
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Purpose
To assess the effect of “N-Acetylation and C-Amidation” on the cellular uptake, cytotoxicity and performance of amphiphilic cell penetrating peptides (CPP) loaded with methotrexate (MTX).
Methods
Several CPPs were synthesized by solid phase peptide synthesis method. Some of these sequences were modified with pyroglutamic acid at N-terminus and benzylamine or memantine at C-terminus. The resultant nanomaterials were prepared due to the physical linkage between CPPs and MTX. The internalization and cytotoxicity of both CPP-MTX bioconjugates and unmodified CPPs against MCF-7 human breast adenocarcinoma cells was evaluated.
Results
N-l and C-terminal modification did not alter the toxicity of CPPs. Physical linkage of CPPs with MTX resulted in a lower drug loading efficiency in comparison with chemically conjugated CPP-MTX bio-conjugates. Both nano-complexes increase the toxic effect of MTX on MCF-7 cells. Furthermore, N- and C-terminal modification may cause a tangible reduction in cellular uptake of CPPs.
Conclusion
In conclusion, it was shown that cytotoxicity of modified peptides which were physically linked with MTX, considerably higher than both physically loaded unmodified peptides and chemically conjugated peptides with MTX. Also, cell internalization was reduced after peptide end-protection. These findings confirmed the effectiveness of N- and C-terminal modifications on cell viability and CPPs internalization.
Background
A successful drug delivery toward cancer possesses some promising breakthroughs due to its potential importance and impacts on human life. Thus, the appealing prospect of newly proposed strategies has made the delivery of desired molecules, in the range of small molecules to larger ones, despite their different hydrophilicity, as an interesting issue [Citation1]. Nowadays, hindrance by the lipid bilayer of the cell membrane is considered as a major hurdle in an efficient drug delivery [Citation2]. Nanotechnology seeks the most applicable approaches in order to overcome some of the restrictions in the scope of drug delivery. To date, one of the most well-known vehicles to circumvent natural cellular barriers (i.e. cellular membrane) is a type of peptides called; cell penetrating peptide (CPP) [Citation3]. CPPs are a kind of peptide sequences composed of 5–30 amino acid residues with hydrophobic, cationic or amphiphilic properties [Citation4,Citation5]. These peptides have shown a special ability to pass through biological membranes spontaneously by energy-dependent or energy-independent mechanisms and become promising vectors to facilitate the transport of biological cargoes to the cells [Citation6]. As same as other groups of peptides, CPPs are vulnerable to be degraded via proteolytic enzymes which are seen in the cells, liver and the blood [Citation7]. In order to protect these versatile carriers, a wide variety of options have been developed in the recent years [Citation8]. One of the most common and reliable procedures to stabilize nearly all kinds of peptides is called “N-Acetylation and C-Amidation”. In this method, a compound is attached to the amino terminus of a peptide and another compound is coupled to the carboxy terminus of the same peptide. Therefore, a chemical modification is applied against exo-peptidases [Citation9]. It is noteworthy to consider that a peptide without any modification may exhibit immunogenicity or toxic effects. However, peptides with modifications result in, toxic effects or not is still unanswered. Currently, the ideas about how different classes of CPPs enter the cells is also a controversial issue. The most prevalent amino acid residues, which show extraordinary uptake patterns, are cationic residues (e.g. Arginine (R), Lysine (K)) besides tryptophan (W), whereas other residues do not exhibit more remarkable impacts on internalization [Citation10]. A very common type of CPPs is polyarginine with different number of residues. Arginine-rich peptides with few residues could enter the cells sufficiently, but the presence of at least eight positive charges is proposed for an efficient cellular internalization of cationic or amphiphilic CPPs [Citation11].
Methotrexate (MTX) is a chemotherapy agent and immune system suppressant which acts as an antagonist of folate. This drug competitively inhibits dihydrofolate reductase (DHFR) and finally affects the production of thymidine monophosphate, which is essential for DNA and RNA synthesis [Citation12]. Aiming to consolidate this research process, loading MTX on desired CPP sequences due to delivery to MCF-7 human breast adenocarcinoma cancer cells was carried out in two different patterns; the physical linkage between drug molecules and desired peptides (modified or unmodified sequences) and also chemical conjugation of drug molecules to CPPs. Finally, cell viability and cytotoxicity of blank carriers (e.g. peptide alone), cell viability and cytotoxicity of drug loaded CPPs; both physically linked and chemically conjugated sequences were assessed by3–(4, 5-dimethylthiazol-2-yl)-2,5diphenyltetrazolium bromide (MTT) assay. In the uptake studies, three groups of peptides were chosen and uptake studies were carried out by flow cytometry, fluorescent microscopy, live cell imager and confocal laser scanning microscope (CLSM).
The peptides undergo proteolytic degradation in the body. It has been shown that N-terminal and/or C-terminal modification would increase the biological half-life of peptides. This study was conducted to investigate the effect of N- as well as C-terminal modification on the cellular uptake, cytotoxicity and performance of amphiphilic CPPs loaded with MTX.
Materials and methods
Materials
N, N, N’, N’-tetramethyl-O-(benzotriazole-l-yl) uronium tetrafluoroborate (TBTU), triisopropylsylane (TIS), 1-hydroxybenztriazole (HOBt), 5(6) carboxyfluorescein (FAM), potassium cyanide (KCN), methotrexate (MTX), propidium Iodide (P. I.), ninhydrine and trypsin were obtained from Sigma-Aldrich (St. Louis, MO). 3-(4, 5-dimethylthiazol-2-yl)-2,5diphenyltetrazolium bromide (MTT) was from Roth (Karlsruhe, Germany). Pyridine was obtained from BDH Laboratory Supplies (England). Trifluoroacetic acid (TFA) and piperidine were from Fluka (Buchs, Switzerland). N-ethyldiisopropylamine (DIPEA), triethylamine (TEA), dimethyl sulfoxide (DMSO), methanol (MeOH), acetic acid (AcOH), isopropanol, ethylenediaminetetraacetic acid (EDTA), formaldehyde, paraformaldehyde, phenol and n-butanol were purchased from Merck (Darmstadt, Germany). Diethyl ether (Et2O), methyl tertiary butyl ether (MTBE), dichloromethane (DCM) and N, N-dimethylformamide (DMF) were from Scharlau Chemie (Barcelona, Spain), Caledon and Daejung were procured from Canada and South Korea. 9-fluorenylmethoxycarbonyl chloride (Fmoc)-amino acid derivatives and 2-chlorotrityl chloride (CTC) resin (loading: 1.08 mmol/g) were obtained from AAPPTec (Louisville, KY, USA). Dulbecco’s Roswell Park Memorial Institute medium (RPMI)-1640 medium and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY), Ppenicillin/streptomycin solution was brought from Applichem (Darmstadt, Germany) and trypan blue was obtained from Biosera (UK). RNase (DNase-Free) enzyme was purchased from Thermo-Fisher/Invitrogen (Waltham, MA). Cell Culture T-75 flask and Cell Culture 96 and 6-well plates were procured from Biofil (Canada) or SORFA (China). MCF-7 Cell line was obtained from Pasteur Institute (Tehran, Iran).
