
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
This study was aimed at preparing and characterizing solid lipid nanoparticles (SLNs) of donepezil (DPL) for delivery to brain via nasal route. SLNs were prepared by solvent emulsification diffusion technique using glyceryl monostearate (GMS) as lipid and blend of tween 80 and poloxamer 188 (1:1) as surfactant. Box–Behnken design was applied for optimization by using drug to lipid ratio, surfactant concentration and stirring time as dependent variables and their effect were observed on particles size, entrapment efficiency and drug loading. Optimized formulation was evaluated for particle size, zeta potential, entrapment efficiency, drug loading, morphological analysis, crystallinity, in vitro drug release, in vivo (biodistribution, pharmacokinetic and Gama scintigraphy) studies. For optimized formulation (OD3), value of particle size, zeta potential, percent in vitro release, entrapment efficiency and drug loading was found to be 121.0 nm, –24.1 mV, 89.35%, 67.95% and 12.15%, respectively. Pharmacokinetic and biodistribution studies were performed on albino Wistar rats and value of AUC0–∞ in brain for DPL-SLNs i.n. was found to be nearly 2.61 times higher than that of DPL-Sol i.v., whereas 2.26 times superior than DPL-Sol administered intranasally. The scintigraphy images were taken in rabbit and result revealed the localization of drug in brain.
Introduction
In certain cases, oral route fails to deliver therapeutic amount of drug to brain due to the presence of certain interfaces like blood–brain barrier (BBB), blood–cerebrospinal fluid barrier (BCSFB) and efflux transporters (AET) [Citation1]. These barriers regulate the exchange between peripheral blood circulation and cerebrospinal fluid (CSF) circulatory system. Other factors like physicochemical properties of drug also create hindrance in central nervous system (CNS) delivery [Citation2]. Thus, various approaches like BBB disruption, drug manipulation and alternative route of drug administration like intra cerebroventricular, intrathecal and olfactory pathways (intranasal route) are being used for targeting of drugs to the brain [Citation3]. In the present scenario, the intranasal route to bypass the BBB is an upcoming field, as this route caters a novel, practical, simple and non-invasive approach to bypass the BBB and reduce the systemic exposure and thus systemic side-effects associated with drug [Citation4]. Drug after intranasal administration reaches the olfactory epithelium region of the nasal mucosa that acts as a gateway for substances entering the CNS due to neural connection between the nasal mucosa and the brain [Citation5].
Many formulation strategies have been developed to overcome and/or bypass BBB via intranasal route but all have their own limitations. On this note, the concept of lipid based nanoparticles like solid lipid nanoparticles (SLNs) matured [Citation6].
SLNs were introduced at the beginning of 1991 as submicron colloidal carriers (50–1000 nm). They are used for delivery of both hydrophilic and lipophilic drug(s) being trapped in biocompatible material core made up of a lipid or combination of lipids and stabilized by surfactant which is present at the outer shell [Citation7]. SLNs showed preferential benefits like targeted drug delivery, controlled release delivery and increased bioavailability hence reduced dose and side effects of drug [Citation6]. They have good tolerability and biodegradability, lack of acute and chronic toxicity, scalability to large scale production [Citation8–10]. SLNs furnish an improvement in nose-to-brain drug delivery since they are able to protect the encapsulated drug from biological and/or chemical degradation and may also increase nasal retention time due to the occlusive effect, good application properties and adhesion to mucous membranes [Citation6,Citation8].
Donepezil (DPL) is a piperidine-based, reversible and non-competitive inhibitor of the enzyme acetylcholinesterase (AChE) [Citation11]. It is the second drug approved by the Food and Drug Administration (FDA) for the treatment of mild to moderate dementia of Alzheimer’s type [Citation12]. DPL is postulated to exert its therapeutic effect by enhancing cholinergic function. This is accomplished by increasing the concentration of acetylcholine (ACh) through reversible inhibition of its hydrolysis by AChE.
DPL exhibited good bioavailability after oral administration. Currently, tablets or capsules (5 or 10 mg/day) of DPL are available in the market for its oral delivery. However, these dosage forms provide non targeted delivery resulting in numerous gastrointestinal side effects such as diarrhoea, nausea, anorexia, gastric bleeding, muscle convulsions, etc. [Citation13]. Another limitation of oral delivery is restricted entry of DPL into brain due to its hydrophilicity (freely soluble in water) [Citation14], thereby entailing frequent dosing resulting in severe cholinergic side effects. As per the previous findings, DPL exhibited hepatotoxicity (but less as related to its processor tacrine) [Citation15] and undergo first pass metabolism, again a restraint for oral delivery [Citation16].
As a result, it will be beneficial to formulate a non-oral delivery system of DPL to restrict the side effects associated with oral delivery, prevention of systemic exposure as well as distribution of drug to non-targeted sites. As the target site for DPL is brain, thereby developed system should also provide the therapeutic concentration of drug at the site of action. Consequently, intranasal route was selected to deliver DPL to target site in the form of SLNs. Moreover, DPL possesses acceptable log p values (4.14), molecular weight (379.492 g mol−1) and small dose (5–10 mg/day) necessary for brain targeting by means of intranasal administration.
In the present study, we report the development of DPL-loaded SLNs (DPL-SLNs) for brain targeting via nasal route. Box–Behnken design was applied for optimization of formulation and physicochemical characterization of developed SLNs was performed using transmission electron microscopy (TEM), differential scanning calorimetry (DSC), X-ray powder diffraction (XRD) and Fourier transform infrared (FTIR) and evaluated them for particle size, poly dispersity index (PDI), zeta potential, entrapment efficiency (%), drug loading (%), in vitro release. In vivo (biodistribution and pharmacokinetic) studies were performed on albino Wistar rat model. The localization of DPL from the SLNs was further studied by gamma scintigraphic analysis.
Materials and methods
Materials
DPL was received as a gift sample from Jubilant Life Sciences, Noida, India. Compritol ATO 888, glyceryl monostearate (GMS) and Precirol ATO 5 were obtained as a gift sample from Gattefosse (Witten, Germany). Stearic acid, palmitic acid, acetonitrile (ACN), triethylamine (TEA), o-phosphoric acid (o-PA), tween 80 and poloxamer 188 along with all other chemicals were purchased from Sigma-Aldrich (New Delhi, India). Dialysis membrane (molecular weight cut off 12,000–14,000 Da), membrane filter and syringe filter (0.22 µm) were purchased from HiMedia Laboratories Pvt. Ltd. (Mumbai, India). ACN, TEA, o-PA were of HPLC grade while all other solvents and chemicals used were of analytical grade.
