonin increases nestin expression and cell viability in PSCs. (A) Nestin expression was tested using Western blotting and RT-PCR after PSCs were exposed to various melatonin concentrations. Results demonstrated that nestin levels increased in a concentration-dependent manner and that PSCs incubated with 10 μM melatonin had significantly increased expression levels compared with other groups. (B) The role of melatonin receptors in nestin expression. Melatonin receptors, including MT1 and MT2, belong to the family of G-protein–coupled receptors. For scanning biological functions of MT1 and MT2, siRNAs were used to knock MT1 or MT2 down when PSCs were exposed to 10 μM melatonin, and protein levels were analyzed using Western blotting. Results indicated that MT2 is an important melatonin receptor for the induction of nestin expression in PSCs. Protein abundance was analyzed using Image J tools and normalized using endogenous control. (C) PSC proliferation was assessed using Ki67-marked methods after treatment with various melatonin concentrations for 72 h.
Role of MT, ERK and TGF-β/SMAD pathways in melatonin-treated cell proliferation
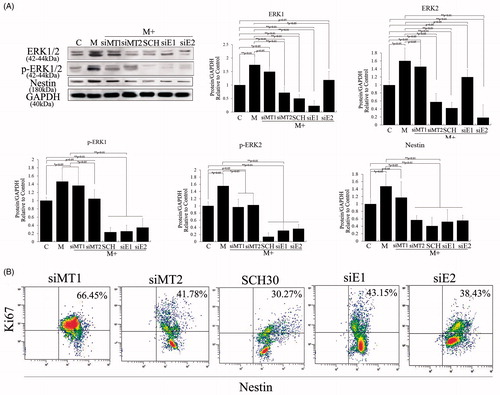
Melatonin receptors, which include MT1 and MT2 and which belong to the family of G-protein-coupled receptors and binding sites (putative receptors), have also been found in the nuclei of many cell types [Citation19,Citation20]. To explore the potential mechanisms of action for melatonin, siMT1 and siMT2 were overexpressed in 10 μM melatonin-treated PSCs to determine which receptor plays a role. Our results indicated that MT2 is an important melatonin receptor for the proliferation of PSCs (). The ERK pathway is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell, including ERK1/2, MEK, JNK and P38. Other studies have shown that ERK1/2 is activated by phosphorylation in response to melatonin in cell cultures, it was found that melatonin can promote cell proliferation by activating the ERK pathway. In the current study, we also demonstrated that melatonin regulates ERK pathways that control cell proliferation. As shown in , phosphorylation and expression of ERK1/2 were significantly upregulated after 10 μM melatonin treatment. Phosphorylation and expression of ERK1/2 were also negatively correlated with overexpressed siMT2, and cell proliferation analyses indicated decreased proliferation efficiency after 10 μM melatonin treatment and siMT2 overexpression, compared with melatonin and siMT1 overexpression. We then used an EKR pathway inhibitor (SCH772984) and siERK1 (siE1) and siERK2 (siE2) to analyze cell proliferation and nestin expression singly after 10 μM melatonin treatment to illuminate the functions of ERK pathway members in the self-renewal of PSCs. Our results showed that SCH772984, siERK1 and siERK2 effectively repressed cell proliferation and nestin expression in PSCs (p < .01; ), and demonstrated ERK1/2 played an important role in melatonin-induced cell proliferation and nestin expression.
Figure 3. Role of melatonin receptors in melatonin-induced PSC proliferation. Melatonin promoted cell proliferation by activating the ERK pathway. To detect the roles of MT1, MT2 and ERK1/2 in melatonin-treated PSCs, we exposed siMT1, siMT2, siERK1, siERK2 and SCH772984 to melatonin-treated PSCs. (A) Western blotting revealed that melatonin activated the ERK pathway via MT2, and nestin expression levels were positively correlated with activation of the ERK pathway and decreased when siERK1, siERK2 or SCH772984 was exposed to melatonin-treated PSCs. C: control, without melatonin; M: 10 μM melatonin added to PSC culture; siMT1: siRNA of MT1; siMT2: siRNA of MT2; SCH: SCH772984, ERK pathway inhibitor; siE1: siRNA of ERK1; siE2: siRNA of ERK2. Protein abundance was analyzed using ImageJ tools and normalized using endogenous controls. (B) PSC proliferation was assessed using Ki67-marked methods after treatment with different factors for 72 h.
