
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Employing targeting ligands with high affinity to tumour receptors is an important strategy to increase treatment efficacy. The use of aptamers as targeting agent is increasingly prevalent in drug delivery systems. Mucin1 (MUC1) is a glycoprotein that is over-expressed on the surface of several cancer cells and plays an important role in metastasis and invasion. 5TR1-aptamer is a DNA aptamer, which targets MUC1 receptors. The present study investigated the anti-tumour activity and therapeutic effectiveness of 5TR1-aptamer-PEGylated liposomal doxorubicin (PLD) delivery system in C26 tumour-bearing mice. The in vitro experiments demonstrated enhanced cytotoxicity and cellular uptake of PLD at the presence of 5TR1 aptamer into MUC1+C26 cell line. Biodistribution study indicated that aptamer conjugation increased tumour accumulation of PLDs. Pharmacokinetic analysis showed despite higher clearance rate, selective delivery of doxorubicin to tumour tissue was increased in the 5TR1-Doxil group. In C26-bearing tumour mice, treatment with 5TR1-Doxil exhibited significant deceleration in tumour growth and enhanced survival. The results suggested that 5TR1 aptamer is promising ligand for active targeting which improves therapeutic efficiency of PLD in cancer therapy.
Introduction
The use of targeted cancer delivery systems has become the focus of many types of research. Using nanoparticle-based systems is an effective way to target chemotherapeutic agents selectively to cancerous cells [Citation1]. Among nanoparticle-based systems, liposomal formulation is one of the most established technologies for improving the efficiency of anti-cancer agents [Citation2,Citation3].
The development of targeted cancer delivery systems needs to find proper targets. Proteins that are specifically expressed on the surface of cancer cells would be potential targets for selective cancer therapy. Mucin1 (MUC1) is a transmembrane glycoprotein, which is overexpressed in most carcinomas. MUC1 overexpression is related to tumour growth and invasion. MUC1 improves angiogenesis and chemoresistance [Citation4,Citation5]. In the United States, about 65% of yearly diagnosed tumours overexpress MUC1 [Citation4]. MUC1 in normal cell is highly glycosylated but in neoplastic tissue is under-glycosylated. The difference of glycosylation allows us to discriminate cancerous cells against normal cells in target delivery [Citation6]. In recent years, the MUC1 has become a topic of concern in cancer treatment [Citation7–10].
Aptamers have shown great potential for novel therapeutic delivery applications. Due to high-target specificity, small size, ease of synthesis, thermodynamic stability and lack of immunogenicity, aptamers are gaining more interest in targeted drug delivery systems [Citation11]. The anti-MUC-1 DNA aptamers were first developed by Ferreira et al. [Citation7]. 5TR aptamer series have been developed to target five consecutive mucin MUC1 tandem repeats (MUC1-5TR). 5TR1 aptamer can be internalized into cancer cells [Citation7,Citation9]. In the present study, we decided to attach 5TR1 aptamer as targeting ligand to PLDs. So, 5TR1 aptamer conjugated PLDs were prepared by post-insertion method. Cell cytotoxicity and cellular binding of 5TR1-functionalized PLDs were assessed on C-26 colon carcinoma cells (MUC1+) and CHO-K1 (Chinese Hamster Ovarian) cells (MUC1-). In vivo biodistribution and therapeutic efficacy of formulations were evaluated using the mice bearing C-26 colon carcinoma.
Experimental
Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy (polyethylene glycol)-2000] (DSPE-PEG(2000) carboxylic acid) was purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol, doxorubicin hydrochloride (Dox), Dowex® and phenazine methosulphate (PMS) were purchased from Sigma-Aldrich (St. Louis, MO). MUC1 (5TR1) GAAGTGAAAATGACAGAACACAACA, 5′-NH2 and/or 3′-FAM aptamer and scrambled version of 5TR1 aptamer (ScApt) as negative control were synthesized by MicroSynth (Balgach, Switzerland) and were PAGE purified. MTS (3-(4,5-dimethylthiazol-2-yl)-5–(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium was purchased from Promega (Madison, WI). Trypan blue, isopropanol and chloroform were purchased from Merck (Darmstadt, Germany). N-Hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-n-ethyl-carbodiimide hydrochloride (EDC) were purchased from Fluka (Buchs, Switzerland). Acidified isopropyl alcohol (90% isopropanol/0.075 M HCl) was prepared by the addition of 7.5 mL HCl 1 M and 2.5 mL water to 90 mL isopropanol (Merck, Darmstadt, Germany). Commercially available Doxil® was purchased from Behestan Darou Company (Tehran, Iran).
All other solvents and reagents were used as chemical grades.
Cell lines and cell culture
C26 murine colon carcinoma cells were purchased from Cell Line Service (Eppelheim, Germany) and CHO-K1 (C111, Chinese Hamster Ovarian cell) were purchased from Pasteur Institute of Iran, Tehran, Iran. RPMI 1640 (Euroclone) supplemented with 10% foetal bovine serum (FBS, heat inactivated, Gibco, Carlsbad, CA) and 100 units/mL penicillin streptomycin (Sigma, St. Louis, MO) were used as the culture medium.
Synthesis of 5TR1 aptamer-conjugated DSPE-PEG (2000) carboxylic acid micelles
DSPE-PEG (2000) carboxylic acid lipids were dissolved in chloroform and then dried. Thin film lipids were rehydrated in water. Micelles were prepared by heating in a 65 °C water bath solicitor followed by stirring. Carboxyl groups of micelles were activated by incubating with 400 mM EDC and 100 mM sulpho-NHS for 15 min at room temperature with gentle stirring. 5′-NH2-modified 5TR1 aptamer or ScApt were covalently attached to end-functionalized PEG in micelles by 2 h incubation at room temperature [Citation12,Citation13].
Preparation of aptamer-modified liposomes
In order to prepare aptamer-linked Doxil, DSPE-PEG(2000)-5TR micelles or DSPE-PEG(2000)-ScApt micelles were co-incubated with 1 mL Doxil formulation for 30 min at 60 °C; thereafter, cooled down slowly. In order to separate aptamer functionalized PEGylated-liposomal doxorubicin (5TR1-Doxil and ScApt-Doxil) from micelles and unincorporated aptamers, liposomes were dialyzed overnight against dextrose 5% [Citation14], using dialysis bags with 3.5 KD molecular weight cut off (MWCO) were employed (Pierce, Rockford, IL).