Methods
Selection of excipients and other parameters
An array of excipients either alone or in the blend were screened for the selection of optimized excipients composition. Lipid was selected on the basis of drug solubility in lipid [Citation17,Citation18]. The preliminary selection of the best composition of excipients was done on the basis of particle size and entrapment efficiency. Excipients like surfactant, organic solvent and their concentration were screened out to obtain the small particle size and high entrapment efficiency. Other parameters like homogenization time, stirring speed and sonication time were also screened. Factors like ratio of drug to lipid, surfactant concentration, stirring time were further optimized in this study.
Compatibility study of selected excipients with drug
The compatibility between the lipid, surfactant and drug was identified by Fourier transform-infrared spectrophotometer (Alpha model Bruker ATR-FTIR spectrophotometer, Billerica, MA). IR spectra of drug, lipid, surfactant and physical mixture of drug, lipid and surfactant were scanned from 4000 cm−1 to 400 cm−1 and recorded.
Preparation of DPL loaded SLNs
DPL-loaded SLNs were prepared by solvent emulsification–diffusion technique [Citation19]. Accurately weighed lipid was dissolved in mixture of ethanol and chloroform (1:1, 5 mL) as the internal oil phase. Drug (drug to lipid ratio 1:4 to 1:6) was dispersed in the above solution. This reaction mixture was heated above the melting point of lipid (65 °C). This organic phase was then poured drop by drop into a homogenizer tube containing 20 mL of hot aqueous surfactant solution of tween 80:poloxamer 188 (1:1, 1.5–2.5%). The mixture was then homogenized (Remi Instruments Pvt. Ltd, Mumbai, India) for 30 min at 3000 rpm to form primary emulsion (o/w). The above emulsion was poured into 80 mL of ice-cold water (2–3 °C) containing surfactant (mixture tween 80:poloxamer 188 in 1:1 ratio, 1.5–2.5%) and stirred to extract the organic solvent into the continuous phase and for proper solidification of SLNs. The stirring was continued (2–3 h) at 3000 rpm to get SLN dispersion. The dispersion was then centrifuged (Remi Instruments Pvt, Ltd, Mumbai, India) at 18,000 rpm (20 min) to separate the solid lipid material containing the drug. These SLNs were washed with deionized water and then re-dispersed in aqueous surfactant mixture (Tween 80:Poloxamer 188, 1:1) and sonicated for 5 min (one cycle, 100% amplitude, Bandelin Sonopuls, Berlin, Germany) to obtain the SLN dispersion of uniform size.
Experimental design for optimization
A three factors three levels Box–Behnken experimental design was used for optimization and to evaluate the relationship between the independent variables like drug to lipid ratio (A), surfactant concentration (B) and stirring time (C) and dependent (responses) variables, i.e. particle size (R1), entrapment efficiency (R2) and drug loading (R3). Therefore, by fixing the homogenization time (30 min), stirring speed (3000 rpm) and sonication time (5 min), the selected variables (A), (B) and (C) were studied at three different levels as low (–1), medium (0) and high (+1). Different batches (D1–D17) were prepared and data were substituted in design expert software (version 8.0.7.1). The generated polynomial equation was used to draw the conclusion after considering the magnitude of coefficient and mathematical sign it carries, i.e. positive or negative. The polynomial regression results were expressed using 3D plots. The criteria of selection of optimum formulations were primarily based on minimum particle size, maximum entrapment efficiency and drug loading.
Optimization of data and validation of response surface methodology (RSM)
Finally, D13 formulation was selected and its composition was further slightly modified to validate RSM. Three new combinations (OD1, OD2 and OD3) were selected as check point. These formulations were again prepared and evaluated for responses. The resulting observed responses were compared with the predicted responses and percentage error was calculated [Citation20].
Characterization of SLNs
Optimized DPL-SLN formulation (OD3) was characterized for the following parameters.
Particle size, zeta potential and morphological study
Average particle size, PDI and zeta potential were measured by photon correlation spectroscopy (PCS; Zetasizer, HAS 3000; Malvern Instruments, Malvern, UK). Measurements were carried out at an angle of 90° at 25 °C [Citation21]. Surface morphology of optimized SLN formulation was done by using TEM (TEM, Philips CM 10, Holland, The Netherlands) and scanning electron microscope (Quanta 200 ESEM, FEI, Hillsboro, OR). Digital micrograph and soft imaging viewer software were used to perform the image capture and analysis.
Determination of entrapment efficiency (%) and drug loading (%)
A fixed quantity of DPL-SLNs dispersion (10 mL) was centrifuged (Remi Instruments, Pvt. Ltd, Mumbai, India) at 18,000 rpm for 20 min at 20 °C. The supernatant was analysed spectrophotometrically at λmax 270.5 nm (Shimadzu 1800, Kyoto, Japan) for determination of unencapsulated drug [Citation22]. The drug loading (%) and drug entrapment efficiency (%) were calculated by using EquationEquations (1)(1)
(1) and Equation(2)
(2)
(2) .
(1)
(1)
(2)
(2)
where Wt is the total weight of drug used, Ws is the weight of drug in the supernatant and WL is the weight of the lipid used in preparing the SLNs.
DSC analysis
The thermograms of drug, lipid and optimized DPL-SLNs were recorded with DSC (Pyris 6 DSC Perkin Elmer, Shelton, CT) under an inert atmosphere. Sufficient amount of samples (5 mg) was loaded into an aluminium pan and an empty aluminium pan was used as a reference. Samples were heated at a scanning rate of 10 °C/min over a temperature range of 40–300 °C and the thermograms were recorded [Citation23].
X-ray diffractometric analysis
X-ray powder diffraction patterns of drug, lipid and optimized DPL-SLNs were measured by X-ray diffractometer (PANanalytical, X’Pert Pro model, Kassel, Germany) equipped with an X-ray generator. Analysis was performed on the samples using Cu Kα radiation (45 kV, 40 mA) and scanned from 5° to 90° (2θ) [Citation24].
In vitro release and release kinetic studies
In vitro release studies from DPL-SLNs were carried out to evaluate the release of drug from the optimized formulation and comparing it with the pure drug. It was performed by dialysis bag diffusion technique employing a dialysis membrane [Citation25]. An accurate amount of DPL-SLNs and DPL solution (DPL-Sol.) each containing the drug equivalent to 5 mg was transferred to dialysis bag and sealed at both ends. The sealed bag was then suspended in a beaker containing 100 mL of phosphate buffer (pH 7.4, corresponding to CSF pH) and stirred at a constant speed (50 rpm) at 37 ± 0.5 °C. Aliquots (5 mL) were withdrawn at predetermined time intervals up to 24 h from receiver compartment (beaker) and replaced with an equal volume of fresh medium to maintain sink condition. The samples were analysed spectrophotometrically at λmax of 270.5 nm. Comparison between release profile of DPL-Sol. and optimized DPL-SLNs was made by simple Student’s t test at p < .05.