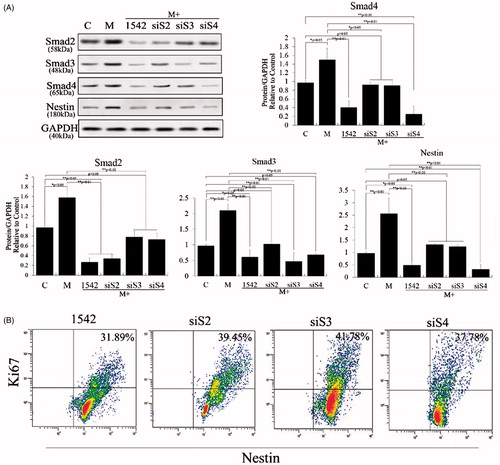
TGF-β signaling initiates as the ligand binds to the receptors followed by the dimerization of type I and type II receptors. The activation of the type I receptor is subsequently followed by the phosphorylation of Smad proteins including Smad2, Smad3 and Smad4, which then translocates into the nucleus and regulates the transcription of related genes. The TGF-β/SMAD pathway plays an important role in cell proliferation, which has been extensively explored over recent years; however, there has been no study of TGF-β1 pathway–altered cell proliferation and nestin expression in PSCs. Smad2, Smad3 and Smad4 are essential components of the intracellular TGF-β/SMAD pathway, and we found that melatonin can promote the expression of all 3 to increase proliferation of PSCs (). We also used TGF-β pathway inhibitor SB431542 and siS2, siS3 and siS4 to detect the role of the TGF-β1 pathway and its members in proliferation and nestin expression of PSCs. Our results showed that SB431542, siS2, siS3 and siS4 effectively repressed cell proliferation and nestin expression in melatonin-induced PSCs (p < .01; ). These results demonstrated the TGF-β/SMAD pathway played critical role in melatonin-induced cell proliferation and nestin expression.
Figure 4. Role of TGF-β/SMAD pathway in melatonin-induced PSC proliferation. TGF-β1 pathway inhibitor (SB431542), siS2, siS3 and siS4 were used to test the functions of this pathway. (A) Western blotting revealed that melatonin promotes expression of Smad2, Smad3 and Smad4 and that nestin expression levels were positively correlated with expression of Smad2, Smad3 and Smad4. These levels were significantly decreased when siS2, siS3, siS4 or SB431542 was applied to melatonin-treated PSCs. (B) PSC proliferation was assessed using Ki67-marked methods after treatment with different factors for 72 h.
Crosstalk between the ERK and TGF-β/SMAD pathways in melatonin-treated cell proliferation
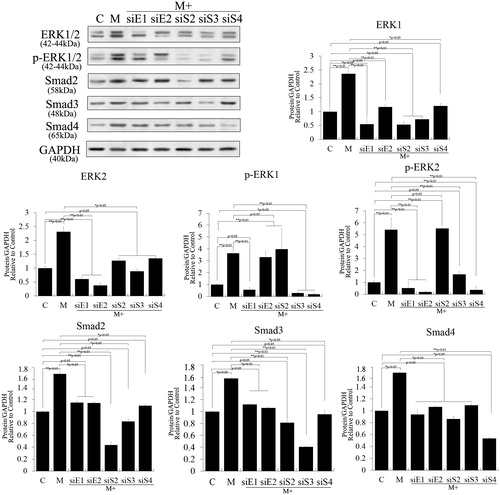
Crosstalk between the ERK and TGF-β/SMAD pathways has been demonstrated in many cell types [Citation21,Citation22], but crosstalk between the pathways to promote proliferation of stem cells remains poorly understood. In the current study, we knocked ERK1, ERK2, Smad2, Smad3 and Smad4 down respectively using small interfering RNAs in melatonin-induced PSCs to analyze the effects of these factors, and we detected protein levels using Western blotting. As shown in , Western blotting revealed that ERK1 knockdown significantly suppressed melatonin-induced ERK1/2 and Smad2/3 expression (p < .01), but ERK2 knockdown did not significantly change them (p > .05). Meanwhile, Smad2/3 knockdown also significantly suppressed melatonin-induced ERK1/2 and Smad2/3 expression (p < .01), but Smad4 knockdown did not do so (p > .05). Hence, phosphorylations of ERK1/2 were significantly decreased in ERK1/2 and Smad3 knockdown experiments (p < .01). Together, these results suggest that Smad2/3 may be involved in melatonin-induced phosphorylation and expression of ERK1/2, which is positive feedback in the interaction between Smad2/3 and ERK1/2.