TR1–doxil conjugation confirmation
Gel electrophoresis was employed to validate the attachment of aptamers on the surface of PLDs. Tris-Acetate-EDTA 1 M (TAE) buffer solution with 2% (w/v) agarose containing 0.5 µg/mL ethidium bromide was used as running gel. Empty PEGylated liposomes conjugated with 5TR1 aptamer were prepared as described before. Diluted samples in loading buffer containing empty liposomes functionalized with 5TR1 aptamer, free 5TR1 aptamer and empty liposomes without surface functionalization were loaded onto the gel. The samples were subjected to 110 V for 20 min [Citation13,Citation14]. Then the gel was visualized under UV light by UV documentation device (Alliance, London, UK).
Liposome characterization
The particle size, zeta potential and polydispersity index of liposomes were measured by dynamic light scattering (Nano-ZS; Malvern, UK) [Citation15].
In order to determine the concentration of doxorubicin loaded in liposomes, aliquots of 5TR1-Doxil were dissolved in acidified isopropyl alcohol bellow doxorubicin self-quenching concentration, and afterwards the concentration of doxorubicin was measured by spectrofluorometry (λex: 470 nm; λem: 590 nm) (Shimadzu RF5000U, Tokyo, Japan). Serial dilution of Doxil® was used to prepare standard curve [Citation16]. All measurements were performed in triplicate. To assay doxorubicin loading efficiency, the concentration of doxorubicin was measured before and after purification. The percent of doxorubicin loaded was determined using the following equation:
Release study of liposomes
Dialysis method was employed to assay release of Doxorubicin from PLDs in the presence of 30% FBS. Liposomal formulations were diluted in RPMI 1640 media containing 30% FBS and added to hydrated dialysis bag (3.5 kD molecular weight cut off (MWCO)) (Pierce, Rockford, IL) and immerged into 100 mL of RPMI 1640 containing 30% FBS and 2% NaN3 at 37 °C while stirring. Samples were collected at predetermined time points and dialysate was refreshed with dextrose 5% [Citation16].
The amount of encapsulated doxorubicin was calculated by determining released doxorubicin and compared with Doxil®.
In vitro experiments
Cytotoxicity study
Cytotoxicity of liposomal preparations was assessed by MTS assay. C26 and CHO-K1 cells were seeded at 5000 cells/well in 96-well plates at 37 °C in complete medium. After overnight incubation, liposomal formulations and free doxorubicin solution were serially diluted in FBS free medium and added to cell cultures.
After 4 h incubation at 37 °C, the cells were washed with pre-warmed PBS and incubated for 72 h at 37 °C in complete culture medium. Then, the viability of cells was determined using MTS/PMS assay as described by manufacture (Promega CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay, Promega, Corp., WI). Finally, the optical densities (ODs) were measured using a spectrometric absorbance of 490 nm against a background of 630 nm on Stat-Fax 2100 microplate reader (Awareness Technology Inc., Palm City, FL). Relative cell death (R) was calculated by R = 1 − [(Atest − Ablank)/(Acontrol − Ablank)]. Atest and Acontrol are the absorbance of the cells treated with sample solutions and culture medium without sample, respectively. Ablank represents the absorbance of MTS/PMS solution added in cell free wells [Citation17]. The IC50 values of liposomal formulations were then calculated using CalcuSyn version 2 software (BIOSOFT, Cambridge, UK).
Cell association study
Flow cytometry method was used to investigate binding specificity and selectivity of 5TR1-Doxil to C26 cell line. CHO-K1 cell line has been used as negative cell for MUC1. About 5 & 105 cells were suspended in PBS buffer containing 10% FBS and incubated with the same concentration of 5TR1-Doxil, ScApt-Doxil and Doxil for 30 min at 4 °C or 37 °C with gentle stirring. After incubation, the cells were washed and resuspended in PBS. The fluorescence signals were recorded on BD FACSCalibur cytometer and the resulting data were analyzed using FCS Express (DENOVO Aoftware, Los Angeles, CA).
The specificity of 5TR1-Doxil binding MUC1 receptors was appraised by competitive assay. 5TR1-Doxil was introduced to C26 cells pre-treated with excess amounts of free anti-MUC1 antibody and the geometric mean (GM) changes were compared with untreated cells.
The amount of doxorubicin accumulated in C26 cells was quantified by the following method: 106 C26 cells were seeded in a 24-well plate and incubated overnight. Afterward medium was replaced with FCS-free medium containing 5TR1-Doxil or ScApt-Doxil or Doxil at a lipid concentration of 100 nmol phospholipid/mL and incubated at 4 °C for 1 h and at 37 °C for 1, 3 and 6 h. After incubation time, cells were washed with cold PBS and dislodged by 100 µL of trypsin–EDTA solution (Gibco, Carlsbad, CA). Detached cells were incubated with acidified isopropanol, to extract cell-associated Doxorubicin, at 4 °C overnight [Citation17]. Then cell debris was pelleted by centrifugation and doxorubicin in supernatant was measured by spectrofluorimeter. The percentage of internalized doxorubicin was calculated as described before [Citation14,Citation16].
In vivo experiments
Animal
Four to six weeks aged female BALB/c mice were purchased from the Pasteur Institute (Tehran, Iran). All animal experiments were complied with standards of the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences (Education Office, dated 26 February 2008; permission number: 87848), based on the Specific National Ethical Guidelines for Biomedical Research issued by the Research and Technology Deputy of Ministry of Health and Medicinal Education (MOHME) of Iran that was issued in 2005. Housing conditions of mice was the same as described before (at 21 °C with free access to tap water and laboratory pellet chow) [Citation14].
Biodistribution study
Suspension of C26 tumour cells (3 × 105 cells per mouse) were injected subcutaneously in the right flank of 4–6 week aged female BALB/c mice [Citation18]. Tumours were allowed to grow to approximately 5 mm wide and then Doxil, ScApt-Doxil and 5TR1-Doxil (as commonly used therapeutic dose of 15 mg/kg of doxorubicin) were administered via tail vein. Control mice received 200 µL of dextrose 5%. Blood samples were collected via the orbital sinus 6 and 12 h after drug administration. Heart puncture method was used to collect blood samples at 24 and 48 h post-administration from sacrificed mice.
