
Abstract
The natural flavonoid fisetin (FS) has shown anticancer properties but its in-vivo administration remains challenging due to its poor aqueous solubility. The aim of the study was to develop FS loaded pluronic127 (PF)-folic acid (FA) conjugated micelles (FS-PF-FA) by the way of increasing solubility, bioavailability and active targetability of FS shall increase its therapeutic efficacy. FA-conjugated PF was prepared by carbodiimide crosslinker chemistry. FS-PF-FA micelles were prepared by thin-film hydration method and evaluated in comparison with free FS and FS loaded PF micelles (FS-PF). The smooth surfaces with spherical in shape of FS-PF-PF micelles displayed smaller in size (103.2 ± 6.1 nm), good encapsulation efficiency (82.50 ± 1.78%), zeta potential (−26.7 ± 0.44 mV) and sustained FS release. Bioavailability of FS from FS-PF-PF micelles was increased by 6-fold with long circulation time, slower plasma elimination and no sign of tissue toxicity as compared to free FS. Further, the FS-PF-FA micelles demonstrated active targeting effect on folate overexpressed human breast cancer MCF-7 cells. The concentration of the drug needed for growth inhibition of 50% of cells in a designed time period (GI50) was 14.3 ± 1.2 µg/ml for FS while it was greatly decreased to 9.8 ± 0.78 µg/ml, i.e. a 31.46% decrease for the FS-PF. Furthermore, the GI50 value for FS-PF-FA was 4.9 ± 0.4 µg/ml, i.e. a 65.737% decrease compared to FS and 50% decrease compare to FS-PF. The results indicate that the FS-PF-FA micelles have the potential to be applied for targeting anticancer drug delivery.
Introduction
There is a crisis in fighting against cancer. The effectiveness of a cancer therapy is understood by its ability to reduce and eliminate tumours without damaging healthy tissues. Therefore, a distinct capacity to target tumours with limited effect on healthy tissues is the most essential for the success of cancer therapy. In order to increase the probability of a drug delivery formulation entering the pharmaceutical market, it should fulfil the main requirements. This means that such drugs need to be formulated with smart drug delivery technologies to make them acceptable for the treatment of patients. In this context, targeting nanoparticles has received a great deal of attention particularly in cancer treatment [Citation1].
Recently, there is a mounting interest in fisetin (FS) (3,7,3′,4′-tetrahydroxyflavone) which is a flavonoid dietary ingredient usually found in the smoke tree (Cotinus coggygria) and is also widely spread in fruits and vegetables, such as strawberry, apple, grape, onion and cucumber [Citation2]. It has been shown to exert antioxidant, anti-inflammatory, anti-angiogenic, hypoglycaemic and anticonvulsant activity [Citation3–6]. It has been shown to possess antiproliferative effect in a wide variety of tumour cells including prostate cancer, liver cancer, colon cancer and leukaemia cells [Citation7–11], where it induces G2/M arrest, decrease the activity of CDK2 and CDK4, suppress retinoblastoma protein phosphorylation, decrease the levels of cyclin D1 and cyclin E, increase the levels of p21 and p53, activate caspase-3 and inhibit DNA methyltransferases [Citation12,Citation13].
The great therapeutic interest of fisetin is however marred by its low oral bioavailability (44.1%) probably due to its high lipophilicity (log p 3.2) and low (10.45 μg/ml) aqueous solubility [Citation14]. Moreover, the fisetin molecule bearing 4 hydroxyl substituents, including a catechol on the phenyl B ring, is extensively metabolized in-vivo [Citation15]. The above factors lead to high/frequent dosing to achieve optimum therapeutic efficacy which often causes severe side effects. To overcome these problems, lipid-based delivery system, such as liposomes, nanoemulsion, lipidic spherulites and nanocochleates have been previously investigated [Citation14–18]. Liposomes, though advantageous, seem to be complicated, expensive and possess poor mechanical stability and low drug-loading capacities. Whereas nanoemulsion was unstable at room temperature displaying increasing particle diameter over the time along with phase separation [Citation19]. Our research group developed FS loaded nanocochleates which markedly improved anticancer efficacy, bioavailability and safety of FS [Citation20]. However, it requires specific storage condition, aggregation and high manufacturing cost. This juncture demands an effective, controlled release with long duration of action and safe drug delivery system of fisetin would be a significant advance for the treatment of cancer.