Figure 5. Crosstalk between ERK and TGF-β/SMADs pathways in melatonin-induced PSCs. ERK1, ERK2, Smad2, Smad3 and Smad4 were knocked down using RNAi to analyze their effects in PSCs after melatonin treatment. The results revealed that ERK1 knockdown significantly suppressed melatonin-induced ERK1/2 and Smad2/3 expression (p < .01), but ERK2 knockdown did not do so (p > .05). Smad2/3 knockdown also significantly suppressed melatonin-induced ERK1/2 and Smad2/3 expression (p < .01), but Smad4 knockdown did not (p > .05). These results suggest that Smad2/3 may be involved in melatonin-induced phosphorylation and expression of ERK1/2, which is positive feedback in the regulation of Smad2/3 and ERK1/2.
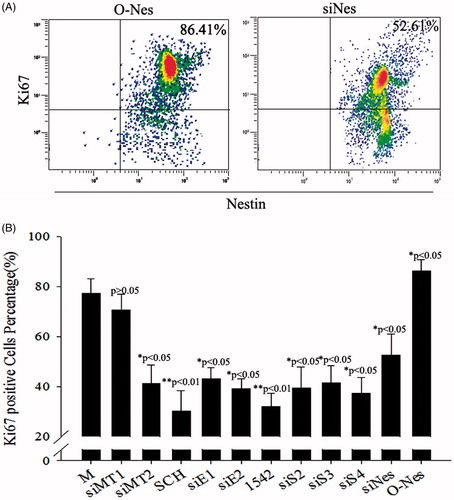
Smad4 plays an important role in the expression of nestin via activation of the TGF-β/SMAD pathway; moreover, increased nestin expression is a positive feedback regulator of the TGF-β pathway [Citation23]. We thus overexpressed nestin in PSCs to test feedback with Smad4. As shown in , Western blotting revealed that lentivirus expressing nestin significantly increased that expression compared with control cells. Phosphorylation and expression of ERK1/2, Smad2 and Smad3 were negligibly affected by nestin overexpression (p > .05), but the expression of Smad4 changed significantly (p < .05). In contrast, knockdown of nestin in PSCs caused no significant changes in the expression of ERK1/2, Smad2, Smad3, or Smad4 (). These results suggest that increased nestin expression was a positive feedback regulator of the TGF-β/SMAD pathway via promoting SMAD4 expression. Our studies have indicated that TGF-β/SMAD–induced nestin expression is promoted via activation of the ERK pathway after melatonin treatment in PSCs. Our assessment of cell proliferation after overexpression or knockdown of nestin in melatonin-treated PSCs demonstrated proliferation and inhibition of PSCs, respectively ().
Figure 6. Crosstalk among nestin and the ERK and TGF-β/SMADs pathways in PSCs. Nestin was overexpressed in PSCs to test feedback of the TGF-β/SMADs pathway. Lentivirus expressing nestin significantly increased nestin expression compared with control cells in PSCs. However, nestin overexpression negligibly affected phosphorylation and expression of ERK1/2, Smad2 and Smad3 (p > .05). The expression level of Smad4 changed significantly (p < .05).
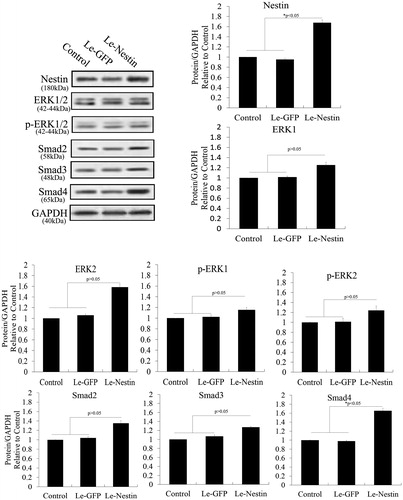
Figure 7. Knockdown of nestin to test feedback of the ERK and TGF-β/SMADs pathways in PSCs. Nestin was knocked down using RNAi. Western blotting revealed that lentivirus expressing siRNA of nestin (siNes) significantly decreased nestin expression in PSCs compared with controls (p < .05). However, nestin knockdown negligibly affected the phosphorylation and expression of ERK1/2, Smad2, Smad3 and Smad4 (p > .05).