After 24 and 48 h post drug administration, three mice from each group were euthanized and spleen, tumour tissue, lung, heart, kidney, lung and a portion of liver and muscle tissue were dissected and rinsed in normal saline. Each tissue was weighed and homogenized in 1 mL acidified isopropanol by zirconia beads (Mini-Beadbeater-1, Biospec, Belfast, UK). After coagulation of blood samples, the blood sera were isolated by centrifugation and diluted in 1 mL acidified isopropanol.
The homogenized samples and the sera were stored at 4 °C overnight to extract doxorubicin. The samples were then centrifuged and doxorubicin concentration in supernatants was assayed by spectrofluorimetry (λEx: 470 nm, λEm: 590). The calibration curves were prepared using serial dilutions of doxorubicin in both tissue and sera extracts of the control mice [Citation14,Citation18,Citation19].
Pharmacokinetic study
The plasma elimination profiles of doxorubicin were determined after i.v. injection of formulations. After 6, 12, 24 and 48 h injection, serum samples were collected and analyzed for doxorubicin concentration.
Pharmacokinetic analysis was performed using non-compartment analysis. The serum concentration of doxorubicin (µg/mL) versus time was plotted. For each formulation, the area under the concentration–time curve (AUC) and the area under the first moment curve (AUMC) was calculated. The mean residence time (MRT) was calculated by dividing the AUMC values to the AUC values. The average elimination rate constant (ke) derived from 1/MRT. Half-life (t1/2) was calculated using the formula t1/2 = 0.693/ke. Total clearance (Cl) was also determined (dose/AUC). The volume of distribution (Vd) was calculated by the multiplying Cl by MRT. Since individual animals were scarified in each time point to collect samples and subsequent sampling over time was not possible, the difference in mean pharmacokinetic parameters was calculated with two-way analysis of variance (General Linear Model, Univariate, SPSS 11.5 software, SPSS, Chicago, IL). In all cases, p < .05 was considered to be statistically significant.
Therapeutic efficacy
Tumour inoculation was performed as described in biodistribution section before. A week after tumour inoculation, mice owing palpable tumour size were received a single dose of 15 mg/kg doxorubicin encapsulated in liposomal formulations via vein tail injection. One group received 5% dextrose solution as negative control.
The weight and tumour size of mice were monitored up to 60 d post-injection. Three orthogonal diameters (a, b and c) were measured every other day with callipers and the corresponding tumour volume was calculated as (a × b×c) × 0.5 mm3. The volumes were estimated at 1 mm3 at first day of post-injection. The exclusion criteria regarding prevailing ethical standards were (1) mice body weight dropped below 15% of their initial mass; (2) the tumour size reach 2.0 cm across in any dimension; (3) they became moribund [Citation18,Citation19]. The mean tumour volume ± SE for each group was plotted versus time. Survival probability of each group was estimated by Kaplan–Meier analysis. The time to reach end point (TTE) for each mouse was calculated from the equation of the line obtained by logarithmic regression of the tumour growth curve. Subsequently, the percent of tumour growth delay (%TGD) was calculated based on the difference between the mean TTE of treatment group (T) and the mean TTE of the control group (C) [Citation14,Citation20]:
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 6 (GraphPad Software, San Diego, CA). Survival data were analyzed by the log-rank test. The tumour volumes of all groups at each time point were examined using one-way ANOVA.
Results
Physicochemical characterization of liposomes
The effect of 5TR1 conjugation on the size of liposomes was investigated. The size of Doxil was 112 nm. The size of ScApt-Doxil and 5TR1-Doxil showed the increase in the diameter of liposomes compared with Doxil. Further zeta potential measurement of liposomes was performed and as shown in aptamer conjugation has decreased the zeta potential of liposomes from −11 to −20 mV. In addition, provides the percent of loading efficiency of formulations, indicating aptamer conjugation had no impact on the loading efficiency of PLDs and Doxorubicin was loaded into liposomes with loading efficiency of >90%. The release kinetic of PLDs also studied and, as shown in , the release profile of Doxorubicin from liposomes had a steady rate and presence of aptamer on the surface of PLDs has no meaningful impact on the leakage stability of PLDs.
Figure 1. Doxorubicin content leakage profile from ScApt-Doxil and 5TR1-Doxil compared to Doxil® at 37 °C at the presence of 30% FCS. Diluted liposomes were dialyzed at 37 °C in 100 mL of the media. At various time points, the percent of doxorubicin retained encapsulated was measured (n = 3).
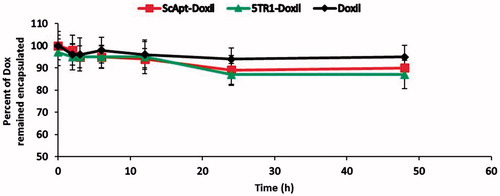
Table 1. Properties of liposomal preparations. Each value represents mean ± SD (n = 3).
Conjugation of 5TR1 aptamer to liposomes
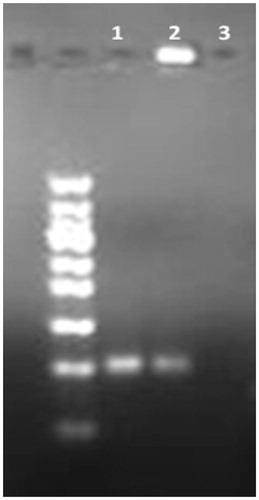
Conjugation of 5TR1 aptamer on liposomes was confirmed by agarose gel electrophoresis assay. As it shown in , 5TR1-Doxil trapped in the second well (lane 2) and could not move thorough the gel as free 5TR1 aptamer (lane 1). Lane 3 shows untargeted liposomes have no signal under UV light.
Figure 2. Confirmation of liposome–aptamer conjugation. Samples are loaded onto the agarose gel. After running time, the gel was visualized by UV light. Lane 1: free 5TR1 aptamer; 2: 5TR1-liposomes; 3: liposome without conjugation.