Polymeric micelles (PMs) have gained significant attention in the last two decades as a multifunctional nanotechnology-based delivery system for poorly water-soluble drugs. Due to their nanoscopic size, ability to solubilize hydrophobic drugs and along with the ligand-mediated active targeting can achieve site-specific delivery, thus PMs hold promise to obtain desirable biopharmaceutical and pharmacokinetic properties of drugs and enhance their bioavailability in treating tumour [Citation21]. FS loaded monomethyl poly(ethylene glycol)-poly(ε-caprolactone) (MPEG-PCL) copolymeric micelles was developed to improve the anticancer activity, however, it achieved passive targeting to cancer cells. There is need of active targeting micelles which could reduce side effects and a lower damage to normal cells due to their specificity and high local concentration in tumour cells.
Pluronic (PF127) (PF) is tri-block copolymers of polyethylene oxide (PEO) and polypropylene oxide (PPO), can assemble themselves to form micelles where the core is made up of hydrophobic PPO segment while the outer parts termed shell or corona is made up of PEO which is hydrophilic [Citation22]. The hydrophobic compartment is used to load hydrophobic drugs inside the micelles whereas the presence of PEO on the surface of micelles acts to protect the micelles from recognition by the immune system and unwanted interactions with serum proteins; thus, the micelles could go undetected enhancing the drug retention times in plasma [Citation23]. An important property of Pluronic micelles is to increase the solubility of sparingly soluble substances in water and simultaneously increase its bioavailability. In this context, solubilization is the spontaneous dissolving of a substance by reversible interaction with the micelles of an amphiphilic Pluronic polymer in water to form a thermodynamically stable isotropic solution with reduced thermodynamic activity of the solubilized material. Usually, the solubility is very low until the Pluronic polymer concentration reaches the cmc. At polymer concentrations above the cmc, the solubility increases linearly with the concentration of Pluronic polymer, indicating that solubilization is related to micellization [Citation23].
Folic acid (FA) as an active targeting ligand has been widely investigated [Citation24] and is a useful approach for active targeting as the polymers conjugated with it could be preferentially taken up into cells overexpressing FA receptors [Citation21,Citation25]. This specific approach of uptake could limit the off-targeted toxic effects of anticancer drugs [Citation26]. There are numerous advantages of FA as a targeting ligand. It is easily available, has low molecular weight and has high affinity towards the receptor. Further, its stability towards organic solvents makes it an attractive choice for further synthesis [Citation27]. Furthermore, through the strategy of synthesis, reactive functional groups can be introduced by selecting functionalized polyethylene oxide (PEO) at the polymer termini for further attachment of target moieties. Literature revealed that FA, an oxidized form of folate was efficiently internalized into cells through the receptor-mediated endocytosis when conjugated with a wide variety of bioactive molecules. Folate receptors are overexpressed in most types of human cancer cells, such as ovarian, breast and prostate cancers while only minimally distributed in normal tissue [Citation21].
Keeping in view the aforementioned characteristics, PF was functionalized by conjugation with FA adding value to selective cancer cell targeting. In this study, we hypothesized that the FS loaded PF-FA conjugated micelles by the way of increasing solubility, stability, bioavailability and targetability of FS shall increase its therapeutic efficacy. To the best of our knowledge, no previous studies have been attempted FS-loaded PF-FA nanomicelle system to improve its anticancer efficacy. Therefore, this study was undertaken to investigate the potential of PF-FA conjugated micelles as vehicle for systemic delivery of FS.
Materials and methods
Materials
Fisetin was purchased from Research Organic, Chennai, India. Pluronic F127 (PF) was purchased from BASF, India. Folic Acid was obtained from Merck Chemicals, Mumbai, India. Carbonyl di-imidazole (CDI), Di-cyclohexyl carbodiimide (DCC), N-hydroxy succinimide (NHS), Dimethyl sulphoxide (DMSO) was occupied by Loba Chemie Pvt. Ltd., Mumbai, India. Dichloromethane (DCM), Acetonitrile (HPLC grade), Methanol (HPLC grade), 1,2-ethylene diamine, Potassium dihydrogen phosphate, Diethyl ether were purchased from Merck Chemicals, Mumbai, India. Dialysis bag (molecular weight cut off 12,000) was obtained from Sigma-Aldrich Chemical private Ltd (Bangalore, India). All other reagents and chemicals were of analytical grade.