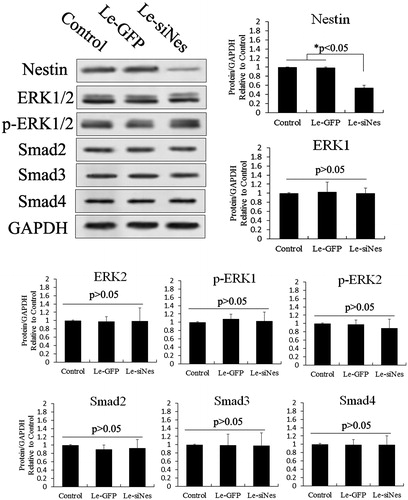
Figure 8. Proliferation assessed after treatment with different siRNAs in melatonin-treated PSCs. (A) Ki67-marked analysis of proliferation after overexpression of nestin in melatonin-treated PSCs, as well as Ki67-marked analysis of proliferation after RNAi-nestin treatment in melatonin-treated PSCs. (B) Statistical analysis of the proliferation effect of gene silencing combined with melatonin treatment in PSCs using Ki67-marked methods. These results showed that melatonin induced cell proliferation by activating the MT2/ERK/SMAD/nestin axis in PSCs. Ki67-positive stem cells were counted from 3 random culture dishes in each of 3 independent PSC samples.
Melatonin weakens differentiation of beta cells in PSCs
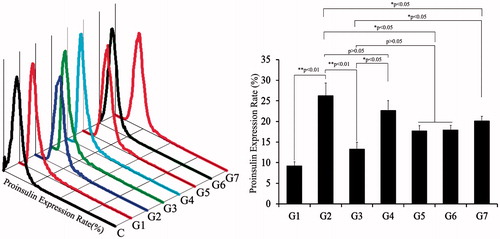
Proliferation effects of melatonin in PSCs were summarized in above research, and then, to assess the roles of melatonin, MT1, MT2, the TGF-β/SMAD pathways, and the ERK pathway in differentiation of beta cells from PSCs, we used siRNAs of MT1 and MT2 and pathway inhibitors SB431542 and SCH772984. We added each of siMT1, siMT2, SB431542 and SCH772984 to PSCs in combination with melatonin. Effects on the differentiation of beta cells are shown in . Single exposures of siMT1, siMT2, SB431542 and SCH772984 in PSCs after 10 μM melatonin treatment showed that melatonin was an inhibitor, while siMT2, SB431542 and SCH772984 were antagonists, of melatonin in the differentiation of beta cells from PSCs (p < .05). FCM data showed decreased efficiency of beta cells differentiation from PSCs following a combination of melatonin and siMT2 overexpression, compared with melatonin and siMT1 overexpression. These results suggested melatonin weakened differentiation of beta cells via MT2-ERK-TGF-β/SMAD pathways. Taken together, our results suggest that melatonin inhibited the differentiation of beta cells, promoted cell proliferation, and increased expression of the stem cell marker nestin through activation of the ERK and TGF-β1 pathways.
Figure 9. Insulin expression in beta cells derived with different combinations of inducers. Analyses of proinsulin expression using FCM. The percentage of cells expressing insulin was highest in G2. C: control; G1: group 1, PSCs; G2: group 2, PSCs induced with a cocktail of growth factors; G3: group 3, melatonin-treated PSCs induced with growth factors; G4: group 4, melatonin-treated PSCs overexpressing siMT1 and induced with growth factors; G5: group 5, melatonin-treated PSCs overexpressing siMT2 and induced with growth factors; G6: group 6, melatonin plus SCH772984-treated PSCs induced with growth factors; G7: group 7, melatonin plus SB431542-treated PSCs induced with growth factors.
