
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
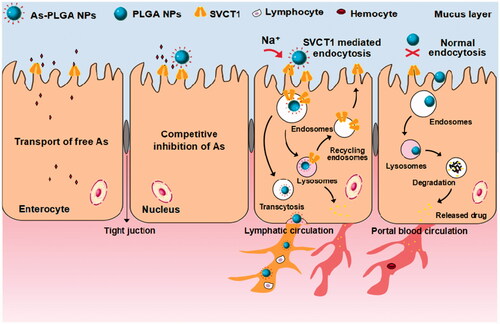
Oral administration is the most convenient route for most patients. However, many therapeutics in clinical applications are limited due to the poor solubility and permeability as well as low stability in the intestinal tract. It still remains a significant challenge of how to improve the oral drug bioavailability. In this study, we have designed functional ascorbate-conjugated NPs (As-PLGA NPs) to offer the interaction with sodium-dependent vitamin C transporter 1 (SVCT1) on the epithelial cells. Cellular uptake study indicated that conjugation of 20% ascorbate to the surface of PLGA NPs might achieve a maximum efficacy in Caco-2 cells. Besides, the majority of As-PLGA NPs were predominantly internalized into cells via caveolae-mediated pathway and thereby circumnavigated the lysosomal compartment. Furthermore, the competitive inhibition test and Na+-dependent study provide strong evidence that SVCT1 was involved in the internalization of As-PLGA NPs. Finally, the biodistribution and perfusion study demonstrated that As-PLGA NPs accumulated in the villi and penetrated to the basolateral side, thus significantly enhanced the intestinal absorption. In summary, it showed that SVCT1 expressed in the apical side of epithelial cells might be conceivably exploited as a potential target for oral delivery of therapeutic drug-loaded pharmaceutical nanocarriers.
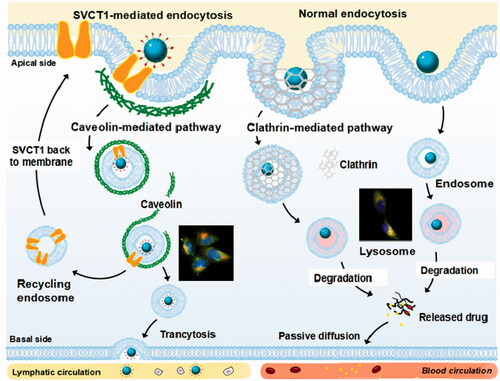
By means of the interaction of SVCT1 and ascorbate on the surface of PLGA NPs, the nanoparticles and transporters accumulated in coated pits, internalizing as ligand-transporter complexes and switched the subcellular sorting of NPs.
Graphical Abstract
Introduction
Oral delivery of anticancer agents is preferred by most patients for its convenience, cost-effectiveness, and favourable adaptability. However, the low water-solubility and poor permeability of therapeutics across the intestinal sites may lead to limited oral bioavailability and thus make it a challenging goal to achieve the therapeutically acceptable level. Consequently, designing and formulating a novel drug delivery system to improve the oral absorption through the gastrointestinal (GI) tract requires to be innovatively and practically investigated.
The solute carrier family (SLC) transporters located at the apical side of the intestinal epithelial cells primarily contribute to the influx of a wide variety of relevant substrates across biomembranes [Citation1–3]. In recent years, SLCs are attracting attention because they mediated nutrient–drug interactions. There are new approaches focusing on the coupling of active drugs with natural substrates to synthesize prodrugs by using the absorptive characteristics of relevant transporters. Compared with the parent drugs, the prodrugs might gain several-fold enhanced oral bioavailability due to their recognition by one or more of these transporters in the GI tract. By taking advantage of the activity of intestinal transporters, like peptide transporter 1 (PepT1, SLC15A1), monocarboxylate transporter 1 (MCT1, SLC16A1), sodium-dependent multivitamin transporter (SMVT) and so on, the systemic exposure of orally administrated parent drugs had been significantly increased [Citation4–7].
Moreover, transporter-assistant nanoparticles (NPs) have also made some achievements in the past few years. NPs enhance efficacy of drugs by improving solubility, stability, release circulation time and passive targeting [Citation8–10]. Additionally, together with the specific modification of functional groups with high selectivity holds great prospects with the promise that all benefits collectively can make a significant synergistic effect [Citation11–13]. Research work has indicated that NPs modified with endogenous substrates targeted to the transporters on the surface of certain tumour cells are proposed to facilitate the cellular uptake and targeting efficiency. This strategy involves facilitative glucose transporter 1 (GLUT1and SLC2A1), Na+/Cl−-coupled amino acid transporter (ATB0,+ and SLC6A14), facilitative amino acid exchanger large neutral amino acid transporter (LAT1 and SLC7A5) and so on [Citation14–19]. What’s more, in the latest progress of our research, we found that l-carnitine-conjugated nanoparticles (LC-PLGA NPs) selectively targeted to organic cation transporter 2 (OCTN2 and SLC22A5) on the enterocytes for enhancing oral delivery of drugs [Citation20–22]. The results greatly encouraged us that transporter-mediated NPs with specific ligands as a formulation hold potential for the application of orally administrated therapeutics. However, the data to further support the role of transporters on the mediation of NPs absorption is still limited, which deserves further investigation, especially on understanding the value of transporters in the disposition of a broad range of nano-sized particles. It is also important to exploit more effective intestinal transporters that could provide high transport capability and at the same time possess high efficiency for the recognition of modified NPs.