Methods
CPP synthesis
All peptides were synthesized manually by solid phase peptide synthesis (SPPS) method on CTC resin by Fmoc strategy using 60 ml innovative polypropylene syringes equipped with poly-tetrafluoroethylene (PTFE) filters, a Teflon stopcock and stopper [Citation13–16]. The Fmoc protected amino acids with different side chain protecting groups used as the following derivatives: Fmoc-Arg (Pbf)-OH, Fmoc-Trp (Boc)-OH, Fmoc-Lys (Boc)-OH, Fmoc-Gly-OH, Fmoc-Gln (Trt)-OH and Fmoc-Glu (OtBu)-OH. The resin was swollen in dry DCM for 1.5 h under dry nitrogen. The first amino acid was loaded on the resin at C-terminal and the resin was end-capped by adding 0.8 ml of MeOH for 1 g of the resin (0.2 ml for 250 mg), then washed with DCM (2 × 3 ml) and DMF (2 × 3 ml). The Fmoc protecting group was removed with 2 ml of 20–40% (v/v) piperidine/anhydrous DMF solution for about 1 h. The resin was then washed with DCM (2 × 3 ml) and DMF (2 × 3 ml), respectively. The effectiveness of the deprotection was monitored by testing for free amine groups with the ninhydrin test. Peptide derivatives were cleaved and deprotected by adding 10 ml of cleavage cocktail (TFA/H2O/Phenol/TIS: 88/5/5/2 v/v/v/v; ). The reaction vessel was stirred for about three hours, then the volume was reduced to one-third of the initial volume by N2 sparge. The crude peptides were precipitated by cold MTBE and an excess amount of ice-cold diethyl ether and harvested by centrifugation. Afterwards, the supernatant was decanted, the resulting solid peptides were dried and finally lyophilized by freeze dryer. CPP/polyglutamate nano-complexes were prepared at N:P ratios (the molar ratio of positively charged amino acids (e.g. Lysine (K) and Arginine (R)) in the peptide to negatively charged amino acids (e.g. Glutamic acid (E) in the synthesized polyglutamate sequence)) ranging from 1–10 by adding appropriate volumes of peptide solution to 200 µg Poly E (E8). Optimized ratio (1:5) was chosen and peptide nano-complexes were prepared. The complexes were incubated at room temperature for at least 10 min to allow the formation of the nano-complexes.
Figure 1. Scheme of solid phase peptide synthesis and final cleavage.
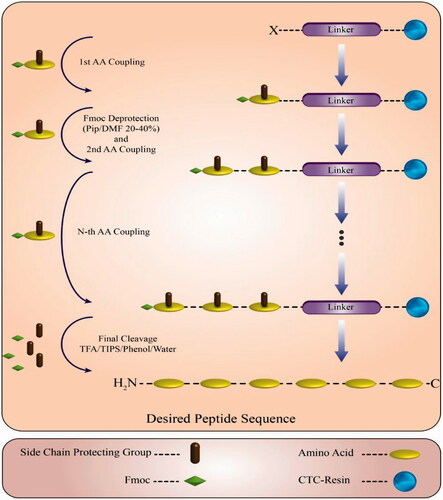
Modification of CPPs
An old well-known method to modify a peptide sequence or protein motif is N- and C-terminal modification to fulfil some ideas like resistance against exo-peptidases cleavage. N-terminal modification of peptides was done with 5(6)-FAM in the case of uptake studies. On the other hand, pyroglutamic acid was chosen to N-terminal modification of another group of peptides to use in cytotoxicity and cell viability assays. The amino protecting groups were removed by piperidine/DMF 20–40% Solution (2 ml). The resin was washed with DMF, then rinsed with DCM (3 × 2 ml) to remove DMF and dried by a vacuum pump. The desired amount of peptide-resin was weighed and poured into a reaction vessel. In a separate vessel, FAM was dissolved in DMF, then TBTU and DIPEA were added and mixed gently. The solution was added to slurry the resin and vessel was stirred for three hours at room temperature. A Kaiser test was performed to confirm end point. The resin was washed with DMF (3 × 4 ml), isopropanol (3 × 4 ml) and DCM (3 × 4 ml). On the other hand, pyroglutamic acid (pyrrolidone carboxylic acid; pGlu) was found as the N-terminal amino acid in various proteins and polypeptides. N-terminal modification with pyroglutamic acid was performed in cytotoxicity and cell viability assays. The coupling procedure was as same as FAM labeling procedure. C-terminal modification was done by applying memantine (Mem) in sequences which were used in uptake studies, also benzylamine (BA) was used in cytotoxicity and cell viability assays. Based on the dried amount of peptide-resin, the produced N-acetylated peptides were cleaved from the resin by treatmenting TFA in DCM (1–2%) with the final volume of 5 ml and each reaction vessel was stirred two hours at room temperature. Neutralization process was performed by calculating the amount of excess DIPEA, then C-amidation of the synthesized peptides was carried out using Mem or BA and HOBt in the presence of TBTU as a coupling reagent, using DIPEA as the base and DMF/DCM mixture as solvent at room temperature. The mixture in a three-neck glass flask was stirred by a magnetic stirrer overnight under vacuum.
Nuclear magnetic resonance (NMR) studies
1 H NMR (500.13 MHz) spectra was recorded on a Bruker spectrometer (Bruker, Ettlingen, Germany) at 500 MHz using TMS as an internal standard and DMSO/D2O as a solvent. For the NMR measurements, 5–10 mg of Mem-[WK]4-FAM and BA-QGR-[WR]3-pGlu sequences were dissolved in 1 ml of DMSO/D2O solution and experiments were carried out at 298 K.
Dynamic light scattering (DLS) and ζ-potential studies
DLS experiments were carried out on a Zetasizer Nano ZS apparatus (Malvern Instruments, Malvern, UK). Measurements were performed in a disposable 1 cm-path length acryl cuvette at 25 °C. Peptides were dissolved in DMSO and polyglutamate was added in 1:5 ratio (E8: CPP) to form nano-conjugates. CPPs and their nano-complexes (i.e. CPP/E8) were diluted to 1 ml with distilled water before the particle size and zeta potential measurements.
Scanning electron microscopy (SEM)
One day prior to the examination, samples were prepared by drop casting a 5 mM aqueous solution (20 μl) of peptides/nano-complexes dissolved in DMSO onto the aluminum foils. Samples were dried in air at room temperature, then were lyophilized to ensure complete drying, also were sputtered with an extra-thin layer of gold in high vacuum mode and finally analyzed by scanning electron microscopy (SEM, Mira3 FEG-SEM Tescan 5.0 kV) to study the surface morphology and sizes of peptides/nano-complexes.