Functional feature of beta cells from PSCs
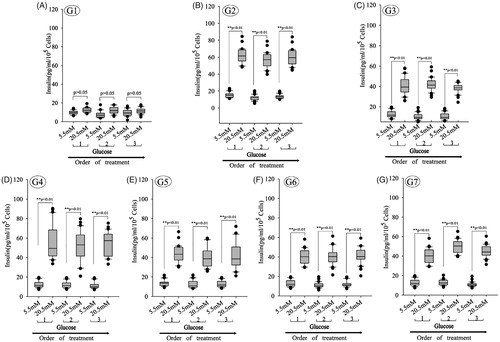
A key functional feature of beta cells is its ability to repeatedly perform glucose-stimulated insulin secretion. Beta cells challenged sequentially with 5.5, 20.5, 5.5, 20.5, 5.5 and 20.5 mM glucose, with a 30 min incubation for each concentration. After sequential low/high-glucose challenges, insulin concentration were analyzed in supernatant using ELISA. And glucose-induced insulin secretion demonstrated that PSCs derived from beta cells could release insulin at glucose concentrations of 5.5 mM and 20.5 mM. Analysis of glucose-induced insulin secretion demonstrated that beta cells derived with different combinations of inducers could release insulin, while undifferentiated progenitor cells (G1) did not release insulin. The box plots show that insulin secretion increased when the glucose concentration increased in G2–G7, when the secreted insulin amounts of IPCs differentiation from PSCs was assessed, the combination of nicotinamide, HGF, and active-A-induced IPCs secretion at a higher efficiency compared with combinations of melatonin (). These validation results were largely in agreement with the data of IPCs differentiation.
Figure 10. Glucose-induced insulin secretion in beta cells derived with different combinations of inducers. (A–G) Glucose-induced insulin secretion in beta cells derived with different combinations of inducers. The box plots show that insulin secretion increased when the glucose concentration increased in all groups except group 1 (n = 30; paired 2-tailed t test). G1: group 1, PSCs; G2: group 2, PSCs induced with a cocktail of growth factors; G3: group 3, melatonin-treated PSCs induced with growth factors; G4: group 4, melatonin-treated PSCs overexpressing siMT1 and induced with growth factors; G5: group 5, melatonin-treated PSCs overexpressing siMT2 and induced with growth factors; G6: group 6, melatonin plus SCH772984-treated PSCs induced with growth factors; G7: group 7, melatonin plus SB431542-treated PSCs induced with growth factors.
Discussion
Type I diabetes is caused by the autoimmune destruction of beta cells in pancreatic islets. While replacing pancreatic beta cells would be an ideal therapy [Citation24,Citation25], generation of beta cells from stem or other cells is an attractive alternative [Citation26–28]. In pancreatic islets, only a small subset of cells expresses nestin, and these have been proposed to be precursors of differentiated pancreatic endocrine cells [Citation13]. These nestin-positive cells are called pancreatic stem cells (PSCs), and it has been proposed that they play an important role in the growth and maintenance of pancreatic islets [Citation14–16,Citation29].
Previous studies have indicated that melatonin has multiple important physiological functions within the neuroimmunoendocrine system and that its wide extracellular and intracellular distribution indicates potentially broad physiological functions. Previous research has reported that persistent activation of melatonin signaling can attenuate pancreatic beta cell loss and dysfunction associated with oxidative and endoplasmic-reticulum stress [Citation30,Citation31]. However, no reports on melatonin promotion of pancreatic progenitor cells are available. Our research on melatonin confirmed it can promote the proliferation of pancreatic progenitor cells. Given that melatonin is a well-known activator of the ERK pathway, Western blotting showed dramatic upregulation of protein expression and phosphorylation levels compared with controls. The ERK1/2 cascade, activated by mitogenic stimuli, is critical for cell proliferation and survival and required for normal progression into mitosis [Citation32,Citation33]. ERK pathways are responsible for melatonin’s proliferative actions in various cells, including neural cells, induced pluripotent stem cells and pancreatic beta cells [Citation9,Citation31]. ERK1/2 activation has been well documented as necessary for cell proliferation and is partly involved in G1 and G2/M phase progression [Citation34–36]. As melatonin increased the expression of p-ERK1/2 and ERK1/2, we tested whether SCH772984, a selective inhibitor of the ERK pathway that disrupts activation of ERK1/2, would inhibit the proliferative effect of melatonin in pancreatic progenitor cells. Our results demonstrated that SCH772984 significantly repressed that proliferative effect after melatonin treatment. These findings strongly suggest that ERK1/2 activation plays a central role in the observed melatonin-mediated effects.