In vitro release data of optimized formulation was fitted to zero order, first order and Higuchi release model [Citation26]. To find out the mechanism of drug release, data were fitted in the Korsmeyer–Peppas model (Mt/M∞ = Ktn) and value of n (exponent) was determined.
In vivo studies
In vivo studies for DPL-SLNs and DPL-Sol. include pharmacokinetic and biodistribution studies.
Animals for in vivo study
In vivo studies of optimized DPL-SLNs and DPL-Sol. were performed on male albino Wistar rats (adult/weighing 200–250 g). A protocol for studies was approved by Institutional Animal Ethical Committee (IAEC). The animals were kept under standard laboratory conditions, i.e. temperature of 22 ± 3 °C and relative humidity of 30–70%. The animals were housed in polypropylene cages, six animals per cage with free access to standard laboratory diet and water ad libitum.
Pharmacokinetic and biodistribution studies
To conduct pharmacokinetic studies, rats were divided into three different groups: Group A, positive control for intravenous (i.v.) drug administration (DPL-Sol.); Group B, positive control for intranasal (i.n.) drug administration (DPL-Sol.); and Group C for intranasal (i.n.) SLN formulation administration (DPL-SLNs). Each group was divided into seven subgroups on time basis as given below and each subgroup contained six animals.
Subgroup 1: drug was administered at time 0 and sacrifice was done after 0.5 h.
Subgroup 2: drug was administered at time 0 and sacrifice was done after 1 h.
Subgroup 3: drug was administered at time 0 and sacrifice was done after 2 h.
Subgroup 4: drug was administered at time 0 and sacrifice was done after 4 h.
Subgroup 5: drug was administered at time 0 and sacrifice was done after 6 h.
Subgroup 6: drug was administered at time 0 and sacrifice was done after 8 h.
Subgroup 7: drug was administered at time 0 and sacrifice was done after 24 h.
Procedure of drug administration
Drug solution (positive control), containing 0.09 mg (for 200 g of rat) of DPL (equivalent to 0.45 mg/kg body weight), was injected through the tail vein (10 µL) in one group of Wistar rats. Similarly, drug solution and drug formulation (DPL-SLNs) containing 0.09 mg of DPL were administered in each nostril in the other two groups with the help of micropipette (10–100 µL) with 0.1 mm internal diameter at the delivery site. The rats were anaesthetized prior to nasal administration by pentobarbital sodium (35–50 mg/kg, i.p.) and held firmly from the back in a slanted position during nasal administration.
Blood sampling, separation and processing other organs
The rats were killed humanely by overdose of pentobarbital sodium at different time intervals (0.5, 1, 2, 4, 6, 8 and 24 h) and the blood was collected by cardiac puncture in EDTA coated Eppendrof tubes. The blood was centrifuged at 5000 rpm for 15 min and aliquots of the supernatant separated and stored at –21 °C until drug analysis was carried out using high performance liquid chromatography (HPLC) [Citation27].
At the same interval of blood collection, the rats were sacrificed to separate the brain and other organs (intestine, kidney, liver, lungs and spleen). Each organ was rinsed twice with normal saline to make free from adhering tissue/fluid and weighed. Cold normal saline solution was added (organ weight:normal saline, 1:5) to each organ and homogenized on ice. Separate process was applied for each organ. Each organ homogenate was centrifuged at 5000 rpm for 15 min at a temperature of 4 °C and aliquots of the supernatant were separated and stored at –21 °C until drug analysis was carried using HPLC.
Extraction of DPL from plasma and other organs
Chromatographic separation was achieved with a Cosmosil C18 column (250 mm × 4.6 mm, particle size 5 μm). Before quantification of drug by HPLC, the method was developed and validated. The mobile phase consisted of 0.02 M phosphate buffer (pH 7.4)–methanol–ACN, 40:50:10 v/v/v and the pH was adjusted with o-PA. 0.1% TEA was added to reduce tailing. The mobile phase was sonicated for 15 min and filtered through 0.22 μm membrane filter before use. Flow rate of mobile phase was maintained at 1.2 mL/min and eluents were monitored at 280 nm. Twenty microlitres of sample was injected using a HPLC injector. All determinations were performed at ambient temperature for a run time of 10 min. The method was linear in drug concentration range of 25–600 ng/mL with a correlation coefficient of 0.992. % RSD value of intraday and interday precision was found to be 0.73–1.27 and 0.85–1.89, respectively. The accuracy (recovery) of the developed method was 97.68–102.39%.
For the determination of DPL, plasma sample and homogenized brain/other organ tissues (100 µL) were taken in test tube separately and mixed with 20 µL (1 µg/mL) internal standard (Ondansetron) solution. The tubes were vortexed (S.M. Scientific Instruments Pvt. Ltd., Delhi, India) and 4 mL of 1% acetic acid was added. The content of each tube was vigorously shaken for 10 min and then centrifuged at 5000 rpm until a clear organic layer was separated (approximately 5 min). Organic layer from each tube was evaporated to dryness at room temperature. After complete removal of organic solvent, the dried samples were then reconstituted separately with 100 µL of mobile phase and evaluated by HPLC for the presence of DPL.
Analysis of pharmacokinetic parameters and brain targeting study
Plasma concentration–time profiles of DPL after i.n. and i.v. delivery were evaluated by pharmacokinetic software (PK Functions for Microsoft Excel, Pharsight Corporation, Mountain View, CA). Various pharmacokinetic parameters as Cmax, Tmax, AUC0–∞, AUMC0–∞, elimination rate constant (Ke) and mean residence time (MRT) were calculated. The data were statistically compared by one-way ANOVA.
The extent of nose-to-brain delivery, bypassing the BBB, due to direct connection between the nose and the brain via the olfactory region of the nasal cavity could be evaluated by many parameters [Citation28]: (i) the brain/blood ratio, at 0.5 h, following intranasal and intravenous administration; (ii) the relative bioavailability percentage following the intranasal administration in the blood and brain; (iii) the drug targeting index (DTI); (iv) the drug targeting efficiency (DTE) percentage; and (v) the nose-to-brain direct transport percentage (DTP) [Citation29]. All five parameters were determined to check the authenticity of developed formulation for brain targeting.
Gama scintigraphy imaging study
The healthy New Zealand rabbits (2.00–2.50 kg) were selected for gamma scintigraphy imaging using gamma camera [Citation30]. The study was performed by direct labelling method using technetium (99mTc) and stannous chloride dihydrate solution (1 mg/mL in ethanol, and 100 µL of this solution) as radioactive compound and reducing agent, respectively. The rabbits were anaesthetized prior to nasal administration with the help of pentobarbital sodium (35–50 mg/kg, i.p.) and held from the back in slanted position during intranasal administration. The radiolabelled formulations (both DPL-SLN and DPL-Sol./100 μCi/50 μL) containing 0.09 mg DPL (equivalent to 0.45 mg/kg body weight) were administered in each nostril with the help of micropipette at the delivery site (deep area of nose) and placed on the imaging platform. Imaging was performed after 0.5 h of drug administration using Single Photon Emission Computerized Tomography (SPECT-CT, Siemens Med. Solutions, Malvern, PA) gamma camera.