Synthesis of folate-conjugated pluronic (PF-FA)
Synthesis of CDI-activated pluronic (PF-CDI)
Pluronic (12.6 g, 1 mmol) was dissolved in dry acetonitrile (15 ml) and added dropwise to an excess amount of N,N′-carbonyldiimidazole (CDI) (1.62 g, 10 mmol) in dry acetonitrile (15 ml) at room temperature for 2 h under nitrogen atmosphere. After the addition, the mixture was kept stirring for an additional 4 h. The solution was concentrated in a rotatory evaporator and pour into excess of ethyl ether. This process was repeated 6–8 times to remove unreacted CDI. The PF-CDI was dried under a vacuum dehydration and collected as white powder.
Synthesis of amino-terminated pluronic (PF-NH2)
The PF-CDI (12.7 g, 1 mmol) was dissolved in dry acetonitrile (15 ml) and added dropwise to 10 ml of 1, 2-ethylenediamine at room temperature in 2 h. The mixture was allowed to react overnight. The unreacted ethylenediamine was removed by rotary evaporation and the crude product was poured into an excess amount of ether to obtain the white precipitate. This process was repeated 6–8 times and the PF-NH2 was dried under a vacuum dehydration and collected as white powder.
Synthesis of folate-conjugated pluronic (FA-FA)
The PF-NH2 (91 g, 0.079 mmol), FA (105 mg, 0.237 mmol), NHS (60 mg, 0.522 mmol) and DCC (108 mg, 0.522 mmol) were dissolved in 10 ml DMSO. The mixture was stirred in a nitrogen atmosphere at room temperature in the dark overnight. Then, the mixture was diluted with 20 ml deionized water and centrifuged in order to separate unreacted DCC. The supernatant was further purified by dialysis against deionized water for 2 d followed by lyophilization.
Characterization of PF-FA conjugate
Fourier transform infrared spectroscopy
To investigate the formation of PF-FA conjugate structure, Fourier transform infrared spectroscopy (FTIR) spectra were taken on the Bruker Vertex-70/80 FTIR (Tokyo, Japan) samples were crushed with KBr to produce pellets by applying a pressure of 150 kg/cm2. FTIR spectra of PF127, Folic acid and PF127-FA were scanned in the 4000–400 cm−1.
Proton NMR
The conjugation of FA onto PF was confirmed by recording and analysing 1HNMR spectra of pure PF, FA and PF-FA conjugate. FA was dissolved in DMSO, whereas PF and PF-FA were dissolved in deuterated water (D2O) for acquisition of NMR spectra. Degree of substitution on a molar basis was determined by dividing the total integral values of five FA aromatic protons peaks o–k in region 6.5–9.0 ppm in PF-FA conjugate by methyl group protons of PPO at 1.05 ppm (peak a) in FA.
UV spectrometric analysis
UV–VIS spectra were acquired using spectrophotometer (V630, JASCO, Japan) as a qualitative proof of FA conjugation with PF. The degree of conjugation of FA was determined by UV–VIS spectrophotometer. FA solutions of known concentration were prepared in DMSO and their respective UV absorbance was recorded. A calibration curve was obtained by plotting UV absorbance against FA concentration. To determine the concentration of conjugated FA. Sample of 10 mg was dissolved in DMSO and UV absorbance at 360 nm was recorded. FA present in conjugate was calculated from calibration curve. Degree of conjugation of FA on a weight basis was calculated by using the following equation: Conjugation of FA (wt%) = Weight of FA in conjugate/Total weight of conjugate ×100.
Powder X-ray diffraction spectroscopy
The powder X-ray diffraction (PXRD) patterns of FA, PF and PF-FA were conducted using X-ray diffractometer ((PW 1729; Philips, Almelo, Netherlands) with Cu as a target at a voltage of 40 kV. Samples were analysed in 2Φ angle range of 10–50° at a scanning rate of 3°/2Φ/min.