TGF-β1 is a member of the TGF-β superfamily of cytokines, which play roles in growth, development, immunity, wound healing, inflammation, apoptosis and cancer. Previous studies have indicated that the TGF-β1 pathway acts through SMAD-dependent (canonical) and SMAD-independent (non-canonical) pathways [Citation37,Citation38]. Smad2 and Smad3, the principal receptor-activated proteins involved in the TGF-β pathway, are activated directly by TGF-β type I receptor kinase, then translocated to the nucleus complexed with a common co-SMAD, Smad4, to regulate the transcription of target genes [Citation39,Citation40]. Many signaling pathways interact with the TGF-β1 pathway. The mitogen-activated protein kinase (MAPK) cascade, composed mainly of ERK, p38 MAPK and c-Jun N-terminal kinases (JNKs), has been shown to represent a major signaling pathway for TGF-β-dependent SMAD activation [Citation41,Citation42]. In crosstalk between the SMAD and MAPK pathways, TGF-β-dependent or -independent induction of activated ERK can occur [Citation40]. We tested crosstalk between the TGF-β/SMAD and ERK pathways in melatonin-induced cell proliferation in our research by transfecting siERK1, siERK2, siS2, siS3 and siS4 into pancreatic progenitor cells treated with melatonin. We demonstrated that knockdown of Smad2 or Smad3 effectively reduced expression levels of ERK1, but knockdown of Smad4 did not. However, knockdown of ERK1 or ERK2 did not result in positive feedback, consistent with data reported previously [Citation22].
Nestin is an intermediate filament protein expressed in dividing cells during the early stages of development in the central nervous system, peripheral nervous system, and myogenic and other tissues. Upon differentiation, nestin becomes downregulated and is replaced by tissue-specific intermediate filament proteins, a theory confirmed in pancreatic progenitor cells in previous research [Citation18,Citation43]. In the current study, protein levels of nestin in PCSs were increased in a concentration-dependent manner after melatonin treatment, suggesting that melatonin is involved in maintaining their self-renewal. However, the molecular mechanisms of nestin expression activation after melatonin treatment remain unknown. We found that proliferation and nestin expression of PSCs were enhanced via positive feedback in crosstalk between the ERK and TGF-β/SMAD pathways after melatonin treatment, and siRNA data showed that the TGF-β/SMAD pathway might have induced nestin expression in PSCs predominantly mediated by the Smad4-dependent pathway. Meanwhile, Smad4 could be regulated by nestin expression as positive feedback, consistent with data reported previously [Citation44].
In summary, melatonin promotes the cell proliferation of PSCs due at least in part to activation of the TGF-β/SMAD and ERK pathways. Melatonin enhances nestin expression of PSCs to maintain self-renewal and prevent differentiation, and this may involve crosstalk between the TGF-β/SMAD and ERK pathways. These findings reveal possible novel mechanisms through which the ERK pathway cooperatively interacts with the TGF-β/SMAD pathway to regulate nestin expression.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Melchiorri D, Reiter RJ, Attia AM, et al. Potent protective effect of melatonin on in vivo paraquat-induced oxidative damage in rats [Comparative Study Research Support, U.S. Gov’t, Non-P.H.S.]. Life Sci. 1995;56:83–89.
- Garcia JJ, Lopez-Pingarron L, Almeida-Souza P, et al. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: a review [Research Support, Non-U.S. Gov’t Review]. J Pineal Res. 2014;56:225–237.
- Manchester LC, Coto-Montes A, Boga JA, et al. Melatonin: an ancient molecule that makes oxygen metabolically tolerable [Review]. J Pineal Res. 2015;59:403–419.
- Reiter RJ, Mayo JC, Tan DX, et al. Melatonin as an antioxidant: under promises but over delivers [Review]. J Pineal Res. 2016;61:253–278.