Stability studies
The optimized DPL-SLNs were subjected to stability studies at 4 ± 2 °C (refrigerator), 25 ± 2 °C/60 ± 5% RH and 40 ± 2 °C/75 ± 5% RH in stability chamber (Hicon Instruments, N. Delhi, India). The sample was withdrawn after a period of 0, 1, 3 and 6 months and the effect on particles size, shape, PDI, zeta potential, entrapment efficiency and loading capacity was determined.
Results and discussion
Selection of excipients and other parameters
Result of solubility study indicated that amongst the GMS, compritol 888 ATO, precirol ATO 5, stearic acid and palmitic acid, the GMS effectively solubilized the DPL (). The applicability of GMS in brain drug delivery had been already reported by Joshi et al. [Citation18].
Table 1. Solubility of DPL in various lipids.
In the preliminary screening studies, chloroform and ethanol (1:1) as organic solvent to solubilize the lipid, mixture of tween 80 and poloxamer 188 (1:1) as surfactant, homogenization time (30 min), stirring speed (3000 rpm), sonication time (5 min) were selected.
Compatibility study of selected excipients with drug
The compatibility between the drug, lipid and surfactant was identified by FTIR spectrophotometer. FTIR spectrum of DPL showed the characteristic peaks of the drug structure ().
Figure 1. IR spectra of (a) DPL, (b) GMS, (c) Tween 80, (d) Poloxamer 188 and (e) DPL + GMS + Tween 80 + Poloxamer 188.
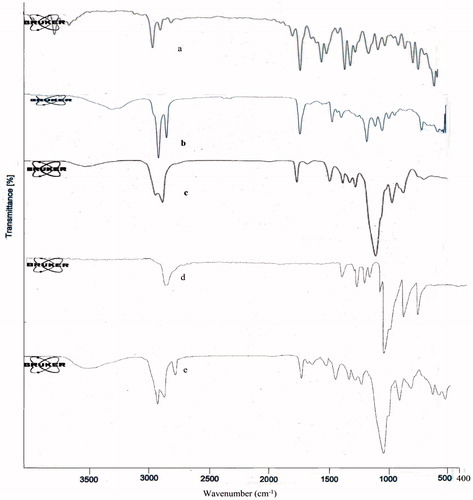
Pure drug (DPL) exhibited the bond variations at 3008 cm−1 (aromatic CH stretch), 2917 cm−1 (aliphatic CH2 stretch), 2850 cm−1 (aliphatic CH stretch), 1680 cm−1 (C=O carbonyl stretch) and 1583 cm−1 (aromatic C=C stretches). Also peaks appeared at 1457, 1360, 1306, 734 cm−1 corresponding to CH2 bending, aliphatic CH bending, C–N stretch, aromatic CH bending, respectively. These characteristic peaks of the drug were also observed in the FTIR spectrum of physical mixture of DPL, GMS, tween 80 and poloxamer 188 without any distinct shift. This fact verified that no chemical interaction between the drug, lipid and surfactant was observed.
Preparation and optimization of DPL loaded SLNs using experimental design
DPL loaded SLNs were prepared by solvent emulsification–diffusion technique [Citation19]. On the basis of results obtained from preliminary and pre-optimization studies, three levels of each independent variable (factors) were decided (). For three factors and three levels, the Box–Behnken design was chosen. On applying three factors and three levels Box–Behnken statistical design, 17 runs with five centre points were obtained. All these batches were prepared and evaluated for responses like particles size (R1), % drug entrapment efficiency (R2) and % drug loading (R3). All the results were placed in Box–Behnken design of Design Expert software (version 8.0.7.1) to obtain the predicted values and final conclusion. All the responses of these runs fitted to first order, second order and quadratic models and the best fit model was quadratic (p < .0001) with insignificant lack of fit (p > .05). Observed responses for 17 runs of DPL-SLNs, summary of results of regression analysis for responses and analysis of variance of calculated model for responses R1, R2, R3 are shown in , respectively. The positive sign in polynomial equation of the factors represent a synergistic effect on the response, while a negative sign mean an antagonistic relationship. Phrases composed of two factors indicate the interaction terms and phrases with second-order factors stand for the nonlinear relationship between responses and variables. Three dimensional plots showed the interaction effects of the independent variables on the responses as well as their usefulness in studying the effects of two factors on one response at a time ().
Figure 2. Response surface plot showing effect of independent variables on particle size.
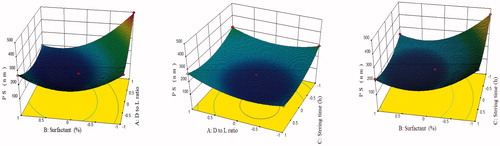
Figure 3. Response surface plot showing effect of independent variables on % entrapment efficiency.
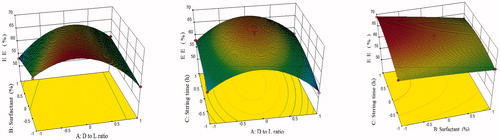
Figure 4. Response surface plot showing effect of independent variables on % drug loading.
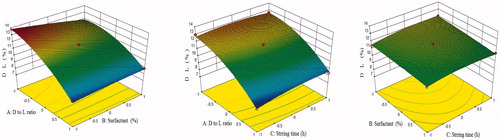
Table 2. Variables and their levels in Box–Behnken design.
Table 3. Observed responses for 17 runs of DPL-SLNs according to Box–Behnken design.
Table 4. Summary of results of regression analysis for responses R1, R2, R3.
Table 5. Analysis of variance of calculated model for responses.
The polynomial EquationEquation (3)(3)
(3) related to particle size (R1) is given as
(3)
(3)
The model F-value of 1515.59 implied that the model was significant (p < .0001). The “lack of fit” (F-value of 1.36) implied that it was not significant (p = .3757). In this case (), A, B, C, AB, AC, BC, A2, B2, C2 in EquationEquation (3)(3)
(3) were significant model terms. Positive coefficients of factor A, AC, BC, A2, B2 and C2 exhibited the synergistic effect on particle size while negative coefficients of B, C, AB and A2 indicated the antagonistic effect on particle size. The Predicted R2 of 0.9955 was in reasonable agreement with the adjusted R2 of 0.9988, indicating the adequacy of the model to predict the response of particle size. The “Adeq Precision” of 122.683 indicated an adequate signal. Therefore, this model was used to navigate the design space. The three-dimensional plots () showed the effect of different formulation factor on the particle size (R1). Average particle size of DPL-SLN formulations with different independent variables varied from 496.78 ± 30.82 nm to 148.83 ± 10.29 nm.