The density of 5TR1 aptamer conjugated to surface of PLDs was determined as described before [Citation14]. To achieve 20 5TR1 aptamer on the surface of each individual liposome, following data have been used: total phospholipid concentration of liposomal preparation was 14 mM. Liposome average size was assumed 100 nm. Then the number of lipid molecule in each liposome will be about 8 × 104. Therefore, there are 1 × 1014 liposomes in each millilitre of formulation. The molecular weight of 5TR1 aptamer was 7734 g/mol. The number of aptamer molecules per each mL aliquots of aptamer-micelles was about 2 × 1015. As a result, at a determined ratio of aptamer-micelles to preformed liposomes (1000 μl of aptamer-micelles with 1000 μl preformed liposomes), the average number of 20 aptamer molecules on the surface of each individual liposome was achieved.
In vitro experiments
Cytotoxicity study
The effect of 5TR1 aptamer on cytotoxicity effect of liposomal formulation was investigated by MTS assay. As shown in , 5TR1-Doxil exhibited significantly higher cytotoxicity than Doxil (p < .001) on C26 cell line, while scrambled sequence did not have a noticeable effect on cytotoxicity of liposomes (p > .05). The results also showed no enhanced cytotoxic effect of 5TR1-aptamer in CHO-K1 cell line ().
Table 2. In vitro cytotoxicity effect (IC50) of liposomal preparations and free DOX against C26 and CHO-K1 cells after 72 h exposure times. Data represented as mean ± SD (n = 3).
Cell association study
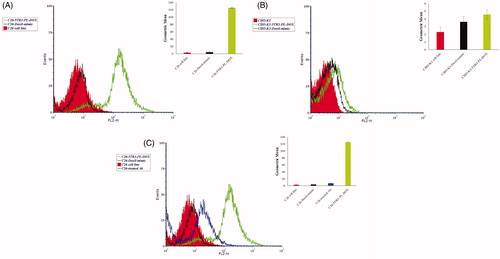
Flow cytometry assay was exploited to measure binding specificity of 5TR1-Doxil in comparison with Doxil. C26 cell line showed higher Geometric mean (GM) when treated with 5TR1-Doxil compared to Doxil (). No significant difference was obtained in the GM between 5TR1-Doxil and Doxil with CHO-K1 cell line (). As shown in , the GM of 5TR1-Doxil groups was reduced after treatment with excess amounts of free anti-MUC1 antibody.
Figure 3. Flow cytometry analysis of doxorubicin cell uptake. C26 cell line (A), CHO-K1 cell line (B) and pre-treated C26 cells with anti-MUC1 antibody (C) were incubated with Doxil and 5TR1-Doxil at 37 °C in the presence of 10% FCS. After 30-min, uptake of doxorubicin was assessed by flow cytometry. Results, expressed as mean fluorescence intensity (MFI), represented mean ± SEM of three independent experiments. There is a significant difference (p < .1) of Doxil compared with 5TR1-Doxil in cellular uptake by C26 cell line. However, cellular uptake by CHO-K1 cells is not affected by 5TR1 targeting. Cellular uptake by C26 cell line significantly decreases after treatment of cell with anti-MUC1 antibody (p < .01).
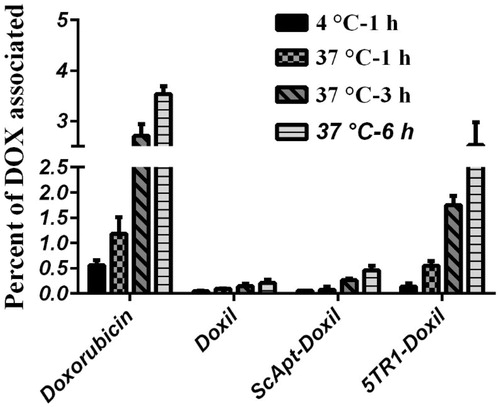
The amount of Doxorubicin uptake was conducted with C26 cell line. The obtained data showed significant enhanced cellular uptake in C26 treated with 5TR1-Doxil compared with other liposomal formulations (p < .001), while there is no remarkable difference between Doxil and ScApt-Doxil-treated groups (p > .05) ().
Figure 4. Liposome–cell association at different time points. C26 cells were treated with different preparations at a concentration of 100 nmol phospholipid/mL for 1, 3 and 6 h of incubation at either 37 °C or 4 °C. Then, the cells were detached, washed and lysed in acidified isopropanol and percent of doxorubicin associated with cells was measured based on the amount of doxorubicin assayed in the medium and amount of doxorubicin detected in cell lysate. Data are expressed as mean ± SD (n = 3).
In vivo experiments
Biodistribution study
Biodistribution data of liposomal formulations were presented in different organs including serum, liver, spleen, tumour, lung, heart and muscle. As indicates, the spleen and liver concentration of Doxorubicin has a marginal rise after aptamer conjugation. Moreover, as shown in , the serum concentrations of ScApt-Doxil and 5TR1-Doxil were slightly decreased. From , however, it can be seen that the ratio of doxorubicin concentration in liver and spleen to serum in the 5TR1-Doxil group was higher and the liver/serum ratio of the 5TR1-Doxil group was significantly (P < .001) higher than the Doxil group. compares the concentration of doxorubicin in the different group in tumour tissue. The tumour level of 5TR1-Doxil was higher than ScApt-Doxil and Doxil. This increase was not significant at 24 h. However, after 48 h, the significant localization of formulation was observed in the 5TR1-Doxil group (p < .01).