The current work is to elucidate the potential utility of this strategy to enhance oral bioavailability of therapeutic drug paclitaxel (PTX). Sodium-dependent vitamin C transporter 1 (SVCT1 and SLC23A1) was chosen as a target, which is proposed to handle the bulk transport of apical ascorbic acids from the intestinal tract (). It is worth mentioning that SVCT1 expresses throughout the whole intestinal tract, providing a long oral absorption window [Citation23]. Better yet, it belongs to the synergistic transporter system so as to mediate the transport of ascorbic acid with high capability [Citation24]. Based on the above, ascorbate-conjugated nanoparticles (As-PLGA NPs) were designed to target the intestinal SVCT1, with an expectation that such interactions will lead to greater cellular uptake and more efficient intestinal delivery of nanoparticles. The resulting As-PLGA NPs were characterized by particle size distribution, morphology, encapsulation efficiency, in vitro release behaviour and stability. In addition, uptake study and endocytosis pathways of NPs on Caco-2 cell monolayer were performed to clarify the relevant mechanisms on cellular level. The transport mechanisms were also explored by competing inhibition test and Na+-dependent study to confirm the pivotal role of SVCT1 during the uptake process. Finally, the intestinal absorption study as well as biodistribution of As-PLGA NPs were performed by in situ perfusion method and observed by laser confocal study.
Figure 1. Schematic of the mechanisms of cellular uptake of As-PLGA NPs in Caco-2 cells, including Na+-triggered SVCT1 transporter-mediated endocytosis, bypassing lysosomal compartments and transcytosis at the basal side of intestine.
Materials and methods
Materials and reagents
Paclitaxel (PTX) was obtained from Xi’an Helin Biological Engineering Co. Ltd. (Xi’an, China). Poly (d, l-lactic-co-glycolic acid) (PLGA, L:G molar ratio: 50:50, Mw: 15,000), polyvinyl alcohol (PVA, 87–89% hydrolyzed), palmitoyl ascorbate (PA) and Coumarin-6 (C6) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rhodamine-labelled phalloidin was purchased from Cytoskeleton (1830 S. Acoma St., Denver, CO, USA). Antibody to SVCT1 was obtained from BD Biosciences (San Diego, CA, USA). TRITC-labelled goat anti-rabbit antibody IgG was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Caco-2 cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Dulbecco’s modified Eagle medium (DMEM, high glucose) and foetal bovine serum were acquired from Gibco (Beijing, China). Penicillin-streptomycin stock solutions, Hank’s buffered salt solution (HBSS), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and concentrated solutions of sodium pyruvate were purchased from Hyclone (Logan, UT, USA). All solvents were of analytical reagent grade.
Preparation of ascorbate-conjugated PLGA nanoparticles (as-PLGA NPs)
As-PLGA NPs with various densities of palmitoyl ascorbate (0%, 10%, 20% and 30%) were prepared by the emulsification/solvent evaporation method. Briefly, PLGA copolymer, PA and appropriate amounts of PTX were dissolved in 5 ml of dichloromethane (DCM), and vortexed for 60 s as organic phase. The resulting solution was slowly added to 30 ml 1% (w/v) PVA aqueous solution to form a crude emulsion and then sonicated into homogeneous emulsion with an energy output of 500 W for 5 min. The organic solvent was removed by evaporation overnight with liphophilic PTX dispersing into the PLGA matrix. The drug-loaded NPs were separated from the free PTX by washing the NPs suspension by ultracentrifugation (Amicon Ultra-4 centrifugal filter). Coumarin 6 (C6)-labelled NPs were prepared by using the same method as described above to observed the behaviour of NPs in cellular uptake and intestinal distribution.
Characterization of ascorbate-conjugated PLGA nanoparticles
Size and zeta potential
The particle size and zeta potential were measured using a Zetasizer (Nano ZX; Malvern Co., Malvern, UK). All samples were analysed in triplicate.
Particle morphology
The morphology of nanoparticles was visualized by transmission electron microscopy (TEM, Tecnai G20; FEI, Hillsboro, OR, USA). NPs suspension was deposited on a carbon-coated copper grid, stained with 1% (w/v) phosphotungstic acid and examined by TEM.
Entrapment efficiency
Entrapment efficiency (EE) was analysed by mini-column centrifugation method. The unloaded PTX was removed by the gel filtration over a Sephadex G-50 column (2.5 cm × 0.8 cm). 0.2 ml of NPs suspension was added onto the column and centrifuged (2000 rpm, 2 min) followed by rinsing with 0.2 ml of distilled water for several times. The eluant was collected and the content of encapsulated PTX was determined by high performance liquid chromatography (HPLC; Shimadzu, Kyoto, Japan) at a wave length of 228 nm.
In vitro release and stability
To determine the in vitro drug release profile, PTX-loaded NPs were placed into a dialysis cartridge (molecular weight cut-off 3 kDa; Thermo Scientific, Rockford, IL, USA). The cartridge was immersed into 50 ml pH 6.8 PBS containing 30% (v/v) ethanol and gently shaken in an orbital shaker (37 °C, 100 rpm). At designated intervals, an aliquot of 1 ml of release medium was removed for determination and replenished with the same volume of fresh PBS. The content of PTX was measured by HPLC analysis.
Simulated gastric fluid containing pepsin (SGF, pH 1.2) and simulated intestinal fluid containing trypsin (SIF, pH 6.8), according to the Chinese Pharmacopoeia, were used to investigated the stability of NPs. 1 ml of NPs was incubated with 100 ml of different mediums under the uniform vibration at 37 °C. At designated intervals, 1 ml of the samples was taken to measure the particle size.
Cell culture
Caco-2 cells were routinely cultured (passages 30–90) in 75 cm2 culture flasks (Greiner, Frickenhausen, Germany) with DMEM medium supplemented with 10% (v/v) foetal bovine serum (FBS), 20 mmol/L HEPES, 1% (v/v) non-essential amino acid, 4 mmol/L L-glutamine, 100 units/mL penicillin and 100 units/mL streptomycin in a 5% CO2 humidified atmosphere (90% humidity) at 37 °C. Cells grown to 80% confluence were trypsinized and sub-cultured in 24-well culture plate (Corning, NY, USA) at a density of 1 × 105 cells/well. The cultures reached confluence within 24 h, uptake was measured seven days after seeding.