The polynomial EquationEquation (4)(4)
(4) related to % entrapment efficiency (R2) is given below:
(4)
(4)
The model F-value 88.69 implied that the model was significant (p < .0001). The “lack of fit” F-value 0.30 implied that it was not significant (p = .8236). In this case (), A, B, C, AB, AC, A2 and C2 were significant model terms. Positive coefficients of A, C, AB and AC in EquationEquation (4)(4)
(4) indicated the synergistic effect, while negative coefficients of B, BC, A2, B2 and C2 indicated the antagonistic effect on % entrapment efficiency. The predicted R2 of 0.9632 was in reasonable agreement with the adjusted R2 of 0.9801, indicating the adequacy of the model to predict the response of entrapment efficiency. The “Adeq Precision” of 28.642 indicated an adequate signal. Therefore, this model was used to navigate the design space. The 3D surface response plots for % entrapment efficiency are shown in . Average entrapment efficiency of DPL-SLN formulations with different independent variables varied from 68.20 ± 3% to 52.17 ± 1.45%.
The polynomial EquationEquation (5)(5)
(5) relating the response of % drug loading (R3) is given as
(5)
(5)
The model F-value of 98.72 implied that the model was significant (p < .0001). The “lack of fit” F-value of 4.77 implied that it was not significant (p = .0827). In this case (), A, B, AB, A2, C2 were significant model terms. Positive coefficients of C, AB and AC in EquationEquation (5)(5)
(5) indicated the synergistic effect, while negative coefficients of A, B, BC, A2, B2 and C2 indicated the antagonistic effect on % drug loading. The predicted R2 of 0.8996 was in reasonable agreement with the "Adjusted R2” of 0.9821 indicating the adequacy of the model to predict the response of drug loading. The “Adeq precision” of 33.728 indicated an adequate signal. Therefore, this model was used to navigate the design space. The 3D surface response plots for % drug loading are shown in . Average drug loading of DPL-SLN formulations with different independent variables varied from 12.11 ± 1.56% to 7.99 ± 0.27%.
Optimization of data and validation of response surface methodology
Point prediction of software was used for optimization of independent variables to obtain the optimum particle size, entrapment efficiency and loading capacity. Four formulations (D13, OD1, OD2, OD3) were selected from point prediction software of design expert and their responses, i.e. particle size, entrapment efficiency and drug loading were evaluated. The composition of all optimum check point formulations, their actual and predicted values for the responses and the % prediction error are shown in . The low value of % prediction error assures the validity of generated equations and thus depicts the domain of applicability of RSM model. Finally, the optimum values of drug to lipid ratio 1:5, surfactant conc. 2.25% w/v and stirring time 2.75 h were obtained (OD3). The formulation (OD3) optimized here showed an average particle size of 121 ± 5.27 nm, entrapment efficiency of 67.95 ± 0.98% and drug loading of 12.15 ± 1.58%. This result showed 96.55%, 99.31% and 99.26% validity of the predicted values of predicted responses. The optimized formulation (OD3) was used for further study.
Table 6. Point prediction check point for optimization, actual and experimental value and % error.
Characterization of optimized formulation
Particle size, zeta potential and morphological study
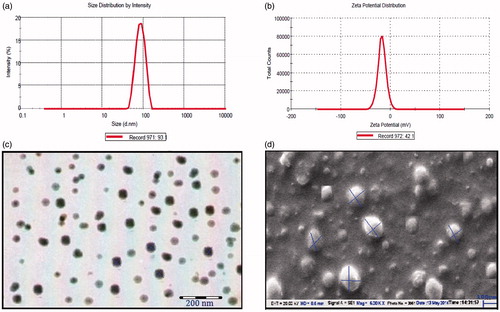
Particle size distribution and zeta potential curve of optimized formulation (OD3) are shown in , respectively. The average particle size of optimized formulation was 121 ± 5.27 nm which was found to be suitable for brain targeting through nasal route. PDI is essentially the ratio of standard deviation to the mean particle size. PDI value 0.3 or less indicates the uniformity in particle size. In this case, PDI value was found to be 0.194 indicating uniformity in particle size of developed SLNs. The zeta potential indicates the degree of charge present on suspended particles in SLNs dispersion. A suitably high value of zeta potential (+30 mV to –30 mV) confers stability because particles resist aggregation. The value of zeta potential (–24.1 mV) of optimized SLN formulation indicated good stability. TEM and SEM images indicated a dense roughly spherical pattern of optimized SLNs (). The result of particle size was found to be in good agreement with the result established by TEM and SEM study.
Figure 5. (a) Particle size distribution curve, (b) zeta potential curve, (c) TEM and (d) SEM image of optimized formulation.
Entrapment efficiency (%) and drug loading (%)
The entrapment efficiency and drug loading of optimized formulation were found to be 67.95 ± 1.58% and 12.15 ± 0.98%, respectively.
DSC analysis
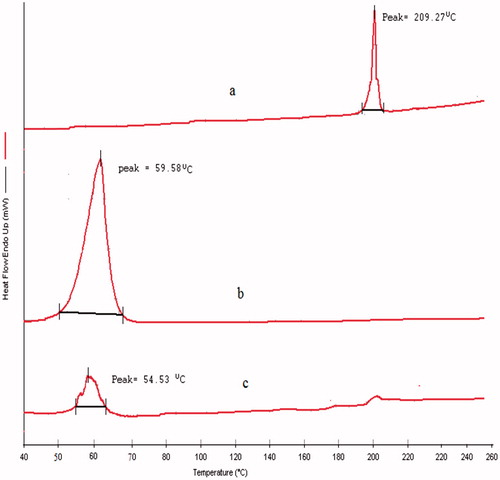
The DSC thermogram of drug (DPL) showed a melting peak of 209.27 °C while lipid (GMS) and optimized DPL-SLNs showed at 59.58 °C and 54.53 °C, respectively (). The thermogram of drug incorporated SLNs did not show the melting peak of crystalline DPL around 209.27 °C, pinpointing complete solubilization of drug inside the lipid matrix and being in amorphous form. Similar finding were observed by Tayade and Kale [Citation31]. Decline in melting peak of GMS in SLNs by 5.05 °C suggested its possible existence in crystalline form [Citation32]. Similar findings were observed by Heike and Tobias [Citation32].
Figure 6. DSC thermogram of (a) DPL, (b) GMS and (c) optimized DPL-SLNs.