Preparation of peptide-MTX conjugates and drug loading efficiency calculations
Peptide-MTX bio-conjugates were prepared by two different procedures in order to compare their efficacy and eligibility. The first one was the chemical conjugation of MTX to some peptide sequences in particular circumstances and another option was a physical linkage of CPPs with MTX. In the chemical conjugation procedure, the required amount of peptide-resin was washed with DCM to remove any remaining DMF (3 × 2 ml), then dried by a vacuum pump under argon flow. Drug conjugation to synthesized peptides was carried out using MTX in the presence of TBTU, DIPEA and DMF at room temperature. The mixture was stirred for about 48 h and a Kaiser test was performed to ensure coupling completion. The resin was filtered and 2 ml of the solution was poured into a micro-tube for spectrophotometry. The peptide-resin was suspended in cleavage cocktail (100 ml/mg of peptide-resin). The reaction vessel was shaken for two hours at room temperature. The resin was filtered and washed with a little additional freshly prepared cleavage cocktail. The combined filtrates in a three-neck glass flask were connected to rotavapor-R to reduce the final volume to one-third of the initial volume under dry nitrogen flow. The combined filtrates in a three-neck glass flask were cooled to 4 °C, then cold diethyl ether was added to precipitate the yellow crude peptide. The peptides containing cleavage cocktail and Et2O were centrifuged at 4000 rpm for three minutes couple of times followed by decantation in order to obtain the solid peptide precipitate. As a method which Cheng et al. [Citation17] introduced and due to presumptions about ionization of some basic residues of synthesized peptides and MTX in a particular pH, the physical linkage between peptides and drug molecules was planned. The desired amount of peptide was weighed and dissolved in a little amount of DMSO. Acetic acid solution (0.1%) was added to attain the final volume of about 1 ml. MTX was dissolved in 1 ml NaOH solution (10−4 M). Aforementioned solutions were mixed together and pH measurement was done carefully. Peptide-MTX conjugate was observed in Falcon tubes. After completion of chemical/physical linkage between each peptide sequence and MTX, the amount of drug loading was calculated by using UV-Vis Spectrophotometer (SHIMADZU, Japan) at λ = 365 for chemical conjugation and at λ = 306 for physical linkage. In physical linkage, the amount of drug was determined after centrifugation (4000 rpm, 10 min). For more accuracy, Amicon® Ultra-centrifugal filters were used in high speed ultra-centrifuge.
Cell culture
Cell viability and cytotoxicity assay. MCF-7 cell line was obtained from Pasteur institute (Tehran, Iran) and were grown in RPMI-1640 supplemented with 10% (v/v) fetal bovine serum ,100 U/ml penicillin and 100 mg/ml streptomycin in a humidified atmosphere at 37 ºC with 5% CO2. The cells were split and dispensed to 25 or 75 cm2 culture flasks every 2–3 days to preserve the exponential growth. One day prior to the assay, the cells were seeded into 96-well plates at a density of 1.5 × 104 cells/well and incubated for ∼16 h at 37 ºC with 5% CO2. Then, media was removed and different concentrations of peptides (25 and 50 µM) and peptide-MTX bio-conjugates (25and 50 nM) were added to each well and the plates were placed back in the incubator for an additional 24, 48 and 72 h, respectively. After definite times, peptide solutions were removed and each well was washed with PBS three times. Afterwards, 100 µl complete growth medium and 50 µl MTT were added to each well. The plates were incubated for 4 h at 37 ºC. The unreacted dye was discarded and DMSO/Sorenson buffer were added to each well to dissolve the purple formazan crystals formed by the living cells. The cells without treatment were used as a control. The absorbance at 570 nm of the solution in each well was read using a microplate reader (TECAN, Austria). Cell viability was calculated relative to control wells according to the EquationEquation (1)(1)
(1) :
(1)
(1)
where A is the absorbance of the control and B is the absorbance of the cells incubated with peptide and peptide-MTX bio-conjugates. Data were collected from at least three independent experiments performed in triplicates and analyzed by Graphpad prism 6 software.
Uptake studies
Live cell imaging. MCF-7 cells were seeded in 6-well plates (5 × 105 cells/well) and cultured for ∼20 h in serum-containing RPMI-1640 and incubated at 37 ºC with 5% CO2. The next day, stock solutions (100 μM) of FAM-labeled CPPs and their nano-complexes in antibiotic-free culture medium were diluted in medium to final concentrations (25 and 50 μM). The culture medium was removed and the peptide solutions were added to each well. After 1.5 h incubation at 37 °C, the cells were washed three times with PBS buffer. Later, the samples were stored protected from light at 4 °C until the plates were placed in a live cell imager apparatus and observed by Cytation 5 (Biotek, Winooski, VT) under bright field and FITC channels.
Confocal microscopy. MCF-7 cells were seeded on coverslips in a six-well plate (5 × 105 cells per well) and cultured for ∼22 h in serum-containing RPMI-1640 and incubated at 37 ºC with 5% CO2. The next day, medium was removed from the plate and the peptide solutions were added to each well. The cells were incubated for 2 h at 37 °C before the peptide solutions were removed. The cells were washed three times with PBS buffer and fixed with 4% paraformaldehyde for 20 min at room temperature. Afterwards, the cells were washed twice with PBS buffer and treated with RNase (DNase free) enzyme to cleave probably double strand molecules except DNA. Propidium iodide was added to each sample for 5 min at room temperature to stain the dead cells. The cells were washed twice with PBS and the coverslips were placed cell-side-down on cytoslides with 4 μL mounting medium. After drying, coverslips were fixed with colorless nail polish. Samples were stored protected from light at 4 °C until measurements using a confocal laser scanning microscope (CLSM) (TCS-SP5 II, Leica, Wetzlar, Germany) equipped with a 20 × and 63 × objective lens were carried out.
Fluorescent microscopy. MCF-7 cells were seeded on coverslips in six-well plates (5 × 105 cells/well) and were cultured for ∼18 h in serum-containing RPMI-1640 and incubated at 37 °C with 5% CO2. The next day, medium was removed and the peptide solutions were added to each well. After 1.5 h incubation at 37 °C, the cells were washed three times with PBS buffer and fixed with 3.7% formaldehyde for 10 min at room temperature Afterwards, the cells were washed with PBS buffer and samples were stored protected from light at 4 °C until the coverslips were placed cell-side-down on cytoslides and observed by an Olympus IX81 fluorescence microscope (Olympus Optical Co., Tokyo, Japan) under bright field and FITC channels.
Flow cytometry. MCF-7 cells were seeded in six-well plates (5 × 105 cells/well) and cultured for ∼20 h in serum-containing RPMI-1640 and incubated at 37 °C with 5% CO2. The next day, medium was removed and the cells were treated with peptide solutions. After 1.5 h incubation at 37 °C, the cells were washed three times with PBS. The cells were digested with 0.25% trypsin-EDTA (0.53 mM) for 5 min to remove any artificial cell surface association and to detect only intracellular uptake. The cells were centrifuged at 3000 rpm (600 g) for 5 min and washed with PBS, then centrifuged two more times. The cells were resuspended in PBS, transferred to fluorescence activated cells sorting (FACS) tubes and were subjected to fluorescence analysis on a FACS calibur flow cytometer using an FL1 filter. Live cells were gated by forwarding/side scattering (FSC and SSC) from a total of 10,000 events.
Results
All peptide sequences were synthesized manually by SPPS method on 2-chlorotrityl chloride resin by Fmoc strategy. To ensure completeness of each coupling and deprotection cycle, Kaiser Test was performed. Peptide modification was applied using desired compounds and several types of peptides were cleaved from the resin, precipitated, lyophilized and stored at −20 ºC (the synthesized peptides are listed in ). Morphology of peptides before and after interaction with polyglutamate was analyzed by SEM. There were two peptide sequences; the first selected unmodified peptide sequence (COOH-QGR-[WK]3-NH2) with its nano-complex (COOH-QGR-[WK]3-NH2/E8) and on the other hand, the second selected modified peptide sequence (BA-QGR-[WK]3-pGlu) with its nano-complex (BA-QGR-[WK]3-pGlu/E8) were studied by SEM. Before adding E8, both unmodified and modified peptides had spherical shaped structures and sizes of about 90 nm. However, the addition of E8 to unmodified peptide resulted in rod-shaped nanostructure and smaller sizes in comparison with peptide alone, the addition of E8 to modified peptides resulted in agglomerated and mixed sphere/rod-shaped nanostructures bigger than peptides alone. Zetasizer was used to measure the size of peptides as well as evaluate size distribution in peptides and their nano-complexes after interaction with polyglutamate. In our study, zeta potential range for unmodified peptide and its nano-complex were between −17.8 to 34.4 mV and −7.68 to 18.4 mV, respectively. On the other hand, zeta potential range for modified peptide and its nano-complex were 15.2 to 97.3 mV and 69.3 to 144 mV, respectively. 1 H NMR spectra were recorded to ensure that amidation was carried out.