Expression of SVCT1 on Caco-2 cells
Caco-2 cells were cultured on coverslips and fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. After washing with TBST, coverslips were incubated with anti-SVCT1 polyclonal antibody at a dilution of 1:100 overnight at 4 °C. Subsequently, TRITC-labelled goat anti-rabbit antibody IgG was applied as a secondary antibody and DAPI was added to stain the nuclei. Finally, the fluorescent signals were visualized by a confocal laser scanning microscope (CLSM; Leica, Heidelberg, Germany).
Uptake studies of as-PLGA NPs on Caco-2 cells
Uptake study
The cellular uptake of nanoparticles was studied by flow cytometry and CLSM on Caco-2 cells. The cells were seeded in 24-well plates at a density of 1 × 105 cells/well and cultured for 24 h. C6-labelled PLGA-NPs and As-PLGA NPs were applied to the cells suspended in serum-free DMEM (C6 final concentration of 100 ng/mL). After 1 h incubation, cells were washed with PBS thrice, trypsinized and collected by centrifugation. Then cells were resuspended in 400 μL PBS for flow cytometry analysis.
For CLSM, Caco-2 cells were cultured on coverslips and incubated with C6-labelled PLGA NPs and As-PLGA NPs, respectively. After 1 h incubation, cells were washed with PBS, fixed with 4% paraformaldehyde (PFA) for 15 min and incubated with 1% triton 100 solution. DAPI and rhodamine–phalloidin were then applied to stain nuclei (blue) and cytoskeleton (red). Subsequently, the cellular monolayer was observed by CLSM.
Endocytosis pathway study
In order to determine the potential endocytotic pathways of PLGA NPs and As-PLGA NPs, Caco-2 cells were first cultured with different specific inhibitors for various kinds of endocytosis for 1 h at 37 °C as follows: indomethacin (15 μg/mL), inhibitor of caveolae-mediated endocytosis; chlorpromazine (8 μg/mL), inhibitor of clathrin-mediated endocytosis; colchicine (20 μg/mL), inhibitor of macropinocytosis; quercetin (20 μg/mL), inhibitor of caveolae- and clathrin-independent endocytosis; sodium azide (20 μg/mL), an energy inhibitor. Subsequently, C6-labelled PLGA NPs and As-PLGA NPs were applied to cells suspended in serum-free DMEM containing one of the above inhibitors for 1 h (C6 final concentration of 100 ng/mL). After incubation, cells were washed, harvested and analysed by flow cytometer.
Mechanistic studies for SVCT1-mediated endocytosis
To explore the effect of the sodium ion on SVCT1-mediated internalization, Na+-free HBSS was used for the culture medium of cellular uptake by replacing NaCl with choline chloride. For competitive inhibition studies, Caco-2 cells were incubated with 500 μM ascorbic acid solution at 37 °C for 30 min prior to and during 1 h uptake process of As-PLGA NPs.
Co-localization of as-PLGA NPs and lysosomes by CLSM
The co-localization of NPs and lysosomes was carried out using double-staining method and visualized by CLSM. The specific probe of LysoTracer Red (Invitrogen, Carlsbad, CA, USA) was diluted according to the manufacturer’s instructions in HBSS. Then C6-labelled PLGA NPs and As-PLGA NPs containing prewarmed LysoTracker working solution (75 nM) were applied to Caco-2 cells at 37 °C, respectively. After 1 h incubation, NPs were removed and cells were washed thrice, fixed with 4% PFA. Finally cells were stained with DAPI and the sample imaging was observed under CLSM.
In situ permeability studies
All animals investigated in this research were executed according to guidelines for Use and Care of Animals approved by China Medical University. SD rats weighing 200–250 g were deprived of food overnight for 12 h with free access to water. Prior to the experiments, anaesthesia was induced by intraperitoneal injection of 20% urethane (1 g/kg) to facilitate the laparatomy of duodenum, jejunum and ileum separation. The intestinal segments (approximately 10 cm) were gently washed with 37 °C saline solution and then perfused with 20 μg/mL PTX-loaded NPs diluted by Krebs Ringer (KR, 7.8 g of NaCl, 0.35 g of KCl, 1.37 g of NaHCO3, 0.02 g of NaH2PO4, and 1.48 g of glucose in 1000 ml of purified water) buffer containing 20 μg/mL phenol red at a flow rate of 0.2 ml/min. The perfusate was collected in tubes every 15 min for analysis and lasted for 120 min. Finally, the length and diameter of each infused segment were accurately recorded. 200 μL of collected perfusion samples were extracted with 1 ml of acetonitrile and centrifuged at 13,000 rpm for 10 min to determine the PTX content. The absorption rate (Ka) and effective permeability (Peff) of both NPs were calculated by the following equations:
where Cin is the concentration of PTX in the donor fluid, Cout is the concentration of PTX in the receptor tube, PRin and PRout are the concentrations of phenol red in the donor fluid and receptor tube, Q is the perfusion flow rate of 0.2 ml/min, L and r refer to the radius and length of the selected intestinal segments.
Biodistribution study in rat small intestine
In order to qualitatively observe the absorption of NPs in the gintestinal tract, C6-labelled NPs were administrated to SD rats by oral gavage (4 mg/kg). After 30 min, the rats were sacrificed and the selected intestinal segments were isolated, everted, and frozen in cryoembedding media at −80 °C. Then, the frozen duodenum, jejunum and ileum were sectioned to 10-μm thick tissue slices (CM 3050 S; Leica, Heidelberg, Germany) and fixed on cationic slides. The slices were stained with DAPI for 5 min and then visualized under CLSM.