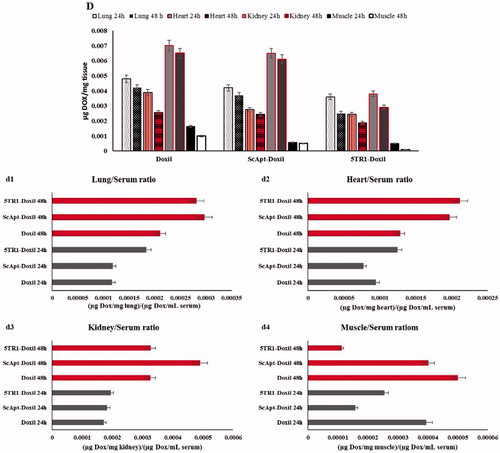
Figure 5. Biodistribution of Doxil, 5TR1-Doxil and ScApt-Doxil at different time points in organs including (A) liver and spleen, (B) tumour, (C) serum and (D) other tissues in BALB/c mice-bearing C-26 tumour after a single dose of 15 mg/kg liposomal doxorubicin administered i.v. 2 weeks after the tumour inoculation. represents ratio of doxorubicin concentration in tumour, liver and spleen to serum at each time point. represents ratio of doxorubicin concentration in tumour to liver and spleen (T/NT ratio) at each time shows ratio of doxorubicin concentration in tumour to serum at each point. represents ratio of doxorubicin concentration in lung, heart, kidney and muscle to serum. Data expressed as mean ± SEM (n = 3).
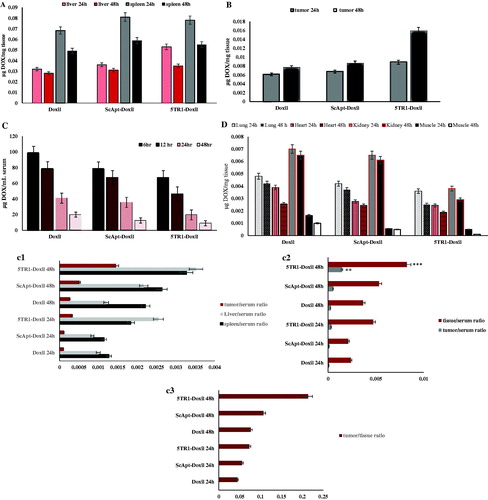
Also after 48 h post injection, the tumour/serum ratio of doxorubicin concentration in the 5TR1-Doxil receiving group was considerably higher than other groups (). The total doxorubicin concentration at serum at 6, 12, 24 and 48 h post-injection are shown in . The mice received ScApt-Doxil and 5TR1-Doxil showed shorter blood circulation time compared with Doxil, although, there was no significant decrease in circulation time (p > .05) (). The tumour/serum ratio, liver/serum, spleen/serum and tumoural/non-tumoural ratio of doxorubicin concentration are reported in . Analysis of other tissues () indicated that there are no remarkable differences in doxorubicin concentration in lung and heart tissues.
Table 3. Pharmacokinetic parameters using non-compartment model in mice following singel intravenous injection of 15 mg/kg of Doxil-mimic, ScApt-PL-DOX and 5TR1-PL-DOX.
At 48 h post-injection, the concentration of doxorubicin in muscle in the 5TR1-Doxil group was 10- and 5-folds lower than Doxil and ScApt-Doxil groups, respectively. Low concentration of doxorubicin in the kidney was observed in 5TR1-Doxil groups at 24 and 48 h post-injection. The amount of doxorubicin in kidney in 5TR1-Doxil at 24 h post-injection was about half of other groups and at 48 h was about 1/3 of other groups. The tissue/serum ratio in all group were increased over the time except in muscle in the 5TR1-Doxil receiving group ().
Pharmacokinetic study
Pharmacokinetics of liposomes were analyzed by taking blood samples at different time points after single i.v. bolus administration of liposomal formulations. The non-compartment model was employed for pharmacokinetic analysis and the pharmacokinetic parameters were determined (). For Doxil and ScApt-Doxil, the elimination of total doxorubicin from plasma was not significantly different (p < .05). At 48 h post-injection, about 15% of injected dose could be found in plasma in Doxil and ScApt-Doxil receiving groups. While this amount was about 9% in the 5TR1-Doxil group. As shows, conjugation to aptamer resulted a decrease of the area under the curve (AUC) (from 3162.77 µg h/mL to 1755.54 µg h/mL) and shortened the half-life of PLDs (t½) (from 18.145 h to 14.52 h). While, the Cl increased (6.323 × 105 (mg)/(μg/mL)/h for Doxil versus 0.00011 (mg)/(μg/mL)/h for 5TR1-Doxil) and volume of distribution (Vd) increased from 0.0016 (mg)/(μg/mL) to 0.0022 (mg)/(μg/mL). For the quantitative evaluation of the selective delivery of Doxorubicin to tumour tissue, “te” was evaluated by following equation [Citation21]:
Or
In which Q is the concentration of drug * weight or volume of tissue.
Because the weight factor was applied in before computations, the weight in above equations was omitted:
The ability of aptamer-targeted liposomes to selective delivery of Doxorubicin was compared with Doxil as R te= or
.
R te for 5TR1-Doxil was 1.25 while for ScApt-Doxil was 1.1. The amount of AUC for each tissue are summarized in .
Table 4. AUC (µh) (24–48 h) of various tissues in mice-bearing C26 carcinoma after injection of 15 mg/kg doxorubicin as Doxil, ScApt-Doxil and 5TR1-Doxil.
Chemotherapy study
Anti-tumour efficacy of 5TR1-Doxil was evaluated in murine C-26 colon carcinoma tumour model. Tumour size, body weight and survival were monitored throughout the time and results are summarized in . The results of animal weight monitoring () indicates the slight weight change in the group receiving 5TR1-Doxil compared with Doxil and Dextrose 5% receiving groups, although the difference in weight changes in all groups was not statistically significant (p > .05). As represented in , the tumour of animals in the control group grew rapidly and before 30 d after injection, all animals required termination because of ethical considerations. Treatment with 5TR1-Doxil has declined the rate of tumour growth compared with Doxil and ScApt-Doxil significantly (p < .01). The survival results were obtained by Kaplan–Meier analysis (). Median survival time (MST), time to reach endpoint (TTE) and percent of tumour growth delay (%TGD) are summarized in . Treatment with 5TR1-Doxil prolonged MST to 50 d, while MST for Doxil and ScApt-Doxil was 33 d. TTE in mice receiving 5TR1-Doxil slightly increased to 45 d; however, the difference between liposomal groups was not significant (p > .05). Targeting liposomes by 5TR1 aptamers almost doubled the percent of tumour growth delay (). The hazard ratio of 5TR1-Doxil groups was about half of the Doxil group. While the hazard ratio of ScApt-Doxil and Doxil groups was almost the same. The number of subjects at risk is illustrated in .