- Dubocovich ML. Melatonin receptors: role on sleep and circadian rhythm regulation [Research Support, N.I.H., Extramural Review]. Sleep Med. 2007;8 (Suppl 3):34–42.
- Hardeland R. Melatonin, hormone of darkness and more: occurrence, control mechanisms, actions and bioactive metabolites [Review]. Cell Mol Life Sci. 2008;65:2001–2018.
- Yang Y, Sun Y, Yi W, et al. A review of melatonin as a suitable antioxidant against myocardial ischemia-reperfusion injury and clinical heart diseases [Research Support, Non-U.S. Gov’t Review]. J Pineal Res. 2014;57:357–366.
- Soleimannejad M, Ebrahimi-Barough S, Soleimani M, et al. Fibrin gel as a scaffold for photoreceptor cells differentiation from conjunctiva mesenchymal stem cells in retina tissue engineering. Artif Cells Nanomed Biotechnol. Forthcoming. [cited 2017 Jul 10]. doi: 10.1080/21691401.2017
- Bai C, Li X, Gao Y, et al. Melatonin improves reprogramming efficiency and proliferation of bovine-induced pluripotent stem cells. J Pineal Res. 2016;61:154–167.
- Gholizadeh-Ghaleh Aziz S, Fathi E, Rahmati-Yamchi M, et al. An update clinical application of amniotic fluid-derived stem cells (AFSCs) in cancer cell therapy and tissue engineering [Review]. Artif Cells Nanomed Biotechnol. 2017;45:765–774.
- Hu W, Lu H, Wang S, et al. Suppression of nestin reveals a critical role for p38-EGFR pathway in neural progenitor cell proliferation. Oncotarget. 2016;7:87052–87063.
- Matsuda Y, Ishiwata T, Yoshimura H, et al. Nestin phosphorylation at threonines 315 and 1299 correlates with proliferation and metastasis of human pancreatic cancer. Cancer Sci. 2017;108:354–361.
- Hunziker E, Stein M. Nestin-expressing cells in the pancreatic islets of Langerhans. Biochem Biophys Res Commun. 2000;271:116–119.
- Lardon J, Rooman I, Bouwens L. Nestin expression in pancreatic stellate cells and angiogenic endothelial cells [Research Support, Non-U.S. Gov’t]. Histochem Cell Biol. 2002;117:535–540.
- Selander L, Edlund H. Nestin is expressed in mesenchymal and not epithelial cells of the developing mouse pancreas [Research Support, Non-U.S. Gov’t]. Mech Dev. 2002;113:189–192.
- Treutelaar MK, Skidmore JM, Dias-Leme CL, et al. Nestin-lineage cells contribute to the microvasculature but not endocrine cells of the islet [In Vitro Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Diabetes. 2003;52:2503–2512.
- Rogelsperger O, Ekmekcioglu C, Jager W, et al. Coexpression of the melatonin receptor 1 and nestin in human breast cancer specimens [Research Support, Non-U.S. Gov’t]. J Pineal Res. 2009;46:422–432.
- Bai C, Li X, Gao Y, et al. Role of microRNA-21 in the formation of insulin-producing cells from pancreatic progenitor cells [Research Support, Non-U.S. Gov’t]. Biochim Biophys Acta. 2016;1859:280–293.
- Pozo D, Garcia-Maurino S, Guerrero JM, et al. mRNA expression of nuclear receptor RZR/RORalpha, melatonin membrane receptor MT, and hydroxindole-O-methyltransferase in different populations of human immune cells [Research Support, Non-U.S. Gov’t]. J Pineal Res. 2004;37:48–54.
- Luchetti F, Canonico B, Bartolini D, et al. Melatonin regulates mesenchymal stem cell differentiation: a review [Research Support, Non-U.S. Gov’t Review]. J Pineal Res. 2014;56:382–397.
- Wang X, Zhu Y, Tao H, et al. Interaction of ERK1/2 and Smad2/3 signaling pathways in TGF-beta1-induced TIMP-3 expression in rat chondrocytes. Arch Biochem Biophys. 2014;564:229–236.
- Zhu Y, Gu J, Zhu T, Jin C, Hu X, Wang X. Crosstalk between Smad2/3 and specific isoforms of ERK in TGF-beta1-induced TIMP-3 expression in rat chondrocytes. J Cell Mol Med. 2017;21:1781–1790.