Results
Preparation and characterization of NPs
On the basis of the traditional emulsion/evaporation technique, an appropriate amount of palmitoyl ascorbate (PA) was added to the organic phase (). It was anticipated that PA would naturally position itself with the fatty acid preferentially inserting into the hydrophobic PLGA matrix, whereas the hydrophilic head groups of ascorbic acids display in the hydrophilic external environment. As listed in , the mean particle sizes of NPs appeared slightly decreased with the increase of the PA content. The zeta potential of non-modified PLGA NPs was close to neutralization, while proportionally reduced with an increase of PA conjugation. The negative electricity on the surface of NPs might derive from the deprotonation effect of carboxyl groups derived from the ascorbic acids, which could indirectly prove that ascorbic acids exists on the surface of As-PLGA NPs. The formation of NPs was then characterized by transmission electron microscopy (TEM), where both PLGA NPs and As-PLGA NPs were morphologically spherical with a relatively uniform size distribution of ∼170 nm (). In another aspect, EE (%) of NPs was reduced to 61.7% when PA reached 30% of the total PLGA matrix, suggesting that an excess of fatty acids might preferentially occupy the drug carrier sites and thus affect the loading efficiency of PTX.
Figure 2. (A) Schematic illustration of palmitoyl ascorbate modified PLGA NPs. (B,C) Transmission electron microscopy (TEM) images of PLGA NPs/PTX (top) and 20%As-PLGA NPs/PTX (down). (Bar = 200 nm) (D) In vitro release profiles of PTX solution, PLGA NPs/PTX, 10%As-PLGA NPs/PTX, 20%As-PLGA NPs/PTX and 30%As-PLGA NPs/PTX in pH 6.8 phosphate buffer (containing 30% ethanol, v/v). (E) Particle size variation of PLGA NPs/PTX, 10%As-PLGA NPs/PTX, 20%As-PLGA NPs/PTX and 30%As-PLGA NPs/PTX after incubation in SGF for 24 h (SGF: simulated gastric fluid containing pepsin); (F) Particle size variation of PLGA NPs/PTX, 10%As-PLGA NPs/PTX, 20%As-PLGA NPs/PTX and 30%As-PLGA NPs/PTX after incubation in SIF for 24 h (SIF: simulated intestinal fluid containing trypsin). Data are shown as mean ± SD (n = 3).
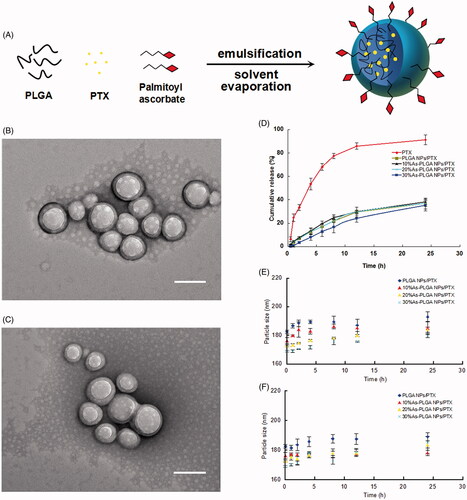
Table 1. Characterization of different PTX-loaded NPs (mean ± SD, n = 3).
In vitro release and stability study
In vitro release profiles of PLGA NPs and As-PLGA NPs with different PA modification were conducted in the media at pH 6.8, simulating the circumstance of intestinal tract. The results showed that the PTX solution was released by nearly 88.2% in 24 h, accompanied by the process of an initial burst effect. In contrast, PLGA NPs and As-PLGA NPs displayed similar release kinetics, with a cumulative drug release of ∼40% in 24 h (). It indicated that the copolymer carrier of PLGA prevented PTX from rapid clearance in the gastrointestinal tract, providing more opportunity for PTX to transport across intestinal epithelia in the form of NPs.
Furthermore, two types of simulated gastrointestinal media, namely simulated gastric fluid containing pepsin (SGF, pH 1.2) and simulated intestinal fluid containing trypsin (SIF, pH 6.8) were used to investigate the stability of PLGA NPs and As-PLGA NPs. All kinds of nanoparticles showed no significant difference in the variation of particle size in both media during 24 h, exhibiting good stability features (). The particle size of NPs in the SIF solution was slightly larger than that in the SGF media, possibly due to the absorption of trypsin on the surface of NPs. The results indicated that the modification of PA did not apparently affected the stability of NPs. On the other hand, PLGA NPs were able to protect drugs from both acid-catalyzed degradation in the stomach and enzymatic degradation in the intestinal tract until they reach the circulatory system.
Expression of SVCT1 on Caco-2 cells
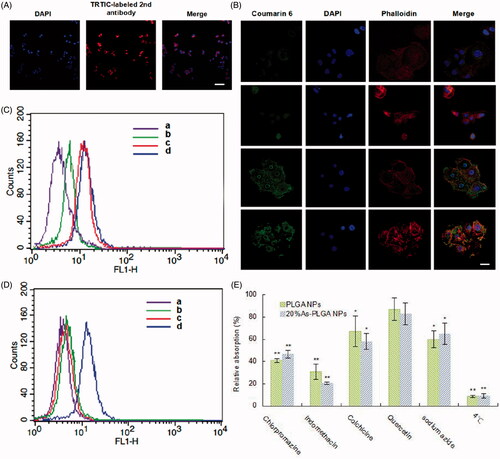
According to the previous researches, SVCT1 was selectively up-regulated during differentiation of the Caco-2 cells [Citation25,Citation26]. Therefore, we qualitatively observed the protein expression and distribution of SVCT1 in Caco-2 cells by immunohistochemistry before the uptake study. As shown in , the red fluorescence of TRTIC-labelled secondary antibody supported the presence of SVCT1 in enterocyte-like Caco-2 cells. The high level of SVCT1 might provide the basis for the following biological experiments, and most importantly, offer an ideal binding site to facilitate the interaction with As-PLGA NPs.