X-ray diffractometric analysis
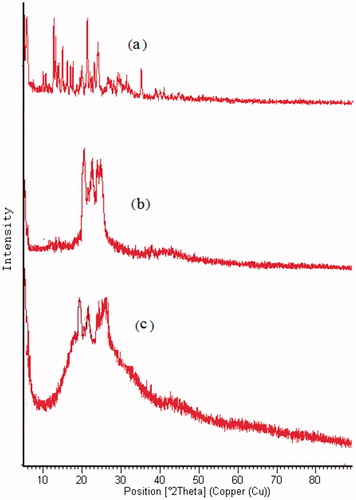
XRD pattern of DPL showed sharp peaks at 2θ-scattered angles of 21° advocating highly crystalline nature of the drug. The XRD pattern of GMS is depicting a peak at about 20°, implying crystalline nature of the lipid (). The principal peak of DPL was absent in drug loaded SLNs XRD spectrum. Furthermore, the principal peak of lipid did not shift but had a reduced intensity as compared to free lipid. This may be attributed to the incorporation of DLP between the parts of the crystal lattice of the lipid, leading to a change in the crystallinity of the DPL-SLNs. Similar findings were observed by Heike and Tobias [Citation32].
Figure 7. XRD of (a) DPL, (b) GMS and (c) optimized DPL-SLNs.
In vitro release and release kinetic studies
Comparative in vitro release study was performed between DPL-Sol. and optimized DPL-SLN formulation (OD3).The dissolution profile from DPL-SLNs indicated an initial burst release, followed by slow release (). The initial burst release may be attributed to the presence of free drug in the external phase and adsorbed drug onto the surface of particles, while the slow release may be owed to the encapsulated drug within the lipid matrix [Citation18]. The optimized DPL-SLNs showed initial burst release of 32.94 ± 6.29%, whereas plain drug showed 52.24 ± 5.95% after 1 h. Thereafter, optimized DPL-SLNs showed sustained drug release with maximum drug release of 89.35 ± 3.27% in 24 h, while aqueous drug suspension exhibit 97.49 ± 4.64% within 5 h. On comparing the observed results, a significance difference (p < .05) was marked between DPL-Sol. and DPL-SLN formulation after 5 h. For optimized formulation (OD3), Higuchi model was found to be the best fitted model () with the highest value of correlation coefficient (R2 = 0.9456). The value of release exponent "n" was found to be 0.352, which appears to indicate diffusion controlled release mechanism, so-called Fickian diffusion. Similar findings were observed by Shazly [Citation33] who prepared ciprofloxacin loaded SLNs.
Figure 8. In vitro release profile of DPL-SLNs and DPL-Sol.
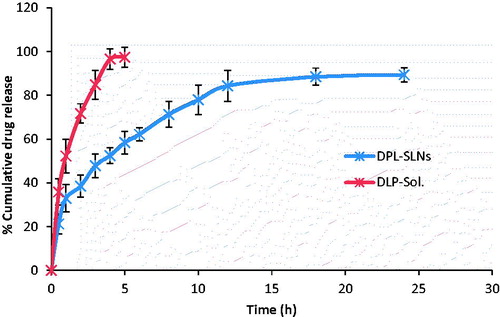
Table 7. Release kinetic models for optimized DPL-SLN formulation.
In vivo studies
In vivo studies involved pharmacokinetic and biodistribution studies.
Pharmacokinetic studies
Various pharmacokinetic parameters were calculated by using PK Functions for Microsoft Excel (Pharsight Corporation, Mountain View, CA) as shown in . The lower value of Tmax for brain (2 h) as compared to blood (4 h) may be attributed to the preferential nose to brain transport following i.n. administration. The significantly (p < .05) higher value of Cmax (133.30 ± 7.96 ng/mL) was obtained for DPL-SLNs administered intranasally as compared to DPL-Sol. administered intranasally (24.15 ± 2.98 ng/mL) and DPL-Sol. administered via intravenous route (17.19 ± 3.20 ng/mL). This might be due to retention ability of SLNs as compared to DPL-Sol. Similar finding was reported by Jose et al. who studied targeting of resveratrol loaded SLNs after intranasal administration [Citation34]. The value of AUC0–∞ in brain for DPL-SLNs administered intranasally was found to be nearly 2.61 times higher than that of DPL-Sol. administered through i.v. route, whereas 2.26 times higher than DPL Sol. administered intranasally. This might be due to direct transport of drug to brain via olfactory pathway.
Table 8. Pharmacokinetic parameters of DPL in brain and plasma after DPL-SLNs i.n., DPL-Sol. i.n. and DPL-Sol. i.v. administration to rats.
Brain targeting study
The extent of nose to brain delivery was evaluated by the following parameters:
The brain/blood ratio, at 0.5 h, was found to be 1.602, 0.098 and 0.014 for DPL-SLNs i.n., DPL-Sol. i.n. and DPL-Sol. i.v., respectively (). The significantly higher brain/blood ratio for DPL-SLNs indicated the brain targeting potential of developed SLNs. Similar findings were observed by Abdelbary and Tadros [Citation28].
Table 9. Results of brain/blood ratio at 0.5 h, DTI, DTE (%) and DTP (%).
Compared to DPL-Sol. administered intranasally, the % relatively bioavailability of DPL-SLNs i.n., in blood and brain were 129.14 ± 5.47 and 257.19 ± 9.79, respectively (). The results revealed a significant (p < .05) enhancement in bioavailability of DPL in the brain following the intranasal administration of DPL-SLNs. These findings are in line with Abdelbary and Tadros [Citation28] who found that micellar nanocarriers increases the relatively bioavailability of olanzapine administered intranasally.
The values of DTI, DTE and DTP for DPL-SLNs and DPL-Sol. were found to be 5.53, 553.95% and 81.94% and 2.43, 243.78% and 58.84%, respectively (). The DTI values >1 could confirm the direct pathway from nose to brain [Citation29]. These findings were in line with Jain et al. [Citation35]. Finally, it was concluded that the higher value of DTI, DTE (%) and DTP (%) speculate better brain targeting efficiency with DPL-SLNs compared to DPL-Sol. i.n. and DPL-Sol. i.v.
Biodistribution studies
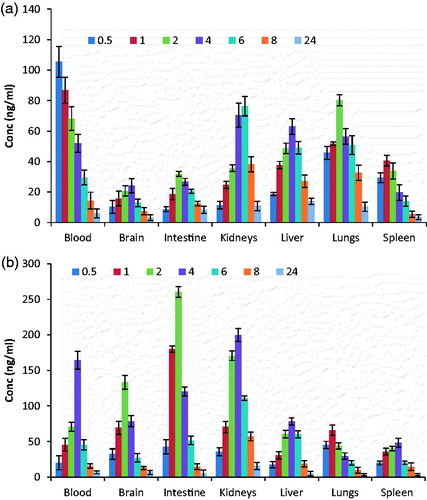
Biodistribution of DPL-SLNs and DPL-Sol. were observed in various organs of interest (intestine, kidney, liver, lungs and spleen) and concentration was estimated at different time intervals of 0.5, 1, 2, 4, 6, 8 and 24 h after intranasal administration ().