Table 1. Synthesized peptide sequences.
Cytotoxicity of unmodified CPPs, modified CPPs, modified CPP-MTX bio-conjugates and unmodified CPP-MTX bio-conjugates (both chemical conjugation and physical linkage) was evaluated in MCF-7 cells. The cells were treated with peptides and their bio-conjugates in concentrations of 25 and 50 µM for 24, 48 and 72 h,respectively. In drug loaded CPPs, the cells were treated with peptides in concentrations of 25 and 50 nM, which were very lower than calculated IC50 concentration for free MTX in MCF-7 cells. Aiming to consolidate this part of the research program, the peptides were divided into several groups. The first group included eight sequences with pyroglutamic acid at N-terminus and benzylamine at C-terminus as well as five sequences without modification. The first subgroup in this group, comprised of modified peptides with benzylamine at C-terminus and pyroglutamic acid at N-terminus in order to assess the safety of modified sequences. It is apparent from that except BA-K10-pGlu at concentration of 50 µM, any remarkable cytotoxicity was not seen, but treatment with the sequences BA-[WR]4-pGlu and BA-R8-pGlu for 48 h at concentration of 25 µM showed increasing cell proliferation which is probably due to the stimulatory effect on the cell growth. The next subgroup comprised of five sequences without modification. After 48 h incubation, the cells treated with this group of peptides showed viability changes but any remarkable toxicity was not seen (). Considerable progress has been gained with regard to this group viability at a concentration of 25 µM, where the maximum cell viability was seen. The second group included five sequences physically linked to MTX and five sequences with chemical conjugated to the drug. The first subgroup in this group, comprised of modified peptides with benzylamine at C-terminus and pyroglutamic acid at N-terminus, which were physically linked with MTX. These bio-conjugates were produced based on self-assembly of poly-cationic peptides in the presence of anionic MTX with two negative charges (). The second subgroup comprised of unmodified peptides, physically linked to MTX as same as the first subgroup (). The third subgroup was related to peptides without modification which was chemically conjugated to MTX (). The cytotoxicity of free MTX was studied in the desired concentrations at 48 h (IC50= 316.346 ± 93.02 (29.4%) nM; ). It is apparent from that after 48 h incubation, the viability was dramatically decreased except for WRWQGRWRW sequence but after 48 h incubation, the viability was reduced in the case of modified peptides especially at the concentration of 50 µM. Among the five physically linked peptide-drug complexes, BA-QGR-[WK]3-pGlu and BA-WRWQGRWRW-pGlu displayed the highest cytotoxicity at 25 and 50 nM. The cytotoxicity of peptide-drug conjugates including QGR-[WK]3-MTX, COOH-WRWQGRWRW-MTX, COOH-[WR]4-MTX, COOH-[WR]3-QGR-MTX and COOH-QGR-[WR]3-MTX was assessed and compared to that of free MTX. The obtained data revealed that among these sequences, COOH-QGR-[WK]3-MTX and COOH-WRWQGRWRW-MTX had toxicity, therefore cell viability was slightly reduced. Drug loaded CPPs at concentrations of 25 and 50 nM except for some sequences especially COOH-[WR]3-QGR-MTX exhibited anti-proliferative activity compared to the free MTX. Free MTX at a concentration of 50 nM showed 82% cell viability. The cell viability value was reduced to 67 and 64% with COOH-WRWQGRWRW-MTX and COOH-QGR-[WK]3-MTX at a concentration of 25 nM, respectively.
Figure 2. Toxicity of (a) modified peptides and (b) unmodified peptides to MCF-7 cells and (c) drug concentrations to calculate IC50. The cells were incubated for 24, 48 and 72 h at 37 ºC with concentrations of 25 and 50 µM, respectively for cell viability assessment by MTT. (i) (P1: BA-[WR]4-pGlu, P2: BA-QGR-[WK]3-pGlu, P3: BA-QGR-[WR]3-pGlu, P4: BA-[WR]3-QGR-pGlu, P5: BA-WRWQGRWRW-pGlu, P6: BA-R8-pGlu, P7: BA-R10-pGlu, P8: BA-K10-pGlu) and (ii) (P1: COOH-WRWQGRWRW-NH2, P2: COOH-QGR-[WR]3-NH2, P3: COOH-QGR-[WK]3-NH2, P4: COOH-[WR]3-QGR-NH2, P5: COOH-[WR]4-NH2).
![Figure 2. Toxicity of (a) modified peptides and (b) unmodified peptides to MCF-7 cells and (c) drug concentrations to calculate IC50. The cells were incubated for 24, 48 and 72 h at 37 ºC with concentrations of 25 and 50 µM, respectively for cell viability assessment by MTT. (i) (P1: BA-[WR]4-pGlu, P2: BA-QGR-[WK]3-pGlu, P3: BA-QGR-[WR]3-pGlu, P4: BA-[WR]3-QGR-pGlu, P5: BA-WRWQGRWRW-pGlu, P6: BA-R8-pGlu, P7: BA-R10-pGlu, P8: BA-K10-pGlu) and (ii) (P1: COOH-WRWQGRWRW-NH2, P2: COOH-QGR-[WR]3-NH2, P3: COOH-QGR-[WK]3-NH2, P4: COOH-[WR]3-QGR-NH2, P5: COOH-[WR]4-NH2).](/cms/asset/95b1d071-5370-498e-b1f8-4fcf8c14f4c5/ianb_a_1414823_f0002_c.jpg)
Figure 3. Cytotoxicity of (a) unmodified and (b) modified peptides interaction with MTX (physical linkage) and MCF-7 cells; (c) Cytotoxicity of unmodified peptides conjugated with MTX. The cells were incubated for 24, 48 and 72 h at 37 °C with concentrations of 25 and 50 nM, respectively for cell viability assessment by MTT. (i) (BA-WRWQGRWRW-pGlu, BA-QGR-[WK]3-pGlu), (ii) (COOH-QGR-[WK]3-NH2, COOH-[WR]3-QGR-NH2, COOH-WRWQGRWRW-NH2) and (iii) (COOH-QGR-[WK]3-MTX, COOH-WRWQGRWRW-MTX, COOH-[WR]4-MTX, COOH-[WR]3-QGR-MTX, COOH-QGR-[WR]3-MTX).