Figure 3. (A) SVCT1 transporter expression on the membrane of Caco-2 cells. Blue: DAPI for nuclei, Red: TRTIC-labelled secondary antibody for SVCT1. Scale bar represents 50 μm. (B) Confocal study of C6-labelled NPs in Caco-2 cells. Caco-2 cells incubated with C6-labelled PLGA NPs, 10%As-PLGA NPs, 20%As-PLGA NPs and 30%As-PLGA NPs (from top to down) for 1 h at 37 °C (Blue: DAPI for nucleus; Red: Rhodamine-labelled phalloidin for cytoskeleton). Scale bar represents 20 μm. (C) Effect of different ascorbate modification on the cellular uptake. Caco-2 cells were treated with C6 labelled (a) PLGA NPs, (b) 10%As-PLGA NPs, (c) 20%As-PLGA NPs and (d) 30%As-PLGA NPs. (D) Effect of free ascorbic acid and sodium ion on the cellular uptake. Caco-2 cells were treated with C6 labelled (a) PLGA NPs, (b) 20%As-PLGA NPs with 500 μM ascorbic acid in the culture media, (c) 20%As-PLGA NPs in Na+-free culture media and (d) 20%As-PLGA NPs. Each sample was incubated with equal C6 concentration of 100 ng/mL after 1 h incubation and investigated by flow cytometry analysis. (E) The endocytosis pathways of C6-labelled PLGA NPs and 20%As-PLGA NPs after incubation with various endocytosis inhibitors for 1 h at 37 or 4 °C. Data are shown as mean ± SD (n = 3). *p < .05, **p < .01 vs control group.
Cellular uptake study of as-PLGA NPs in Caco-2 cells
The cellular uptake was quantitatively calculated by flow cytometry. To investigate the effect of ascorbate density on the targeting efficiency of SVCT1, coumarin-6 (C6) was used as a fluorescent probe and encapsulated into PLGA NPs. As shown in , the results demonstrated that compared to the non-modified PLGA NPs, As-PLGA NPs exhibited a significant enhancement of cellular uptake, which might result from the specific and effective interaction with SVCT1 on the cell membrane. On the other hand, the internalized As-PLGA NPs was gradually elevated with the ascorbate density ranging from 10 to 20% (w/w) and strike a balance point. This implied that the uptake was no longer increased with the extra addition of ascorbate reaching to 30% of PLGA matrix. We concluded that there might be a certain degree of saturated concentration of As-PLGA NPs recognized by SVCT1, which was similar to the interaction between transporters and their small molecule substrates.
The intracellular uptake of NPs in Caco-2 cells was visualized by CLSM to semi-quantify the internalized As-PLGA NPs into cells. The images in depicted the cellular internalization and distribution of C6-labelled NPs in Caco-2 after 1 h incubation. The nuclei and cellskeleton of Caco-2 cells were treated with DAPI (in blue) and rhodamine-labelled phalloidin (in red), respectively. It could be observed that there was slight C6 fluorescence in cells treated with PLGA NPs. However, the internalized As-PLGA NPs have been improved to varying degrees with different ascorbate densities, suggesting that NPs were more efficiently taken up with the action of ascorbate modification. When cells were exposed to 20%As-PLGA NPs, the fluorescence of C6 showed the greatest evidenced by overwhelmingly increasing nuclear and perinuclear accumulation of green signals in the cytoplasm. Coincidentally, an increase in the surface density of ascorbate did not contribute to further enhancement of C6 internalization, which was in good agreement with the results of quantitative study performed by flow cytometry (). These results were combined to illustrate that there is an optimal intermediate ligand density on the surface of the NPs that is needed for the maximal uptake.
Based on different C6 distributions, it was suggested that As-PLGA NPs were internalized into cells via specific transporter-mediated endocytosis. Therefore, competitive inhibition experiments were carried out to verify whether SVCT1 was associated with the improved internalization of NPs. Ascorbic acid, the typical substrate of SVCT1 transporter, was selected as a competitive inhibitor during the process of cellular uptake. As shown in , the uptake of 20%As-PLGA NPs was strongly reduced by two-fold with the treatment of free ascorbic acids in the culture medium, indicating that small molecule substrates and As-PLGA NPs competed for the favourable binding site of SVCT1 and thus transporter-mediated endocytosis might be hampered more or less. In addition, the deprivation of Na+ in the uptake buffer severely restricted the cellular internalization of As-PLGA NPs, while that of PLGA NPs remained the same level. These results demonstrated that the sodium ion constituted an integral part of the normal function of SVCT1 and played a crucial role in initiating the internalization process of As-PLGA Ns. The combined results of the investigations on the uptake mechanisms could help conclude the essentiality of SVCT1 for the enhanced cellular uptake.
Subsequently, the mechanisms involved in the endocytosis pathways of NPs was investigated (). Among various inhibitors for the corresponding endocytosis, the presence of indomethacin and chlorpromazine (inhibitors for caveolin- and clathrin-mediated endocytosis) significantly decreased the cellular uptake of both NPs (p < .01). Beyond that, the uptake process was also inhibited at the presence of colchicine (inhibitor of macropinocytosis) and sodium azide (energy inhibitor) but no change after incubating with quercetin (inhibitor of cavelae- and clathrin-independent endocytosis). The uptake study was also performed at 4 °C, the temperature at which endocytosis was likely to be limited, exhibiting a reduced cellular uptake below to 10%. The results above indicated that endocytosis played an important role in the absorption of NPs.
Co-localization of as-PLGA NPs with lysosomes
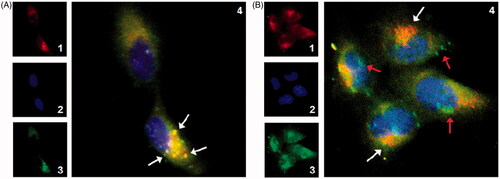
To evaluate the intracellular delivery efficiency of PLGA NPs and As-PLGA NPs, the localization of NPs inside cells was observed by double immunofluorescence. Lysosomes were stained with organelle-selective red dyes of LysoTracker, while C6 was shown in green signals. Immunoblot analysis of PLGA NPs, as illustrated in , showed the co-localization of the NPs and LysoTracker Red appeared overwhelmingly yellow in the cytoplasm (white arrows). These results suggested that PLGA NPs administration fused with lysosomes in the cytoplasm and that clathrin-mediated endocytosis may be one of the main pathways of NPs translocation in the Caco-2 cells. In contrast, judging by the great separation between the green and red fluorescence (), it was deduced that most of the internalized As-PLGA NPs were coated with caveolae, another form of the endocytic vesicle that might avoid the degradation of lysosomal pathway and facilitate the entry into cytoplasm (red arrows). To sum up, the translocation of As-PLGA NPs in cells is beneficial to the transfer of NPs to the basement membrane in an intact form and enter to lymphatic circulation through transcytosis.