Figure 6. Therapeutic efficacy of various liposomal preparations in female BALB/c mice after i.v. administration of a single dose of 15 mg/kg liposomal doxorubicin or dextrose 5% on day 8 after tumor inoculation. (A) Percentage change in animal body weight, (B) tumour growth rate, (C) survival curve and (D) survival curve with error bars. Data represented as mean ± SE (n = 5).
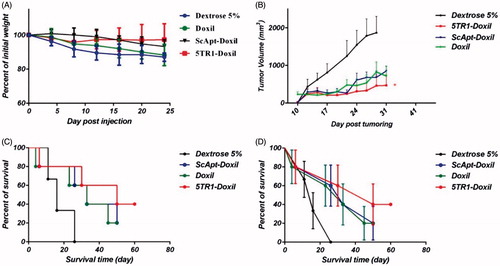
Table 5. Therapeutic efficacy data of liposomal preparations in mice-bearing C26 colon carcinoma.
Table 6. Number of at risk in every group at days 50 and 60 post-injections.
Discussion
Among all nanoparticle-based cancer drug delivery systems, PEGylated-nanoliposomes, owing to high stability, long blood circulation time, low toxicity, high drug loading efficiency and the ability to target the tumours by enhanced permeation and retention (EPR) mechanism, have exploited as a prominent drug carrier [Citation22]. Doxil® is the PEGylated liposomal formulation of Doxorubicin that has been approved for cancer treatment by passive ways based on EPR effect [Citation23]. Surveying the literature indicates that one of the key limitations of Doxil is release profile of drug in tumour site. The release profile of doxorubicin from PEGylated liposomes in tumour site is weak and the efficient concentration in tumour site is achieved after 48 h [Citation23]. The main challenge of stealth liposomes-like Doxil is the low release rate in tumour environment and poor cellular uptake [Citation24,Citation25].
Several attempts have been made to overcome this problem by exploiting ligand targeting [Citation26–28]. Ligand-targeting induced receptor-mediated endocytosis and can enhance the release of liposome content intracellularly. This effect reduces the diffusion back of drug from tumour site to blood circulation and overcome drug resistance [Citation29–31]. While liposomes increase the level of drug at the tumour site, attached ligands promote the cellular uptake [Citation32]. Employing ligands for receptors that are overexpressed by tumour cells compared with the normal cells is a prevalent way for active targeting [Citation33,Citation34]. Employing aptamers has become more interesting for active target delivery. The advantages of aptamers over antibodies have been discussed elsewhere. Briefly, aptamers have high affinity and specificity to their receptors thus a lower dose of a nanoparticle is needed for the treatment. Moreover, they have low toxicity and immunogenicity. The main reason of utilizing aptamers in target delivery studies is their cost effectiveness [Citation11,Citation35,Citation36]. Several aptamers have been employed successfully for the functionalization of liposomes and other nanoparticles [Citation14,Citation35–39].
Our proof of concept study aimed to demonstrate that targeting liposomal doxorubicin by 5TR1 aptamer could be much more effective than non-targeted liposome in tumor growth inhibition in mice model. We have previously reported that aptamers are promising targeting agents for PEGylated liposomal doxorubicin (PLDs) [Citation14]. Also, we proved that 5TR1 aptamers are able to target nanoparticles to C26 colon carcinoma cell lines [Citation10].
In this study, we utilized Mucin 1(MUC1) as a model receptor for target delivery of for Doxil®. MUC1 protein is expressed in the elevated amount in the majority of human adenocarcinomas. MUC1 overexpression is a marker of poor prognosis. The use of MUC1-targeting aptamer has been reported in a number of literature as targeting agents [Citation10,Citation40]. In our study, active targeting of liposomal doxorubicin via 5TR1 aptamer was investigated with C26 cell line and CHO-K1 cell line, the latter lacking the MUC1 receptor for 5TR1 aptamer as the negative control [Citation7,Citation10]. 5TR1 conjugation to the surface of PEGylated liposomes was performed by post-insertion method and the release test confirmed that liposomes remain stable after post-insertion and aptamer attachment had no negative effect on the structure of liposomes ().
The mean diameter of liposomes before and after 5TR1 conjugation was measured to ensure that 5TR1-Doxil had an acceptable range for tumour targeting (). The growth of liposome size and the decrease in zeta potential could be a sign for efficient insertion of negatively charged 5TR1 aptamers to the surface of liposomes [Citation14,Citation37]. Correspondingly, agarose gel electrophoresis demonstrated successful aptamer–liposome conjugation (). The physicochemical properties of functionalized PLDs approved that the post-insertion technique was quite simple but effective method for the post-insertion of aptamer-micelle into preformed liposomes. This method did not have any negative impact on leakage property of liposomes ().
The MTS assay was utilized to compare cytotoxic effect of formulations on C26 and CHO-K1 cell lines. The results reinforced the importance of 5TR1 aptamer on the uptake of liposomes in C26 cells and empowerment of cell killing effect of liposomal doxorubicin ().
To investigate the binding specificity and selectivity of 5TR1 to MUC1 overexpressing cells, cellular uptake of doxorubicin from different formulations was measured. As it has shown in the result section, the affinity of 5TR1-Doxil to C26 cell line was significantly higher than Doxil (). While the affinity to CHO-K1 (MUC1−) cells remains almost the same for both formulations (). The GM of C-26 cells treated with free antibody before the addition of 5TR1-Doxil were significantly lower than that of cells incubated with 5TR1-Doxil, confirming the competition among MUC1 aptamer and antibody for the same binding site (). Correspondingly, the cellular uptake of doxorubicin into C26 cells was quantified (). In the same way, obtained data showed a higher interaction between C26 cells and 5TR1-Doxil compared with Doxil in all exposure times. The amount of doxorubicin uptake in liposomal groups had no significant difference at 4 °C, while intracellular delivery was significantly higher at 37 °C in the 5TR1-Doxil group. The results demonstrated that drug delivery was mediated via ligand–receptor interaction [Citation41]. In vitro results indicated that 5TR1 functionalized PLDs successfully targeted C26 cell line.