Figure 9. Biodistribution studies on organ of interest with (a) DPL-Sol.(i.n.) and (b) DPL-SLNs(i.n.).
Effect of formulation (DPL-SLNs) on nose-to-brain delivery of DPL
The concentration in brain following i.n. administration of DPL-SLNs were found to be significantly higher (p < .01) at all time points as compared to positive control (DPL-Sol. i.n) which could be due to:
Rapid clearance of the administered DPL-Sol. from the nasal cavity by the mucociliary clearance mechanism.
By the active efflux transporter pumps at the apical membrane surface (P-gp) or
Enzymatic degradation in the olfactory epithelium [Citation36].
SLNs may offer an improvement in the nose-to-brain drug delivery since they are able to protect the encapsulated drug from biological and/or chemical degradation and extracellular transport by P-gp efflux proteins. The nasal mucosal contact of DPL-SLNs increases the nasal retention time due to the occlusive nature of lipids in the SLNs [Citation30]. Furthermore, the improved delivery by SLNs may be assigned to the use of tween 80 in the preparation of SLNs which is reported to enhance brain delivery of nanoparticles by (i) solubilization of endothelial cell membrane lipids and membrane fluidization [Citation37], (ii) through the temporary opening of inulin spaces [Citation2], (iii) endocytosis of nanoparticles [Citation37] and (iv) inhibition of efflux system, especially P-gp present on the intranasal membrane [Citation38].
Effect of formulation (DPL-SLNs) on biodistribution of DPL
Biodistribution studies revealed more localization in kidney, spleen and liver for DPL-SLNs as compared to DPL-Sol. The higher level of intranasal DPL-SLNs in different organs may be explained on the basis of their nanoparticulate and lipophilic nature, avoidance from the degrading environment in the nasal and enhancement in permeation/absorption by the surfactant added during the formulation [Citation30]. In addition, different molecules administered in the body are taken by body with different mechanism [Citation39]. The accumulation in the liver and the spleen is generally ascribed to uptake by the reticuloendothelial system (RES) like macrophage cells [Citation39], whereas the presence of lipid nanoparticles in the lungs may be the result of their agglomeration caused by the adsorption of plasma proteins [Citation30]. However, the higher uptake of DPL-SLNs by RES organs can be based on the fact that the GMS containing nanoparticles showed higher uptake by the RES organs as reported by Pandey et al. [Citation40].
Gamma scintigraphy imaging study
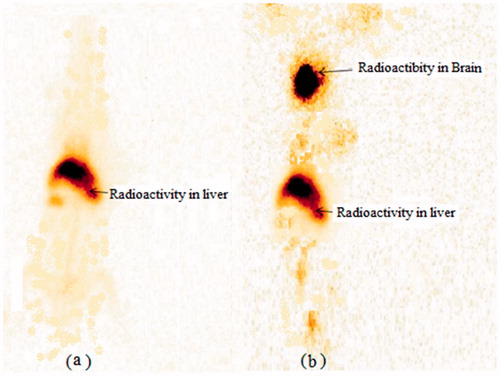
The gamma scintigraphy images exhibited localization of drug in brain as shown in . The increased radioactivity was shown from the radiolabelled DPL-SLNs in the brain region as compared to drug Sol. The scintigraphy images were consistent with the results of biodistribution as well as pharmacokinetic studies and high uptake of DPL-SLNs into the brain was observed. Similar finding were observed by Singh et al. when they performed gamma scintigraphy of alprazolam loaded SLNs on rabbit model [Citation30].
Figure 10. Gamma scintigraphy images of rabbit following i.n. administration of (a) DPL-Sol. (b) DPL-SLNs after 0.5 h.
Stability studies
No significant (p < .05) change was observed in particle size of DPL-SLN formulation when it was stored at 4 ± 2 °C (refrigerator) and 25 ± 2 °C/60 ± 5% RH up to six months, but the particle size increased significantly (p < .001) when it was stored at 40 ± 2 °C/75 ± 5% RH due to aggregation. The average particle size after 6 months at 40 ± 2 °C/75 ± 5% RH was found to be 1650.25 ± 25.36 nm while the PDI was 0.889. Zeta potential plays an important role in physical stability. There was no significant change observed in zeta potential of SLN formulation when they were stored at 4 ± 2 °C (refrigerator) and 25 ± 2 °C/60 ± 5% RH up to six months but a significant drop in zeta potential at 40 ± 2 °C/75 ± 5% RH (p < .001) was found to be a function of time and temperature. This might be due to the fact that at high temperature and relative humidity, the outer surfactant coating get dissolved, leading to aggregation of lipid nanoparticles. The entrapment efficiency (%) and drug loading (%) were also reduced with time, but no significance difference (p < .05) was observed.
Conclusions
SLNs for DPL were developed successfully and evaluated for various in vitro and in vivo parameters. All the parameters were found to be in the acceptable range. In vitro release study revealed that optimized DP-SLN formulation was more sustained as compared to DPL-Sol. Pharmacokinetic and brain targeting studies in rats revealed a significantly high concentration of drug in brain upon i.n. administration of DP-SLNs as compared DPL-Sol. The results of biodistribution studies were in line with the results of pharmacokinetic studies and indicate brain targeting efficiency of developed formulations. Stability studies revealed no significant change in the particle size, zeta potential, entrapment efficiency and drug loading at 4 ± 2 °C (refrigerator) and 25 ± 2 °C/60 ± 5% RH up to six months. The shelf life of optimized DPL-SLN was found to be 2.29 years. Thus, this study demonstrated the utility of SLNs for the delivery of DPL to brain via i.n. route.
Acknowledgements
Authors acknowledge Research Assistantship from I.T.S College of Pharmacy, Muradnagar, Ghaziabad, UP, India, for carrying out this research work. Authors are also thankful to INMASS for providing the facility to conduct gamma scintigraphy study.
Statement of animal rights: All institutional and national guidelines for the care and use of laboratory animals were followed. A protocol for studies was approved by IAEC.
Disclosure statement
All authors declare that there is no conflict of interest between them.
Funding
The project was non-funding but some monetary assistance was provided by management of I.T.S. College of Pharmacy, Muradnagar, Ghaziabad, UP, India.
References
- Mishra A, Ganesh S, Shahiwala A, et al. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6:252–273.
- Gastaldi L, Battaglia L, Peira E, et al. Solid lipid nanoparticles as vehicles of drugs to the brain: current state of the art. Eur J Pharm Biopharm. 2014;87:433–444.
- Illum L. Nasal drug delivery: new developments and strategies. Drug Discov Today. 2002;7:1184–1189.