Figure 4. Intracellular delivery of the C6 labelled (A) PLGA NPs and (B) 20%As-PLGA NPs on Caco-2 cells after 1 h incubation observed by CLSM. The late endosomes and lysosomes were stained by LysoTracker Red. 1. Red fluorescence of lysosomes; 2. Blue fluorescence of nucleus; 3. Green fluorescence of C6; 4: overlay of 1–3. White arrows indicate the occasions of coincidence between the NPs and lysosomes; red arrows indicate NPs in the cytoplasm that circumnavigated lysosomes.
In situ single-pass intestinal perfusion in rats
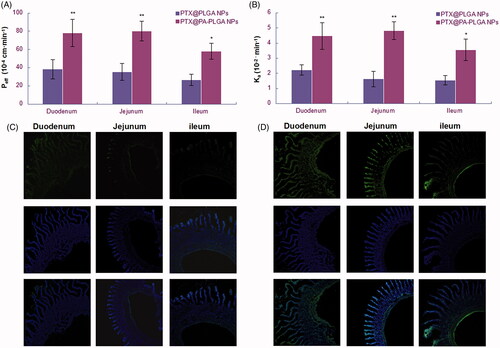
The oral absorption performance of NPs was evaluated by in situ small intestinal perfusion method in rats. Phenol red was added as a nonabsorbable marker to measure the change of water in the perfusate. As shown in , compared with PLGA NPs, the effective membrane permeability (Peff) of As-PLGA NPs was significantly increased by 1.44-, 2.26-, and 2.19-fold in duodenum, jejunum and ileum, respectively. On the other hand, the absorption rate (Ka) of As-PLGA NPs was also improved in the whole intestine, with the jejunum segment producing the highest absorption rate of 4.83 ± 0.59 10−2/min, increased by almost 2.6-fold than that of PLGA NPs (). As a result, As-PLGA NPs effectively improved the absorption of PTX with low water-solubility through the whole intestinal segment, suggesting there might be special mechanisms that was responsible for the oral absorption of PTX-loaded As-PLGA NPs.
Figure 5. (A) Effective permeability (Peff) and (B) Absorption rate (Ka) performed by in situ single-pass intestine perfusion of 20%As-PLGA NPs/PTX compared with PLGA NPs/PTX in rats. (Data are shown as mean ± SD, n = 3. *p < .05, **p < .01.) The fluorescence micrographs of rat intestine (duodenum, jejunum and ileum) 30 min after oral administration of C6 labelled (C) PLGA NPs and (D) 20%As-PLGA NPs. The histological sections were counterstained with DAPI for nuclei and observed under CLSM (10×).
Biodistribution of as-PLGA NPs in the GI tract
Confocal laser scanning microscopy (CLSM) was applied to visualize the biodistribution of NPs in the GI tract of rats (). In the obtained images with a ten-fold expansion of vision, the green fluorescence of C6 could be detected in the enterocytes. Compared with non-modified PLGA NPs, 20%As-PLGA NPs acquired stronger C6 fluorescence not only on the apical side of intestinal tracts but also deeply inside the intestine villi, and even at the basolateral side. Based on this, we could draw a conclusion that the modification of ascorbate on the surface of NPs was likely to penetrate the mucus layer, then recognized and internalized by enterocytes with the assistance of SVCT1, and then enter the systemic circulation. The observed images were in good accordance with the results obtained by the in situ single pass intestinal perfusion experiment.
Discussions
The intestinal transporters bear important physiological functions by controlling the import and export of drugs across intestinal tissues. Armed with the determination of the structure–activity relationships between transporters and substrates, we can create a suitable biological model that accounts for the relevant fluxes. As a result, the previous study of a novel strategy merits attention by using these functional protein targets for promoting oral delivery of therapeutic drugs.
As stated earlier, there are a number of limitations to the oral delivery of therapeutics due to the physicochemical properties of the drug itself and the complex gastrointestinal environment. In this work, a novel transporter-mediated drug delivery system was designed to improve the oral bioavailability of therapeutics. SVCT1 was chosen as a target with ideal properties of high-level expression and transport capability throughout the whole intestinal tract. To achieve this, PLGA NPs were engineered by inserting an amphipathic compound composed of both fatty acid and ascorbate acid groups with an ester linkage. The added component PA, consisted of both hydrophilic and lipophilic parts, might reduce the surface tension of water–oil phase and thus lead to a more uniform dispersion system with the polydispersity index (PDI) around 0.019, compared with non-modified PLGA NPs. More importantly, the ligands on the surface of NPs could be flexibly and precisely adjusted to an optimum quantity in order to achieve the maximization of the value with SVCT1.
Accordingly, a distinct advantage offered by particulate-based active targeting is establishing multivalent interactions with intestinal cells. Once achieved, multivalency can lead to enhanced avidity to target cells and can dramatically improve payload delivery [Citation27,Citation28]. Meanwhile, it is also important to note that achieving this effect is highly dependent on the surface density of ligands. An optimum contact between a carrier and a target on the biological surface is necessary to increase the drug absorption. In this study, an intermediate ascorbate modification density of 20% (w/w) was found to harvest the highest cellular uptake while an additional amount of ascorbate no longer contributed to the increase in the uptake. This was in accordance with our previous experimental results involving the transport characteristics of an amino acid transporter ATB0,+, suggesting that high ligand density was competing for transporters due to the multipoint interaction and was leading to a depletion of available transporters to fully wrap the NPs with a decrease in cellular endocytosis [Citation17]. In addition, a large number of C6-labelled 20%As-PLGA NPs were found to congregate onto the cell membrane, by which the contact between NPs and enterocytes might be beneficial to the next biological process. These data might be interpreted as the fact that the selective moiety (As) was able to be recognized by the corresponding SVCT1 transporter on the cell membrane and to enter cells in the manner of SVCT1-mediated endocytosis.