In present study, tissue distribution of 5TR1-conjugated liposomal doxorubicin was studied in mice model and impact of aptamer conjugation on the biodistribution of liposomes was evaluated. It is well known that pharmacokinetic profile of liposomal doxorubicin includes two separated procedure: pharmacokinetic of lipid carrier and pharmacokinetic of free doxorubicin which was released from liposomes [Citation42]. Since lipid composition and the amount of loaded doxorubicin were the same in formulations, the only factor influences on pharmacokinetic profile was aptamer attachment on the surface of liposomes. As regard, 5TR1 aptamer had the negative charge, an alteration in biodistribution of liposomes was expected. Obtained data indicated despite there was no significant difference in serum, spleen and liver concentration in different groups, the ratio of liver/serum considerably increased in the 5TR1-Doxil group compared with Doxil. It is assumed that the conjugation of aptamers on the surface of liposomes increases their uptake by RES due to the elevation of negative charges on liposomes [Citation43]. Resulting data demonstrated that 5TR1 attachment increased CL of liposomal doxorubicin by 2-fold. The t1/2 and MRT also had a slightly decline. Pharmacokinetic parameters of ScApt-Doxil had no remarkable variation compared with Doxil (). It might be explained by the difference between tertiary structure and charge of scrambled and 5TR1 aptamers. These data are consistent with results of in vitro experiments showing that scrambled aptamer had no effect on cellular up take and cytotoxicity. The data indicated that conjugation of 5TR1 on the surface of liposomes contributed to increase in liposome clearance rate by the liver, likewise reducing circulation time in blood stream [Citation43]. As demonstrates, the distribution pattern of doxorubicin in other tissues follows the same pattern in all groups. The concentration of doxorubicin in all tissues was lower in 5TR1-Doxil groups; however, the difference in heart and lung was not significant. This finding is consistent with obtained pharmacokinetic parameters (). The most noticeable feature of these results was the more tumour localization of the 5TR1-Doxil receiving group compared with other groups. This is a considerable achievement and indicated the active role of 5TR1 aptamer in tumour localization of liposomal doxorubicin. The ligand-dependent step in tumour localization could explain the higher level of 5TR1-Doxil accumulation. After i.v. administration, PEGylated liposomes are confined in the central compartment, with limited redistribution in tissues other than those with high vascular permeability (EPR effect). This step is same in targeted or non-targeted liposomes [Citation44]. After extravasation of liposomes in the solid tumour tissue, the interaction of ligand-targeted liposomes (5TR1 in this study) with cell surface receptor (MUC1) enhances cellular uptake and inhibits re-entering into the central compartment [Citation25,Citation45]. Further, the higher clearance rate in 5TR1-Doxil groups results in lower accumulation in non-tumoural organs. This finding was strengthened by information depicted in . In addition, lower plasma AUC may decrease the incidence of toxicity. The therapeutic efficiency is always not related to the plasma AUC [Citation42]. Vss value in the 5TR1-doxil group is larger than the Doxil group that might ascribe the higher tumour accumulation. In chemotherapy experiment, the body weight reduction was used as the standard for toxicity evaluation [Citation46]. The results reinforced that treatment with 5TR1-Doxil was well tolerated and reduced off-target toxicity. One possible explanation may be that after drug release in extracellular space a percentage of drug diffuses back from tumour site to blood circulation [Citation36]. This may increase toxicity and morbidity of drug. Aptamers on the surface of liposomes enhance liposome–cell interaction and inhibit liposomes diffusion back to blood stream in comparison with non-targeted counterpart. Since most of the injected formulations were accumulated in spleen and liver, we estimated their doxorubicin concentration as the non-tumoural concentration in . The obtained data in this figure indicated that the tissue/serum ratio and tumour/serum ratio were increased in both non-tumoural and tumour tissue concurrently in the 5TR1-Doxil group; however, the higher increase was observed in tumour/serum ratio. The value of te and Rte endorse that selectivity of 5TR1-Doxil to tumour tissue. The selectivity of 5TR1-Doxil to tumour tissue was 25% more than Doxil.
The above information showed that despite the comparable organ distribution of targeted and non-targeted liposomes, the tumour localization in the targeted formulation is higher. These findings were consistent with chemotherapy study which 5TR1-Doxil clearly decelerated tumour growth size. Considering in vitro results, the amount of 5TR1-Doxil entering the tumour was high enough for killing tumour cells.
Survival analysis demonstrated that the therapy with 5TR1-Doxil did not significantly increased survival time. In many studies, MST is reported instead of overall survival because survival time is frequently skewed. Therefore, the MST is a more reliable factor. Results revealed MST in 5TR1-Doxil receiving group remarkably increased. The hazard ratio is described as the slope of survival curve and quantifies the rate of death in every group. Obtained data showed that targeting with 5TR1 decreased hazard ratio of liposomal doxorubicin. This finding consolidates previous results. For all these reasons, it is claimed 5TR1 attachment not only is not likely to cause a loss of Doxil stability but also decreases progression of tumour growth during experiment.
Conclusion
In conclusion, MUC1-targeted PEGylated liposomal doxorubicin was successfully constructed by post-insertion method. The size of functionalized liposomes was 120 nm with high loading efficiency of doxorubicin. The results showed 5TR1 increased cytotoxicity and cellular uptake of liposomes in MUC1 over-expressed cells, while it had no significant effect on MUC1− cell line. Pharmacokinetic analysis endorsed the rule of 5TR1 aptamer in obtaining high drug accumulation in tumour tissue. Moreover, 5TR1 aptamer improved therapeutic efficacy of PLDs in BALB/c mice-bearing C-26 colon carcinoma. Our results supported that 5TR1-Doxil could be a promising delivery system to overexpressed MUC1 cancer cells and treat many types of cancers.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664.
- Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592–599.
- Allen TM, Mumbengegwi DR, Charrois GJR. Anti-Cd19-targeted liposomal doxorubicin improves the therapeutic efficacy in murine B-cell lymphoma and ameliorates the toxicity of liposomes with varying drug release rates. Clin Cancer Res. 2005;11:3567–3573.
- Pillai K, Pourgholami MH, Chua TC, et al. Muc1 as a potential target in anticancer therapies. Am J Clin Oncol. 2015;38:108–118.