![Figure 3. Cytotoxicity of (a) unmodified and (b) modified peptides interaction with MTX (physical linkage) and MCF-7 cells; (c) Cytotoxicity of unmodified peptides conjugated with MTX. The cells were incubated for 24, 48 and 72 h at 37 °C with concentrations of 25 and 50 nM, respectively for cell viability assessment by MTT. (i) (BA-WRWQGRWRW-pGlu, BA-QGR-[WK]3-pGlu), (ii) (COOH-QGR-[WK]3-NH2, COOH-[WR]3-QGR-NH2, COOH-WRWQGRWRW-NH2) and (iii) (COOH-QGR-[WK]3-MTX, COOH-WRWQGRWRW-MTX, COOH-[WR]4-MTX, COOH-[WR]3-QGR-MTX, COOH-QGR-[WR]3-MTX).](/cms/asset/be82e282-fafc-44fa-9190-4c214e74e763/ianb_a_1414823_f0003_c.jpg)
Cellular uptake studies for desired CPPs were designed in three different steps. In the first step, two sequences (R8 and K8) conjugated with 5(6)-FAM at N-terminus and their nano-complexes in concentrations of 25 and 50 µM were prepared (). Our findings based on FACS flow cytometry would seem to show that polyarginine had higher uptake efficacy in comparison with polylysine. Our study has highlighted that adding E8 did not lead to dramatic changes in uptake of polyarginine. Moreover, polylysine demonstrated higher internalization in comparison with polylysine nano-complex in both concentrations (25 and 50 µM). There was not remarkable difference in internalization between R8-FAM and its nano-complex. In comparison with unstained samples, FAM-labeled R8 and its nano-complex had appropriate uptake. On the other hand, K8-FAM and its nano-complex could not enter the cells efficiently. At the concentration of 25 µM, cellular uptake for FAM-labeled K8 was higher in contrast with the concentration of 50 µM. Furthermore, FAM-labeled K8/E8 nano-complex demonstrated lower positive cells in comparison with peptide alone, therefore interaction between E8 and polylysine did not affect its uptake. In the second step, three sequences conjugated with 5(6)-FAM at N-terminus, Mem at C-terminus and their nano-complexes with concentrations of 25 and 50 µM were chosen (). Our findings based on FACS flow cytometry data illustrated a dramatically change in the internalization due to their modification on N- and C-termini in comparison with polyarginine and polylysine. In this step, FAM-labeled modified peptides including Mem-QGR-[WK]3-FAM, Mem-QGR-[WR]3-FAM, Mem-WRWQGRWRW-FAM and their nano-complexes at concentrations of 25 and 50 µM were used. There was not remarkable difference between Mem-QGR-[WK]3-FAM and its nano-complex in cellular uptake. In comparison with unstained samples, FAM-labeled modified peptides had cellular internalization, while in the internalization of Mem-QGR-[WR]3-FAM, Mem-WRWQGRWRW-FAM and their nano-complexes, a determined decline was observed. Whereas internalization of the latter peptide was reduced to less than 60%, uptake of this peptide was similar to its nano-complex. These modified peptides and their nano-complexes could not enter the cells efficiently in comparison with unmodified peptides. In order to prove our findings, two similar sequences and their nano-complexes were used (R8-FAM and Mem-R8-FAM) in the third step. Either sequences were conjugated with 5(6)-FAM at N-terminus, but one of them was end-capped with Mem at C-terminus whereas the other one was unmodified. Polyarginine was chosen due to its great ability to penetrate the cell membrane [Citation18]. Unmodified polyarginine and its nano-complex in both concentrations had appropriate uptake. Different concentrations of this peptide exhibited low uptake in comparison with unmodified peptide and its nano-complex, whereas 25 µM concentration of modified peptide showed various results. Fluorescent images obtained by fluorescent microscopy, live cell imaging system () and CLSM () showed that interaction between E8 and any desired peptides, caused a slight reduction in internalization of nano-complexes into the cells nucleus. The images of FAM-labeled peptides exhibit a dramatic reduction in cellular uptake with C-terminal modification. Although unmodified polyarginine had the highest uptake at a concentration of 50 µM (), its nano-complex showed the highest uptake at 25 µM (). Generally, arginine containing peptides had better uptake than lysine or tryptophan-containing peptides. Arginine containing peptides were mostly distributed around the cells nucleus and it demonstrates homogeneous staining through cellular structures that were morphologically identified as the cell nucleus and nucleoli. Translocation of modified peptides was affected by their C-terminal modification and their uptake was dramatically decreased ( and ). Most of the modified peptides in fluorescent microscopy were distributed around the cell membrane ( and ). In conclusion, fluorescent microscopy data belong to all of the peptides and their conjugates demonstrate that the uptake of unmodified peptides especially octa-arginine is better than modified peptides. However, polyglutamate interaction with peptides probably decreases translocation of CPPs into the cell’s nucleus and intracellular delivery of these peptides could be influenced in the presence of E8.
Figure 4. Live cell images. Row (a) Bright field (40×): A (R8-FAM, 25 µM), D (R8-FAM/E8, 25 µM), G (Mem-R8-FAM, 50 µM) and J (Mem-R8-FAM/E8, 50 µM). Row (b) Fluorescent (40×): B (R8-FAM, 25 µM), E (R8-FAM/E8, 25 µM), H (Mem-R8-FAM, 50 µM) and K (Mem-R8-FAM/E8, 50 µM). Row (c) Merged: C (R8-FAM, 25 µM), F (R8-FAM/E8, 25 µM), I (Mem-R8-FAM, 50 µM) and L (Mem-R8-FAM/E8, 50 µM).
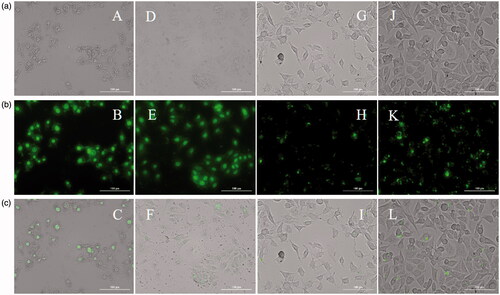
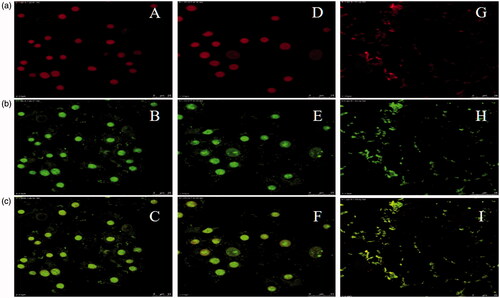
Figure 5. CLSM images. Row (a) Propidium iodide (63×): A (R8-FAM, 50 µM), D (R8-FAM, 25 µM) and G (Mem-R8-FAM/E8, 50 µM). Row (b) Fluorescent (63×): B (R8-FAM, 50 µM), E (R8-FAM, 25 µM) and H (Mem-R8-FAM/E8, 50 µM). Row (c) Merged: C (R8-FAM, 50 µM), F (R8-FAM, 25 µM) and I (Mem-R8-FAM/E8, 50 µM).