Critical micellar concentration
An iodine UV spectroscopy method was used to determine the CMC of PF and PF-FA in double distilled water as previously reported [Citation28]. To prepare a standard KI/I2 solution, 1 g of iodine and 2 g of potassium iodide were dissolved in 100 ml of deionized water. For CMC determination, a set of polymer solutions (concentration ranging from 0.0001 to 0.1%), containing the same amount of KI/I2 (25 ml of the standard solution) was prepared. The final solutions were equilibrated for 12 h in the dark at room temperature before measurement. The UV absorbance value of various polymer concentrations at 366 nm was measured using a UV–VIS spectrometer (Shimadzu UV-2401, Shimadzu, Tokyo, Japan). The absorption intensity was plotted against the logarithm of the polymer mass concentration.
Preparation of FS loaded PF and PF-FA micelles
FS loaded PF and PF-FA micelles were prepared by film hydration method [Citation28]. Briefly, 5 mg of FS and 10 mg of conjugation (1:2) ratio were dissolved in (7 ml) methanol in a round bottom flask. The solvent was evaporated by rotatory evaporator at 50 °C for about 30 min to obtain a thin drug-polymer film. Residual methanol was further removed under vacuum at room temperature overnight. The resultant thin film was hydrated with 20 ml ultrapure water at room temperature to obtain FS loaded PF-FA micelles (FS-PF-FA micelles). The same procedure was adopted to prepare FS loaded PF micelles (FS-PF127 micelles).
Characterization of FS-PF and FS-PF-FA micelles
Particle size and zeta potential determination
The micelles size and size distribution were determined by laser diffraction technique (Malvern 2000 SM; Malvern Instruments, Malvern, UK). The micelles size measurement was carried out at a 90° scattering angle. The samples were dispersed in distilled water and the average micelles size was determined and expressed in terms of d (0.9) μm. The zeta potential was measured with the laser Doppler electrophoretic mobility measurements using Zetasizer 300 HSA (Malvern Instruments Ltd., Malvern, UK) at a temperature of 25 °C.
Entrapment efficiency
Encapsulation efficiency of FS-PF and FS-PF-FA micelles were determined by separating non-encapsulated FS from micellar suspension by centrifugation (Eppendorf 5810 R, Hamburg, Germany) at 12,000 rpm for 2 h at 4 °C. The supernatant was diluted with phosphate buffer pH7.4 and free drug content was determined at 320 nm. The percentage encapsulation efficiency was calculated as actual amount of the drug encapsulated in micelles/initial amount of the drug used in the micelles ×100.
Surface morphology
Surface morphology of the FS-PF-FA micelles was performed by using transmission electron microscopy (TEM; Philips CM-200, Eindhoven, The Netherlands). A drop of FS-PF-FA micelles (1 mg/Ml) was placed on Formvars® coated copper grids (Ted Pella, Inc., Redding, CA) and allowed to equilibrate. Excess liquid was removed with a filter paper and dried at room temperature for about half an hour. The dried grid containing the FS-PF-FA micelles micelle was visualized using TEM.
In-vitro drug release
The in-vitro release of FS from FS-PF and FS-PF-FA micelles was carried out by dialysis bag method using phosphate-buffer saline (PBS) as the release medium at pH7.4 (corresponding to physiological pH) and pH5.3 (to mimic the slightly acidic environment in the tumours). Formulation equivalent to 1 mg of FS or 1 mg FS solution (1 mg/ml in 50%w/w mixture of PEG 400 and water) as control added into the dialysis bag (cellulose membrane, mw cut off 12,000 Da), hermetically sealed and immersed into 50 ml of respective medium. The entire system was kept at 37 ± 0.5 °C with continuous magnetic stirring at 100 rpm. At selected time interval, sample was removed and replaced with fresh medium in order to maintain sink conditions. The amount of FS released was determined from the calibration curve using UV absorbance at 320 nm.