- Apostolopoulos V, Stojanovska L, Gargosky SE. Muc1 (Cd227): a multi-tasked molecule. Cell Mol Life Sci. 2015;72:4475–4500.
- Li Y, Cozzi PJ. Muc1 is a promising therapeutic target for prostate cancer therapy. Curr Cancer Drug Targets. 2007;7:259–271.
- Ferreira CS, Cheung MC, Missailidis S, et al. Phototoxic aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res. 2009;37:866–876.
- Tan L, Neoh KG, Kang E-T, et al. Pegylated anti-Muc1 aptamer–doxorubicin complex for targeted drug delivery to Mcf7 breast cancer cells. Macromol Biosci. 2011;11:1331–1335.
- Ray P, White RR. Aptamers for targeted drug delivery. Pharmaceuticals (Basel). 2010;3:1761.
- Jalalian SH, Taghdisi SM, Shahidi Hamedani N, et al. Epirubicin loaded super paramagnetic iron oxide nanoparticle–aptamer bioconjugate for combined colon cancer therapy and imaging in vivo. Eur J Pharm Sci. 2013;50:191–197.
- Levy-Nissenbaum E, Radovic-Moreno AF, Wang AZ, et al. Nanotechnology and aptamers: applications in drug delivery. Trends Biotechnol. 2008;26:442–449.
- Klibanov AL, Torchilin VP, Zalipsky S. Long-circulating strerically protected liposomes. In: Torchillin VP, Weissig V, editor. Liposomes. London: Oxford University Press; 2010. p. 231–265.
- Gu F, Langer R, Farokhzad OC. Formulation/preparation of functionalized nanoparticles for in vivo targeted drug delivery. Methods Mol Biol. 2009;544:589–598.
- Moosavian SA, Abnous K, Badiee A, et al. Improvement in the drug delivery and anti-tumor efficacy of pegylated liposomal doxorubicin by targeting RNA aptamers in mice bearing breast tumor model. Colloids Surf B Biointerfaces. 2016;139:228–236.
- Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–468.
- Amin M, Badiee A, Jaafari MR. Improvement of pharmacokinetic and antitumor activity of pegylated liposomal doxorubicin by targeting with N-methylated cyclic RGD peptide in mice bearing C-26 colon carcinomas. Int J Pharm. 2013;458:324–333.
- Horowitz AT, Barenholz Y, Gabizon AA. In vitro cytotoxicity of liposome-encapsulated doxorubicin: dependence on liposome composition and drug release. Biochimica Et Biophysica Acta (BBA) Biomembranes. 1992;1109:203–209.
- Huang Z, Jaafari MR, Szoka FC Jr. Disterolphospholipids: nonexchangeable lipids and their application to liposomal drug delivery. Angew Chem Int Ed. 2009;48:4146–4149.
- Huang Z, Szoka FC Jr. Sterol-modified phospholipids: cholesterol and phospholipid chimeras with improved biomembrane properties. J Am Chem Soc. 2008;130:15702–15712.
- Schluep T, Hwang J, Cheng J, et al. Preclinical efficacy of the camptothecin-polymer conjugate It-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–1614.
- Gupta PK, Hung CT. Quantitative evaluation of targeted drug delivery systems. Int J Pharm. 1989;56:217–226.
- Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115:10938–10966.
- Barenholz Y. Doxil®-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–134.
- Bertrand N, Wu J, Xu X, et al. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25.
- Lammers T, Kiessling F, Hennink WE, et al. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–187.
- Béduneau A, Saulnier P, Benoit J-P. Active targeting of brain tumors using nanocarriers. Biomaterials. 2007;28:4947–4967.
- Sakurai Y, Kajimoto K, Hatakeyama H, et al. Advances in an active and passive targeting to tumor and adipose tissues. Expert Opin Drug Deliv. 2015;12:41–52.
- Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010;197:3–53.
- van der Meel R, Vehmeijer LJC, Kok RJ, et al. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65:1284–1298.
- Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740.
- Park JW, Hong K, Kirpotin DB, et al. Anti-Her2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8:1172–1181.
- Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–1626.
- Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760.
- Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine (London, England). 2013;8:1509–1528.
- Farokhzad OC, Biswas S, Torchilin VP. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64:7668–7672.
- Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci. 2006;103:6315–6320.
- Xing H, Tang L, Yang X, et al. Selective delivery of an anticancer drug with aptamer-functionalized liposomes to breast cancer cells in vitro and in vivo. J Mater Chem B Mater Biol Med. 2013;1:5288–5297.
- Alshaer W, Hillaireau H, Vergnaud J, et al. Functionalizing liposomes with anti-Cd44 aptamer for selective targeting of cancer cells. Bioconjug Chem. 2015;26:1307–1313.
- Baek SE, Lee KH, Park YS, et al. Rna aptamer-conjugated liposome as an efficient anticancer drug delivery vehicle targeting cancer cells in vivo. J Control Release. 2014;196:234–242.
- Pieve CD, Perkins AC, Missailidis S. Anti-Muc1 aptamers: radiolabelling with 99mTc and biodistribution in Mcf-7 tumour-bearing mice. Nuclear Med Biol. 2009;36:703–710.
- Kamps JAAM, Scherphof GL. Liposomes in biological systems. In: Torchilin VP, Weissig V, editors. Liposomes. New York: Oxford University Press; 2003. p. 271–272.
- Hong RL, Huang CJ, Tseng YL, et al. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: is surface coating with polyethylene glycol beneficial? Clin Cancer Res. 1999;5:3645–3652.
- Fathi S, Oyelere AK. Liposomal drug delivery systems for targeted cancer therapy: is active targeting the best choice? Future Med Chem 2016;8:2091–2112.
- Allen TM. Pharmacokinetics and biopharmaceutics of lipid-based drug formulations. In: Liposome Technology. vol. 3. NewYork: Informa Healthcare; 2007. p. 49–64.
- Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36–48.
- Tuscano JM, Martin SM, Ma Y, et al. Efficacy, biodistribution, and pharmacokinetics of Cd22-targeted pegylated liposomal doxorubicin in a B-Cell non-Hodgkin's lymphoma xenograft mouse model. Clin Cancer Res. 2010;16:2760–2768.