In relation to the mechanism of SVCT1-mediated transport, the uptake of As-PLGA NPs was in a Na+-dependent pathway. Since SVCT1 belongs to sodium-dependent transporter by harnessing the energy of pre-existing electrochemical gradients of Na+ when mediating an uphill uptake of ascorbic acids. Once the synergistic ions were lost, the executive function of the transporter might be hindered. Furthermore, the uptake was significantly inhibited by the presence of free ascorbic acids that competes for the same binding domains of transporters, thereby impeding the following uptake. Those that mentioned above indicated that SVCT1 was involved in the transport of As-PLGA NPs across the membrane and Na+ ion executed as a prerequisite for the activation of SVCT1.
As for the endocytosis process, the results indicated that ligands conjugation onto the surface can switch the mode of cell trafficking. Based on our study, transport of ascorbate-conjugated PLGA NPs appear to predominantly take place by a route that is inhibited by caveolae-specific inhibitors. This data implied that As-PLGA NPs circumnavigate the lysosomal compartment in the intracellular delivery [Citation29,Citation30]. Following the conjugation to ascorbate, As-PLGA NPs not only internalized into cells to a large extent, but also switched to an efficacious pathway without degradation of hydrolytic enzymes. The microscopy images subsequently reconfirmed the observation with a significant dissociation from lysosomes shown in red and from effective cytoplasmic distributions of As-PLGA NPs in green. On the other hand, according to the previous study, the polypeptides prefer to form an alpha helix in an acidic environment. The conformation enhances the affinity of polypeptides and membranes around, which triggers the rupture of the latter [Citation31]. This may also be responsible for the release of As-PLGA NPs from coated vesicles into cytoplasm. From this point of view, it might be concluded that by means of the interaction between SVCT1 and ascorbate on the surface of PLGA NPs, the nanoparticles and transporters accumulated in coated pits, internalizing as ligand–transporter complexes and converted the subcellular sorting of NPs. The oral absorption of As-PLGA NPs was therefore enhanced since they were more likely to be transported in a complete state and to enter into lymphatic circulations by transcytosis [Citation32].
Sodium-dependent vitamin C transporter 1 protein expresses predominantly on the intestinal tract and leads to more uptake of ascorbic acids as pH increases [Citation33,Citation34]. The small intestine with the alkaline characteristic is clearly an important segment for the absorption of As-PLGA NPs. The results of in situ intestinal perfusion indicated that the enhanced absorption induced by the mediation of SVCT1 occurs through the whole intestinal tract. The decoration of ligands made As-PLGA NPs cluster on the apical side of epithelial cells, enter into enterocytes and even transmit to the basolateral side. The high-level expression and transport capacity of SVCT1, along with the multipoint contact with As-PLGA NPs, may be ascribed to the increase of intestinal absorption.
The results combined are providing strong evidence of the involvement of SVCT1 in the intestinal absorption of As-PLGA NPs. The multipoint contact between ligands and transporters might avoid the clearance from the mucus layers. What’s more, SVCT1 was widely distributed throughout the whole small intestine so that this binding site would offer a long absorption window, which clearly suggest that ascorbate-conjugated nano-drug delivery systems hold great potential for oral drug delivery.
Conclusions
In this study, functional ascorbate-conjugated nanoparticles were designed to target SVCT1 on the apical side of intestine for the enhancement of PTX oral absorption. The optimum formulation with 20% modification of ascorbate displayed spherical particles with uniform size distribution and a maximum of cellular uptake. The competitive inhibition test and ion-dependent study provided strong evidence that SVCT1 transporter was involved in the internalization of ascorbate-conjugated nanoparticles. Moreover, the transport of As-PLGA NPs took place predominantly by caveolae-mediated endocytosis, indicating that As-PLGA NPs could circumnavigate lysosomal compartments accompanied by transcytosis at the basal side of enterocytes. The results of in situ perfusion and biodistribution study revealed that the permeation of As-PLGA NPs across intestinal membrane was markedly enhanced over 2.6-fold compared to PLGA NPs. These findings may provide new insights into the practical use of transporter-mediated nanoparticulate system with promoted oral delivery of therapeutic drugs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Deng D, Xu C, Sun P, et al. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125.
- César A, Snijder B, Frappier-Brinton T, et al. Call for systematic research on solute carriers. Cell 2015;162:478–487.
- Nakanishi T, Tamai I. Solute carrier transporters as targets for drug delivery and pharmacological intervention for chemotherapy. J Pharm Sci. 2011;100:3731–3750.
- Sugawara M, Huang W, Fei YJ, et al. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J Pharm Sci. 2000;89:781–789.
- Thomsen AE, Christensen MS, Bagger MA, et al. Acyclovir prodrug for the intestinal di/tri-peptide transporter PEPT1: comparison of in vivo bioavailability in rats and transport in Caco-2 cells. Eur J Pharm Sci. 2004;23:319–325.
- Anand BS, Patel J, Mitra AK. Interactions of the dipeptide ester prodrugs of acyclovir with the intestinal oligopeptide transporter: competitive inhibition of glycylsarcosine transport in human intestinal cell line-Caco-2. J Pharmacol Exp Ther. 2003;304:781–791.
- Stephen R, Adam M, Kerry K, et al. XP13512 [(+/-)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J Pharmacol Exp Ther. 2004;311:315–323.
- Ghaznavi H, Hosseini-Nami S, Kamrava SK, et al. Folic acid conjugated PEG coated gold-iron oxide core-shell nanocomplex as a potential agent for targeted photothermal therapy of cancer. Artif Cells Nanomed Biotechnol. 2017; [1–11]. doi: https://doi.org/10.1080/21691401.2017.1384384.