Pharmacokinetic study in rats
Animals
Healthy female Wistar rats (National Toxicology Centre, Pune, India) weighing between 150 and 200 g were used for pharmacokinetic study and toxicity studies. Animals were housed together under standard conditions of temperature (24 ± 1 °C), relative humidity (55 ± 10%) and 12 h duration light/dark cycles throughout the experiment. Animals had free access to commercially available standard pellet diet (containing 4% carbohydrates, 22.15% protein, 4.15% fat, 2.46% glucose and 1.8% vitamins) (Pranav Agro 7 Industries, Sangli, Maharashtra, India) and filtered water ad libitum unless otherwise stated. Animals were acclimatized for one week prior to the initiation of experiment. During this acclimatization period, the health status of the animals was monitored daily. A research proposal was prepared according to the guidelines of the Committee for the Purpose of Control and Supervision of Experiment of Animals (CPCSEA). The experiment protocol was approved by Institutional Animal Ethics Committee (IAEC) (1703/PO/c/13/CPCSEA) of Poona College of Pharmacy, Pune, India.
Experimental design and sample collection
The rats were randomly divided into two groups (six animals each). Animals were fasted, but provided free access to water, overnight before the commencement of the experiment. Group I received FS at the dose of 15 mg/kg (formulated in 30% PEG and 70% saline at a final concentration of 3 mg/ml) by intraperitoneal (i.p.) route. Group II received FS-PF-FA micelles at the dose of 15 mg/kg by i.p. After mild ether anesthesia, blood samples were collected using retro-orbital puncture technique at predetermined time intervals (0.5, 1, 2, 3, 4, 5, 6, 7 and 8 h) in EDTA-coated tubes (BD Vacutainer (R) K2 EDTA) serially (0.5–2 ml each). Samples were centrifuged at 10,000 rpm for 15 min at 4 °C (Cryocentrifuge 2810 R, Eppendorf, Westbury, NY) and the plasma obtained was stored in polypropylene tubes below −20 °C and promptly analysed.
Quantification of fisetin in plasma
The plasma FS concentrations were determined by HPLC (reverse phased) analysis according to the method reported earlier with slight modification [Citation20]. Plasma quantification of FS was performed by HPLC (reverse phased). Fifty microlitres of 30 μg/ml methanolic solution of rutin (used as an internal standard) and 200 μl of cold acidified methanol (methanol:perchloric acid 70%, 200:1 v/v; used to precipitate proteins) was added to 100 μl of plasma. The final volume was made up to 550 μl using saline. After vortexing for 15 min, the samples were kept in ice bath for 15 min and centrifuged at 10,000 rpm at 4 °C for 15 min. The resultant supernatant (20 μl) was injected into HPLC system.
The HPLC conditions for analysis of FS were as follows: pump: PU-1580 (JASCO, Japan); injector, autosampler (AS-1555; JASCO); column, Hypersil ODS C18, 150 × 4.6 mm, 5 μm (Thermo Electron Corporation, Waltham, MA) with a Javelin Guard column (10 × 4.6 mm, 5 μm) filled with the same materials; detector, UV/visible (UV-1575; JASCO, Japan). Data acquisition and analysis were carried out using Borwin/HSS 2000 software (LG 1580–04; JASCO, Japan). The mobile phase was a mixture of 25% acetonitrile and 75% acidified water. The column was maintained at temperature of 20 °C and a flow rate of 1 ml/min with the detector set at wavelength of 320 nm. Under these conditions, FS and rutin were eluted at 15.1 and 4.8 min, respectively. Limit of detection and quantification for FS was 50 and 100 ng/ml, respectively. The FS calibration curve was linear (y = 0.096x + 0.053) at a concentration range of 100–3000 ng/ml with its correlation coefficients being 0.999. The ratio of peak area of FS/rutin was used for quantification of plasma samples.
Pharmacokinetic and statistically analysis
The pharmacokinetic parameters were analysed by a non-compartmentalized model with the aid of the programme WinNonlin version 4 (Pharsight Corp., Mountain View, CA). Peak plasma concentration (Cmax) and times to reach peak concentration (Tmax) were determined from the individual plasma concentration-time curves. The terminal elimination half-life (t1/2) was calculated as 0.693/Ke. The area under the plasma concentration–time curve (AUC) was calculated using the log-linear trapezoidal rule. Mean residence time (MRT) was calculated as AUMC/AUC. The apparent clearance (Cl/F) was calculated as dose/AUC. The differences in pharmacokinetic parameters of the study groups were statistically evaluated by the t-test. All values were expressed as their mean ± SD Difference was considered to be significant at a level of p < .05.