- Kiwanuka E, Junker JP, Eriksson E. Transforming growth factor beta1 regulates the expression of CCN2 in human keratinocytes via Smad-ERK signalling. Int Wound J. Forthcoming. [cited 2017 Mar 29]. doi: 10.1111/iwj.12749
- Zulewski H. Stem cells with potential to generate insulin producing cells in man [Research Support, Non-U.S. Gov’t Review]. Swiss Med Wkly. 2006;136:647–654.
- Bazwinsky-Wutschke I, Muhlbauer E, Albrecht E, et al. Calcium-signaling components in rat insulinoma beta-cells (INS-1) and pancreatic islets are differentially influenced by melatonin [Research Support, Non-U.S. Gov’t]. J Pineal Res. 2014;56:439–449.
- Scharfmann R. Alternative sources of beta cells for cell therapy of diabetes [Research Support, Non-U.S. Gov’t Review]. Eur J Clin Investig. 2003;33:595–600.
- Pennarossa G, Maffei S, Campagnol M, et al. Brief demethylation step allows the conversion of adult human skin fibroblasts into insulin-secreting cells [Research Support, Non-U.S. Gov’t]. Proc Natl Acad Sci U S A. 2013;110:8948–8953.
- Saei Arezoumand K, Alizadeh E, Pilehvar-Soltanahmadi Y, et al. An overview on different strategies for the stemness maintenance of MSCs. Artif Cells Nanomed Biotechnol. 2017;45:1255–1271.
- Ali T, Kim MO. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3beta pathway in the mouse hippocampus [Research Support, Non-U.S. Gov’t]. J Pineal Res. 2015;59:47–59.
- Park JH, Shim HM, Na AY, et al. Melatonin prevents pancreatic beta-cell loss due to glucotoxicity: the relationship between oxidative stress and endoplasmic reticulum stress [Research Support, Non-U.S. Gov’t]. J Pineal Res. 2014;56:143–153.
- Costes S, Boss M, Thomas AP, et al. Activation of melatonin signaling promotes beta-cell survival and function [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Mol Endocrinol. 2015;29:682–692.
- Hayne C, Tzivion G, Luo Z. Raf-1/MEK/MAPK pathway is necessary for the G2/M transition induced by nocodazole [Research Support, U.S. Gov’t, P.H.S.]. J Biol Chem. 2000;275:31876–31882.
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases [Review]. Science. 2002;298:1911–1912.
- Chambard JC, Lefloch R, Pouyssegur J, et al. ERK implication in cell cycle regulation [Research Support, Non-U.S. Gov’t Review]. Biochim Biophys Acta. 2007;1773:1299–1310.
- Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition [Research Support, Non-U.S. Gov’t Review]. Oncogene. 2007;26:3227–3239.
- Liu L, Xu Y, Reiter RJ, et al. Inhibition of ERK1/2 signaling pathway is involved in melatonin’s antiproliferative effect on human MG-63 osteosarcoma cells. Cell Physiol Biochem. 2016;39:2297–2307.
- Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily [Review]. Science. 2002;296:1646–1647.
- Lu L, Wang J, Zhang F, et al. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Immunol. 2010;184:4295–4306.
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review]. Cell. 2003;113:685–700.
- Akel S, Bertolette D, Ruscetti FW. Crosstalk between the smad and the mitogen-activated protein kinase pathways is essential for erythroid differentiation of erythroleukemia cells induced by TGF-beta, activin, hydroxyurea and butyrate. J Leuk. 2013;1:109.
- Engel ME, McDonnell MA, Law BK, et al. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. J Biol Chem. 1999;274:37413–37420.
- Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis [Review]. Oncogene. 2005;24:5742–5750.
- Bai C, Gao Y, Li X, et al. MicroRNAs can effectively induce formation of insulin-producing cells from mesenchymal stem cells. J Tissue Eng Regen Med. Forthcoming. [cited 2017 Apr 10]. doi: 10.1002/term.2259
- Su HT, Weng CC, Hsiao PJ, et al. Stem cell marker nestin is critical for TGF-beta1-mediated tumor progression in pancreatic cancer [Research Support, Non-U.S. Gov’t]. Mol Cancer Res. 2013;11:768–779.