Discussion
There is a major hurdle in the case of efficient passage through biological membranes for drug delivery to cancer cells. Without good translocation of drugs, high dose administration and side effects in different tissues, will be expected to that drug. Due to this, application of smart drug delivery systems like CPPs, is a reliable option for efficient drug delivery of suitable quantity of drug to the specific tissues. In this study, two main specific goals including: a) effects of modifications on internalization of synthesized peptide sequences and b) loading efficiency of various strategies besides toxic effects of both modified CPPs and peptide-MTX conjugates on the cells, were designed. As a routine, the surface morphology, particle size, size/diameter distribution and average diameter of the synthesized peptides were assessed by the SEM and DLS. Before adding E8, COOH-QGR-[WK]3-NH2 had spherical shaped structures and a size of about 90–100 nm. Polyglutamate addition, caused changes in the size and morphology of peptide. Actually, E8 addition to COOH-QGR-[WK]3-NH2, resulted in rod-shaped nanostructure, which is related to polyglutamate negative charge culminating in electrostatic interactions with cationic peptides to form rod-shaped nanostructures and bigger average size in comparison with peptides alone. The modified peptide (i.e. BA-QGR-[WK]3-pGlu) had spherical shaped structures and the size of 90 nm before E8 addition, as same as unmodified peptide. Surprisingly, adding E8 to modified peptides (BA-QGR-[WK]3-pGlu/E8) resulted in agglomerated and mixed sphere/rod-shaped nanostructures bigger than peptide alone, also unmodified nano-complexes which were nearly unpredictable and unjustifiable in the presence of polyglutamate. The presumed reason responsible for this circumstance might be steric hindrance of modified peptides. Peptides formed rod-shaped structures in smaller sizes. SEM images and zetasizer analysis reported the size from 32.67 to 372 nm for unmodified peptide and its nano-complex, whereas sizes from 21.04 to 955.4 nm were observed in modified peptide and its nano-complex. Based on ζ-potential studies, our findings would seem to imply that modified peptides were more stable than unmodified peptides in suspension which prevent aggregation in standard condition due to the surface charge and steric hindrance. The results point to the likelihood that polyglutamate addition decreases the average zeta potential in both unmodified and modified peptides. These results, justify agglomeration and mixed sphere/rod-shape of nanostructures in the presence of polyglutamate. Generally, the self-assembled peptides nano-complexes can spontaneously form in an aqueous solution or triggered by pH, ionic strength, enzyme or light, temperature, driving by non-covalent interactions, such as hydrophobic interaction, hydrogen bonding and π–π stacking [Citation19]. Nowadays, there are two main strategies to prepare drug-loaded nanomaterials based on self-assembling amphiphilic peptides. The first strategy is physical linkage (encapsulation) between drug and the self-assembled nanomaterials like peptides. For example, Grigoriou et al. [Citation20] has synthesized a self-assembled nanofiber of peptide amphiphile as the matrix to encapsulate camptothecin, which has shown good performance in in vivo tumor growth inhibition. Liu et al. [Citation21] has also prepared self-assembled nanofibers of D-amino acid-based peptides to encapsulate 10-hydroxycamptothecin for drug delivery. The second strategy is the covalently linkage of the peptide with drug, affording peptide-drug bio-conjugates [Citation22]. The peptide-drug bio-conjugates are often able to self-assemble into nanostructures and possess higher drug loading efficiency as compared to the drug-encapsulated peptide nanomaterials. For instance, a taxol-peptide conjugate consisting of taxol, β-sheet forming peptide and a cleavable linker was designed and synthesized by Lin et al. [Citation23], which could self-assembled into supramolecular filaments with a drug loading content of more than 41%. Moreover, both Gao et al. [Citation24] and Wang et al. [Citation25] have revealed that taxol conjugation with peptides has the ability of self-assembling. The self-assembly capacity of peptides allows the taxol-peptide conjugates to form nano-fibrous hydrogels, which serve as carriers and delivered components. In the current study, we used some amphiphilic peptides with the ability of self-assembly in order to deliver MTX to the cancer cells. MTX is one of the most effective and commonly used therapeutics in cancer chemotherapy. MTX entrance to the cells is handled by the RFC1 and once inside the cell, the enzyme FPGS lead to polyglutamylation [Citation26]. Polyglutamylation of the drug, results in its retention within the cell, therefore binding to its target enzyme (i.e. DHFR) will increase dramatically. The effectiveness of MTX is often hampered by its hydrophilicity which results in poorly diffusion through cell membranes, also development of drug resistance mechanisms such as inhibition of active transport involving the cell folate receptor that mostly internalizes MTX. In order to bypass drug resistance and still retain cytotoxic activity, several strategies have been proposed. One of these strategies consists of chemical modification of MTX to improve lipophilicity of drug. Chemical conjugation of MTX to amphiphilic CPPs is responsible for uptake enhancement due to semi lipophilic characteristics of desired peptides, besides their special ability to cross biological barriers. On the other hand, due to the possibility of charge interaction between MTX and some positively charged residues of synthesized peptides, we chose this drug in the second part of our study to compare with chemically conjugation of MTX with synthesized peptides. Linking cargoes to peptides is an effective procedure to overcome extracellular barriers as well as for achieving intracellular delivery of cargos before they are ultimately trafficked to lysosomes [Citation27]. Covalent coupling is commonly used for conjugation of drugs, fluorescent labels, antibody fragments and targeting moieties to a peptide sequence [Citation28]. Non-covalent complexes are most widely used for delivery of oligonucleotides such as splice correcting oligonucleotides (SCO), siRNA and plasmids [Citation29–31]. The main drawback of non-covalent interaction is its weak attraction which leads to uncontrolled release of cargo before reaching its target in some cases. In current study, both linking methods were used in order to compare different efficient drug delivery methods to targeted cells. MTX was negatively charged with a pH of 7.4 and pKa of 3.8, 4.8 and 5.6, respectively which caused an electrostatic attraction between MTX and the positively charged amino moiety of basic residues such as arginine (α-carboxylic acid (pKa1 = 2.01), α-amino (pka2 = 9.04) and side chain (pka3 = 12.48)) and lysine (α-carboxylic acid (pKa1 = 2.18), α-amino (pka2 = 8.95) and side chain (pka3 = 10.53)) in peptides. The hydrogen bonding might be present between the amino groups and -COO- groups in MTX molecules with the cationic amine (-NH2) of arginine or lysine in synthesized peptides at pH 6–6.5. Therefore large amount of the MTX molecules could be attached into the peptides by physical linkage due to ionization of desired moieties in MTX/peptides at specific pH value. Consequently, the higher the pH of the solution, the more negative charges are on the MTX and the stronger attraction exists between the negatively charged MTX and the positively charged carriers. The selection of these segments in carrier and MTX, as anticancer drug, was to achieve the larger drug loading and pH-dependent drug release. It is preferable that MTX releases at mild acidic pH with its release being prohibited considerably at physiological pH (7.4). In this way, the normal cells will be protected from toxic side effect of MTX and only the cancer cells will be targeted due to the effectiveness of pH-dependent physical linkages between MTX and peptide bio-conjugates. One of the advantages of using CPPs for therapeutic and diagnostic delivery into the cells is the lack (or less) of toxicity in comparison with the other carriers, such as liposomes, polymers, etc. However, one of the problems using exogenous peptides for delivery of various molecules to mammalian cells is their immunogenicity. Due to highly cationic nature of some CPPs, it may be proposed that they could be as toxic as other biopolymers. Therefore several groups evaluated different CPPS in vitro and in vivo to ensure about their cellular toxicity. There was no toxicity observed for different cationic peptides (namely Tat, Antp, Rev and VP22) in Hela or Jurkat cells with up to 20–30 μM concentrations [Citation32]. Zhang et al. [Citation33] reported that the C-terminal conjugated Tat peptide showed higher cellular uptake in relative to the N-terminal analogue. They also showed that the doxorubicin conjugation to C-terminal partially overcame the multi-drug resistance of cervical cancer cells, while the N-terminal conjugate showed no significant improvement in cytotoxicity when compared with free doxorubicin. We have obtained comprehensive results proving that, N- and C-terminal modification at least with mentioned process do not show any great changes in cytotoxicity of synthesized sequences. There is a strong probability that, peptide modification has no toxic effects, therefore safety of these synthesize amphiphilic peptides has been approved. In the case of treatment with the sequences BA-[WR]4-pGlu and BA-R8-pGlu at 48 h incubation time, concentration of 25 µM showed increase in cell proliferation which is probably due to the stimulatory effect on the cell growth. Consequently, in the case of MTX delivery to cancer cells, we believe strongly that toxic effects of MTX were increased in the presence of peptides compared to MTX alone. Obviously drug conjugation may have more loading efficiency, but physical entrapment shows more efficacy in an efficient drug delivery.