- Afsharzadeh M, Hashemi M, Mokhtarzadeh A, et al. Recent advances in co-delivery systems based on polymeric nanoparticle for cancer treatment. Artif Cells Nanomed Biotechnol. 2017; [1–16]. doi: https://doi.org/10.1080/21691401.2017.1376675.
- Chen D, Sun J, Sun K, et al. In vivo evaluation of novel ketal-based oligosaccharides of hyaluronan micelles as multifunctional CD44 receptor-targeting and tumor pH-responsive carriers. Artif Cells Nanomed Biotechnol. 2016;44:898–902.
- Ahmad N, Alam MA, Ahmad R, et al. Preparation and characterization of surface-modified PLGA-polymeric nanoparticles used to target treatment of intestinal cancer. Artif Cells Nanomed Biotechnol. 2017; [1–15]. doi: https://doi.org/10.1080/21691401.2017.1324466.
- Siddhartha VT, Pindiprolu SKSS, Chintamaneni PK, et al. RAGE receptor targeted bioconjuguate lipid nanoparticles of diallyl disulfide for improved apoptotic activity in triple negative breast cancer: in vitro studies. Artif Cells Nanomed Biotechnol. 2017; [1–11]. doi: https://doi.org/10.1080/21691401.2017.1313267.
- Abdolahpour S, Toliyat T, Omidfar K, et al. Targeted delivery of doxorubicin into tumor cells by nanostructured lipid carriers conjugated to anti-EGFRvIII monoclonal antibody. Artif Cells Nanomed Biotechnol. 2017; [1–6]. doi: https://doi.org/10.1080/21691401.2017.1296847.
- Shan XH, Hu H, Xiong F, et al. Targeting Glut1-overexpressing MDA-MB-231 cells with 2-deoxy-D-g1ucose modified SPIOs. Eur J Radiol. 2012;81:95–99.
- Guo Y, Zhang Y, Li J, et al. Cell microenvironment-controlled antitumor drug releasing-nanomicelles for GLUT1-targeting hepatocellular carcinoma therapy. ACS Appl Mater Interfaces. 2015;7:5444–5453.
- Venturelli L, Nappini S, Bulfoni M, et al. Glucose is a key driver for GLUT1-mediated nanoparticles internalization in breast cancer cells. Sci Rep. 2016;6:21629.
- Luo Q, Gong P, Sun M, et al. Transporter occluded-state conformation-induced endocytosis: amino acid transporter ATB0,+-mediated tumor targeting of liposomes for docetaxel delivery for hepatocarcinoma therapy. J Control Release. 2016;243:370–380.
- Luo Q, Yang B, Tao W, et al. ATB0,+ transporter-mediated targeting delivery to human lung cancer cells via aspartate-modified docetaxel-loading stealth liposomes. Biomater Sci. 2017;5:295–304.
- Li L, Di X, Wu M, et al. Targeting tumor highly-expressed LAT1 transporter with amino acid-modified nanoparticles: toward a novel active targeting strategy in breast cancer therapy. Nanomedicine. 2017;13:987–998.
- Kou L, Hou Y, Yao Q, et al. L-Carnitine-conjugated nanoparticles to promote permeation across blood-brain barrier and to target glioma cells for drug delivery via the novel organic cation/carnitine transporter OCTN2. Artif Cells Nanomed Biotechnol. 2017; [1–12]. doi: https://doi.org/10.1080/21691401.2017.1384385.
- Kou L, Yao Q, Sun M, et al. Cotransporting ion is a trigger for cellular endocytosis of transporter-targeting nanoparticles: a case study of high-efficiency SLC22A5 (OCTN2)-mediated carnitine-conjugated nanoparticles for oral delivery of therapeutic drugs. Adv Healthcare Mater. 2017;6:1700165.
- Kou L, Yao Q, Sivaprakasam S, et al. Dual targeting of l-carnitine-conjugated nanoparticles to OCTN2 and ATB0,+ to deliver chemotherapeutic agents for colon cancer therapy. Drug Deliv. 2017;24:1338–1349.
- Wohlrab C, Phillips E, Dachs GU. Vitamin C transporters in cancer: current understanding and gaps in knowledge. Front Oncol. 2017;7:74.
- Marc B, Yoshiro S, Daniel A, et al. The sodium-dependent ascorbic acid transporter family SLC23. Mol Aspects Med. 2013;34:436–454.
- James CB, Christine EC, Wade JS, et al. Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem Biophys Res Commun 2005;19:150–156.
- Maulén NP, Henríquez EA, Kempe S, et al. Up-regulation and polarized expression of the sodium-ascorbic acid transporter SVCT1 in post-confluent differentiated CaCo-2 cells. J Biol Chem. 2003;278:9035–9041.
- Davis ME, Chen Z, Shin DM, et al. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782.
- Montet X, Funovics M, Montet-Abou K, et al. Multivalent effects of RGD peptides obtained by nanoparticle display. J Med Chem. 2006;49:6087–6093.
- Du W, Fan Y, Zheng N, et al. Transferrin receptor specific nanocarriers conjugated with functional 7-peptide for oral drug delivery. Biomaterials. 2013;34:794–806.
- Robyn F, Driton V, Francisco FT, et al. Nanoparticle transport in epithelial cells: pathway switching through bioconjugation. Small. 2013;9:3282–3294.
- Lee MT, Sun TL, Hung WC, et al. Process of inducing pores in membranes by melittin. Proc Natl Acad Sci U S A. 2013;110:14243–14248.
- Xiang S, Tong H, Shi Q, et al. Uptake mechanisms of non-viral gene delivery. J Control Release. 2012;158:371–378.
- Liang WJ, Johnson D, Jarvis SM. Vitamin C transport systems of mammalian cells. Mol Membr Biol. 2001;18:87–95.
- Tsukaguchi H, Tokui T, Mackenzie B, et al. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75.