- Krishnamoorthy R, Mitra AK. Prodrugs for nasal drug delivery. Adv Drug Deliv Rev. 1998;29:135–146.
- Khan S, Patil K, Bobade N, et al. Formulation of intranasal mucoadhesive temperature-mediated in situ gel containing ropinirole and evaluation of brain targeting efficiency in rats. J Drug Target. 2010;18:223–234.
- Blasi P, Glovagnoli S, Schoubben A, et al. Solid lipid nanoparticles for targeted brain drug delivery. Adv Drug Deliv Rev. 2007;59:454–477.
- Schwarz C, Mehnert W, Lucks JS, et al. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. Production, characterization and sterilization. J Control Release. 1994;30:83–96.
- Kaur IP, Bhandari R, Bhandari S, et al. Potential of solid lipid nanoparticles in brain targeting. J Control Release. 2008;127:97–109.
- Helgason T, Awad TS, Kristbergsson K, et al. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN). J Colloid Interface Sci. 2009;334:75–81.
- Kumar R, Yasir M, Saraf SA, et al. Glyceryl monostearate based nanoparticles of mefenamic acid: fabrication and in vitro characterization. Drug Invent Today. 2013;5:246–250.
- Areosa SA, Sherriff F. Memantine for dementia. Cochrane Database Syst Rev. 2003;20:154–160.
- Zhiyong X, Qiongfeng L, Xinjun X, et al. Rapid and sensitive determination of donepezil in human plasma by liquid chromatography/tandem mass spectrometry: application to a pharmacokinetic study. Rapid Commun Mass Spectrom. 2006;20:3193–3198.
- Christodoulou C, Melville P, Scherl WF, et al. Effects of donepezil on memory and cognition in multiple sclerosis. J Neurol Sci. 2006;245:127–136.
- Khan S, Patil K, Yeole P, et al. Brain targeting studies on buspirone hydrochloride after intranasal administration of mucoadhesive formulation in rats. J Pharm Pharmacol. 2009;61:669–675.
- Guo W, Quan P, Fang L, et al. Sustained release donepezil loaded PLGA microspheres for injection: preparation, in vitro and in vivo study. Asian J Pharm Sci. 2015;10:405–414.
- Tiseo PJ, Perdomo CA, Friedhoff LT, et al. Metabolism and elimination of 14 C-donepezil in healthy volunteers: a single dose study. Br J Clin Pharmacol. 1998;46:19–24.
- Shah KA, Date AA, Joshi MD, et al. Solid lipid nanoparticles (SLN) of tretinoin: potential in topical delivery. Int J Pharm. 2007;345:163–171.
- Joshi AS, Patel HS, Belgamwar VS, et al. Solid lipid nanoparticles of ondansetron HCl for intranasal delivery: development, optimization and evaluation. J Mater Sci: Mater Med. 2012;23:2163–2175.
- Trotta M, Debernardi F, Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsification–diffusion technique. Int J Pharm. 2003;257:153–160.
- Maurya DP, Yasmin S, Aqil M, et al. Formulation and optimization of alkaline extracted ispaghula husk microparticles of isoniazide in vitro and in vivo assessment. J Microencapsul. 2011;28:472–482.
- Jores K, Mehnert W, Drechsler M. Investigations on the structure of solid lipid nanoparticles (SLN) and oil loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95:217–227.
- Varshosaz J, Tabbakhian M, Mohammadi MY. Formulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailability. J Liposome Res. 2010;20:286–296.
- Chadha R, Bhandari S. Drug-excipient compatibility screening—role of thermoanalytical and spectroscopic techniques. J Pharm Biomed Anal. 2014;87:82–97.
- Tamjidi F, Shahedi M, Varshosaz J, et al. Design and characterization of astaxanthin-loaded nanostructured lipid carriers. Innov Food Sci Emerg Technol. 2104;26:366–374.
- Chen DB, Yang TZ, Zhang O. In vitro and in vivo study of two types of long-circulating solid lipid nanoparticles containing paclitaxel. Chem Pharm Bull Discipline. 2001;49:1444–1447.
- Yasir M, Sara UVS. Solid lipid nanoparticles for nose to brain delivery of haloperidol: in vitro drug release and pharmacokinetics evaluation. Acta Pharm Sin B. 2014;4:454–463.
- Pappa H, Farru R, Vilanova PO, et al. A new HPLC method to determine Donepezil hydrochloride in tablets. J Pharm Biomed Anal. 2002;27:177–182.
- Abdelbary GA, Tadros MI. Brain targeting of olanzapine via intranasal delivery of core–shell difunctional block copolymer mixed nanomicellar carriers: in vitro characterization, ex vivo estimation of nasal toxicity and in vivo biodistribution studies. Int J Pharm. 2013;452:300–310.
- Wang X, Chi N, Tang X. Preparation of estradiol chitosan nanoparticles for improving nasal absorption and brain targeting. Eur J Pharm Biopharm. 2008;70:735–740.
- Singh AP, Saraf SK, Saraf SA. SLN approach for nose-to-brain delivery of alprazolam. Drug Deliv Transl Res. 2012;2:498–507.
- Tayade PT, Kale RD. Encapsulation of water-insoluble drug by a crosslinking technique: effect of process and formulation variables on encapsulation efficiency, particle size, and in vitro dissolution rate. AAPS PharmSci. 2004;6:1–8.
- Bunjes H, Unruh T. Characterization of lipid nanoparticles by differential scanning calorimetry, X-ray and neutron scattering. Adv Drug Deliv Rev. 2007;59:379–402.
- Shazly GA. Ciprofloxacin controlled-solid lipid nanoparticles: characterization, in vitro release, and antibacterial activity assessment. BioMed Res Int. 2017;2017:2120734. DOI:10.1155/2017/2120734
- Jose S, Anju SS, Cinu TA, et al. In vivo pharmacokinetics and biodistribution of resveratrol-loaded solid lipid nanoparticles for brain delivery. Int J Pharm. 2014;474:6–13.
- Jain R, Nabar S, Dandekar P, et al. Micellar nanocarriers: potential nose-to-brain delivery of zolmitriptan as novel migraine therapy. Pharm Res. 2010;27:655–664.
- Illum L. Nasal drug delivery – possibilities, problems and solutions. J Control Release. 2003;87:187–198.
- Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47:65–81.
- Awasthi S, Hallene LK, Fazio V, et al. RLIP76, a non-ABC transporter, and drug resistance in epilepsy. BMC Neurosci. 2005;6:61–67.
- Dobrovolskaia MA, Aggarwal P, Hall JB, et al. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5:487–495.
- Pandey R, Sharma S, Khuller GK. Oral solid lipid nanoparticle-based antitubercular chemotherapy. Tuberculosis (Edinb). 2005;85:415–420.