For CPPs, it is widely accepted that the interaction of positively charged peptides (e.g. polyarginine, etc.) with negatively charged cell membrane components like heparan sulfate and phospholipids is prerequisite for cell penetration [Citation34]. Generally, two major mechanisms including non-endocytotic and endocytotic mechanism have been proposed depending on CPPs own features (e.g. cationic residues, concentration, etc.), the cargo molecule (size and physicochemical nature of cargo), the treated cell type and the membrane components (i.e. heparan sulfate and phospholipids) [Citation35]. Hirose et al. [Citation36] showed that conjugation of hydrophobic small molecules to arginine-rich CPPs resulted in the indirect translocation through the cell membrane at sites where the peptides formed unique “particle-like” structures composed of multiple vesicles on the plasma membrane. We investigated the effect of N- and C-terminal modification on uptake of CPPs into live cells with fluorescently labeled peptides utilizing FACS flow cytometry, Fluorescent microscopy, live cell imaging and confocal microscopy to quantify cellular uptake of the peptides. For the study by fluorescent microscopy, the cells were fixed to reveal nuclear distribution of peptides, qualitatively. However, flow cytometry could not distinguish between inter-(i.e. membrane-bounded) and intracellular fluorescence. Hence cell treatment and digestion with 0.25% trypsin/EDTA (0.53 mM) for 5 min was done to remove any artificial cell surface association so that only intracellular uptake could be detected. On the other hand, unfixed cells were used to quantify cell internalization besides cytoplasmic distribution. Different studies suggest the presence of at least eight positive charges for an efficient internalization of cationic or amphiphilic CPPs [Citation11]. Octa-arginine (R8) is a sequence that we used to evaluate cell internalization efficacy in the presence of Mem at C-terminus and 5(6)-FAM at N-terminus in comparison with no modification at C-terminus of the analogue peptide. Conjugating a hydrophobic peptide sequence FFLIPKG to either terminal of a homogeneous oligomer of arginine like R8, showed a significant effect on cell internalization ability of desired peptide [Citation37]. Due to this findings, we decided to use several positively charge basic residues as well as tryptophan in some sequences for better evaluation of interaction with cell membrane and internalization. Our findings seemed to show that polyarginine had higher uptake efficacy in comparison with polylysine. Peptides containing arginine and lysine interact with anionic/acidic components of the cell membrane in a receptor-independent manner [Citation13–15]. Polycationic peptides like polylysine did not show efficient internalization as same as polyarginine in the study conducted by confocal microscopy. Studies have provided further evidence that polyglutamate (E8) addition to peptides resulted in improving cellular uptake [Citation38,Citation39], however our study has highlighted that adding E8 did not lead to dramatic change in uptake of polyarginine. Moreover, polylysine demonstrated higher internalization in comparison with its nano-complex in both concentrations (25 and 50 µM). Our findings illustrated that internalization of N- and C-terminally modified tryptophan-containing peptides () dramatically changed due to their modifications on both termini in comparison with aforementioned polyarginine and polylysine sequences. Even though the presence of tryptophan makes changes in uptake of some CPPs, herein end-capping resulted in cellular uptake decrease. To investigate accuracy of previously discussed data, we decided to use a unique sequence (Herein; R8) and its counterpart without C-terminal modification (). We have demonstrated that some synthesized peptides in this research, strongly interacts with membrane lipids, mainly through its Trp-Arg-rich domains, that was shown to be critical for insertion of the peptide into the membrane. Mandal et al. [Citation40] showed the amphipathic nature of [WR]n (n = 4,5), also interactions of arginine and tryptophan residues with the corresponding negatively charged phospholipids and hydrophobic residues in the lipid bilayer may have played a role for initial entry into the cell membrane. Hydrophobic interactions between tryptophan residues and the lipids result in possible distortion of the outer phospholipid monolayer, thus leading to peptide internalization and enhanced cellular uptake of the cargo. Hence, a dramatic contradiction is seen here, which can be attributed to specific conformational change in secondary structure of synthesized peptides (α-helix and β-sheet). We have obtained comprehensive results proving that peptide N- and C-terminal modification will result in reducing cell internalization and direct site localization inside a cell near the nucleus as well.
Conclusions
In conclusion, the coupling of anticancer drug to CPPs provides a powerful tool in drug delivery with increased solubility, intracellular uptake and improved bio-distribution as well as pharmacokinetic profiles. We have obtained satisfactory results demonstrating that, the use of CPPs with a chemotherapeutic agent could lead to improved therapy. Some modified peptides interacted with MTX which included BA-QGR-[WK]3-pGlu and BA-WRWQGRWRW-pGlu, that could successfully increase toxic effects of the drug in comparison with their unmodified counterparts. The cytotoxicity of modified peptides which were physically linked to MTX, showed higher cytotoxicity than both physically linked unmodified peptides and chemically conjugated peptides with MTX, even though they had lower loading. Also, we have obtained comprehensive results proving that peptide end-capping will result in reduced internalization as well as direct site localization inside a cell. In contrary, increased drug toxic effects was seen in the case of drug delivery using these sequences. Uptake studies also approved the negative effects of our designed peptides in cell internalization, by increasing concentration or E8 addition. Finally, a number of potential shortfalls need to be considered. Firstly, the present study has only investigated the effects of unmodified peptides as well as N- and C-terminal modifications without peptide purification which can be responsible for some unpredictable errors and secondly, this study has only investigated all these procedures in vitro. Consequently, to obtain more accurate and reliable results, we recommend doing this research in vivo. Also, peptide stability assessment procedures should be considered before modification of CPPs.
Disclosure of interest
The authors report no conflicts of interest.
Acknowledgements
This article is a part of a dissertation (No. 93/2–11/3) submitted by Mehdi Soleymani-Goloujeh for MSc degree in Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran.
Additional information
Funding
Related Research Data
References
- Foerg C, Weller KM, Rechsteiner H, et al. Metabolic cleavage and translocation efficiency of selected cell penetrating peptides: a comparative study with epithelial cell cultures. AAPS J. 2008;10:349–359.
- Rennert R, Wespe C, Beck-Sickinger AG, et al. Developing novel hCT derived cell-penetrating peptides with improved metabolic stability. Biochim Biophys Acta. 2006;1758:347–354.
- Najafi-Hajivar S, Zakeri-Milani P, Mohammadi H, et al. Overview on experimental models of interactions between nanoparticles and the immune system. Biomed Pharmacother. 2016;83:1365–1378.
- Niazi M, Zakeri-Milani P, Najafi Hajivar S, et al. Nano-based strategies to overcome p-glycoprotein-mediated drug resistance. Expert Opin Drug Metab Toxicol. 2016;12:1021–1033.
- Jin C, Bai L, Lin L, et al. Paclitaxel-loaded nanoparticles decorated with bivalent fragment HAb18 F(ab’)2 and cell penetrating peptide for improved therapeutic effect on hepatocellular carcinoma. Artif Cells Nanomed Biotechnol. 2017 [cited Aug 4]; [9 p.]. DOI:https://doi.org/10.1080/21691401.2017.1360325
- Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587:1693–1702.
- Tang L, Persky AM, Hochhaus G, et al. Pharmacokinetic aspects of biotechnology products. J Pharm Sci. 2004;93:2184–2204.
- Elmquist A, Langel U. In vitro uptake and stability study of pVEC and its all-D analog. Biol Chem. 2003;384:387–393.
- Sato AK, Viswanathan M, Kent RB, et al. Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol. 2006;17:638–642.
- Rydberg HA, Matson M, Amand HL, et al. Effects of tryptophan content and backbone spacing on the uptake efficiency of cell-penetrating peptides. Biochemistry. 2012;51:5531–5539.
- Futaki S, Suzuki T, Ohashi W, et al. Arginine-rich peptides: an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840.
- Vila A, Sanchez A, Janes K, et al. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm. 2004;57:123–131.
- Farkhani SM, Johari-Ahar M, Zakeri-Milani P, et al. Enhanced cellular internalization of CdTe quantum dots mediated by arginine- and tryptophan-rich cell-penetrating peptides as efficient carriers. Artif Cells Nanomed Biotechnol. 2016;44:1424–1428.
- Mussa Farkhani S, Asoudeh Fard A, Zakeri-Milani P, et al. Enhancing antitumor activity of silver nanoparticles by modification with cell-penetrating peptides. Artif Cells Nanomed Biotechnol. 2016;45:1029–1035.
- Zakeri-Milani P, Mussa Farkhani S, Shirani A, et al. Cellular uptake and anti-tumor activity of gemcitabine conjugated with new amphiphilic cell penetrating peptides. EXCLI Journal. 2017;16:650–662.
- Mohammadi S, Zakeri-Milani P, Golkar N, et al. Synthesis and cellular characterization of various nano-assemblies of cell penetrating peptide-epirubicin-polyglutamate conjugates for the enhancement of antitumor activity. Artif Cells Nanomed Biotechnol. 2017 [cited Sep 21]; [14 p.]. DOI:https://doi.org/10.1080/21691401.2017.1379016
- Cheng H, Cheng YJ, Bhasin S, et al. Complementary hydrogen bonding interaction triggered co-assembly of an amphiphilic peptide and an anti-tumor drug. Chem Commun. 2015;51:6936–6939.
- Wang H, Sun M, Li D, et al. Redox sensitive PEG controlled octaarginine and targeting peptide co-modified nanostructured lipid carriers for enhanced tumour penetrating and targeting in vitro and in vivo. Artif Cells Nanomed Biotechnol. 2017 [cited Mar 31]; [10 p.]. DOI:https://doi.org/10.1080/21691401.2017.1307214
- Grigoriou S, Johnson EK, Chen L, et al. Dipeptide hydrogel formation triggered by boronic acid–sugar recognition. Soft Matter. 2012;8:6788–6791.
- Soukasene S, Toft DJ, Moyer TJ, et al. Antitumor activity of peptide amphiphile nanofiber-encapsulated camptothecin. ACS Nano. 2011;5:9113–9121.
- Liu Y, Ran R, Chen J, et al. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials. 2014;35:4835–4847.
- Wang H, Yang Z. Molecular hydrogels of hydrophobic compounds: a novel self-delivery system for anti-cancer drugs. Soft Matter. 2012;8:2344–2347.
- Lin R, Cheetham AG, Zhang P, et al. Supramolecular filaments containing a fixed 41% paclitaxel loading. Chem Commun. 2013;49:4968–4970.
- Gao Y, Kuang Y, Guo ZF, et al. Enzyme-instructed molecular self-assembly confers nanofibers and a supramolecular hydrogel of taxol derivative. J Am Chem Soc. 2009;131:13576–13577.
- Wang H, Wei J, Yang C, et al. The inhibition of tumor growth and metastasis by self-assembled nanofibers of taxol. Biomaterials. 2012;33:5848–5853.
- Volk EL, Rohde K, Rhee M, et al. Methotrexate cross-resistance in a mitoxantrone-selected multidrug-resistant MCF7 breast cancer cell line is attributable to enhanced energy-dependent drug efflux. Cancer Res. 2000;60:3514–3521.
- Park JS, Han TH, Lee KY, et al. N-acetyl histidine-conjugated glycol chitosan self-assembled nanoparticles for intracytoplasmic delivery of drugs: endocytosis, exocytosis and drug release. J Control Release. 2006;115:37–45.
- Stewart KM, Horton KL, Kelley SO. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org Biomol Chem. 2008;6:2242–2255.
- Mäe M, Andaloussi SE, Lundin P, et al. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J Control Release. 2009;134:221–227.
- Singh A, Trivedi P, Jain NK. Advances in siRNA delivery in cancer therapy. Artif Cells Nanomed Biotechnol. 2017 [cited Apr 19]. [10 p.]. DOI:https://doi.org/10.1080/21691401.2017.1307210
- Prabha S, Vyas R, Gupta N, et al. RNA interference technology with emphasis on delivery vehicles-prospects and limitations. Artif Cells Nanomed Biotechnol. 2016;44:1391–1399.
- Sugita T, Yoshikawa T, Mukai Y, et al. Comparative study on transduction and toxicity of protein transduction domains. Br J Pharmacol. 2008;153:1143–1152.
- Zhang P, Cheetham AG, Lock LL, et al. Cellular uptake and cytotoxicity of drug-peptide conjugates regulated by conjugation site. Bioconjug Chem. 2013;24:604–613.
- Ferrari A, Pellegrini V, Arcangeli C, et al. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther. 2003;8:284–294.
- Madani F, Lindberg S, Langel Ü, et al. Mechanisms of cellular uptake of cell-penetrating peptides. Biophys J. 2011;2011:414729. Available from: https://www.hindawi.com/archive/2011/414729/cta/
- Hirose H, Takeuchi T, Osakada H, et al. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Molec Ther. 2012;20:984–993.
- Takayama K, Nakase I, Michiue H, et al. Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas). J Control Release. 2009;138:128–133.
- Mohammadi S, Mojarrad JS, Zakeri-Milani P, et al. Synthesis and in vitro evaluation of amphiphilic peptides and their nanostructured conjugates. Adv Pharm Bull. 2015;5:41.
- Farkhani SM, Shirani A, Mohammadi S, et al. Effect of poly-glutamate on uptake efficiency and cytotoxicity of cell penetrating peptides. IET Nanobiotechnol. 2016;10:87–95.
- Mandal D, Nasrolahi Shirazi A, Parang K. Cell‐penetrating homochiral cyclic peptides as nuclear‐targeting molecular transporters. Angew Chem Int Ed. 2011;50:9633–9637.