In-vivo toxicity assessment in rat
Healthy 8 weeks old female rat weighing 200 g fasted overnight with free access to water before the study. To investigate the toxicity of free FS, FS-PF-FA micelles, 18 female rats were randomly divided into three groups (six rats in each). Group I (vehicle-treated group) received distilled water by i.p route. Group II received free FS at the dose of 15 mg/kg (formulated in 30% PEG and 70% saline at a final concentration of 3 mg/ml) by i.p route. Group III received FS-PF-FA micelles at the dose of equivalent to 15 mg/kg by i.p route for 15 consecutive days. The animals were sacrificed by excess ether inhalation and necropsy was performed to analyse the macroscopic external features of vital organs, such as heart, liver, spleen and kidney. These organs were carefully removed and fixed in 10% buffered formalin and embedded in paraffin. Histology sections (5 μm thick) were stained with haematoxylin and eosin dye and examined under a light microscope (Olympus CH02; Olympus, Tokyo, Japan).
In-vitro anticancer study
In-vitro anticancer activity of free FS, FS-PF, FS-PF-FA and blank PF (B-PF-FA) micelles were evaluated against on folate overexpressed human breast cancer MCF-7 cells using in-vitro sulforhodamine B assay (SRB assay) [Citation29]. The cells were cultured in RPMI1640 medium, supplemented with 10% v/v foetal bovine serum (FBS) and 2 mM L-glutamic acid. Cells were seeded at the density of 5 × 103 cells per well in 96-well plates using in-situ fixing agent trichloroacetic acid (TCA). After 24 h of incubation at 37 °C with 100% relative humidity (RH), the growth medium was replaced with 100 μl of fresh medium containing various concentrations (1–15 μg/ml) of free FS in DMSO (the final concentration of DMSO should below 0.2%), FS-PF, FS-PF-FA and blank FS-PF-FA micelles. The culture media without any drug formulation was used as a control. After 48 h incubation, assay was terminated by adding 50 μl of the cold TCA and incubated for 60 min at 4 °C. The media was removed and washed with sterile PBS and dried. SRB of 50 μl solution (0.4% w/v in 1% acetic acid) was added to each well and further incubated for 20 min at room temperature. After staining, unbound dye was removed by washing with 1% acetic acid and plates were air dried. Bound stain was eluted with 10 mM trizma base and the absorbance was measured on an ELISA plate reader at a wavelength of 540 nm with 640 nm reference wavelength. Percent growth was calculated on a plate-by-plate basis for test results relative to control wells using the following equation. Cell growth (%) = (Avg absorbance of the test well/Avg absorbance of the control wells) × 100.Using the six absorbance measurement (time zero (Tz)), control growth (C) and test growth in the presence of drug at various concentration level (Ti), the percentage growth was calculated at each of the drug concentration levels, percentage growth inhibition was calculated as: [(Ti–Tz)/(C-Tz)] × 100 for concentration where Ti ≥ Tz or (Ti–Tz) is positive or zero; as [(Ti–Tz)/Tz] × 100 for concentration where Ti ≤ Tz or (Ti–Tz) is negative. Growth inhibition of 50% (GI50) was calculated from equation [(Ti–Tz)/(C-Tz)] × 100 = 50 as the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation.
Results and discussion
Over the past few decades, nanomedicine-based drug delivery systems have provided tremendous applicability in improving the reliability and safety of existing drugs. Naturally derived bioactives are preferred over synthetic drugs because of their relative safe and biocompatible nature. However, the low aqueous solubility, poor bioavailability, environmental and physiological instability hamper their clinical use. Many diseases like cancer need targeted drug delivery release which can be achieved by using novel drug delivery system. Therefore, nanomedicine-based drug delivery systems have been developed for various natural bioactives to improve safety, efficacy and patient compliance [Citation30–33]. Several edible plant-derived compounds have been linked to the chemoprevention and treatment of cancer [Citation34]. Among these natural compounds, the naphthoquinones have shown several pharmacological properties of interest in the prevention and treatment of cancer. FS, a naturally occurring bioactive, inhibits the growth of different types of cancers. Its poor aqueous solubility, targetability and toxicity continue to be highlighted as a major challenge in developing formulation for clinical efficacy. In this study, we developed PF and PF-FA conjugated micelles as a carrier for fisetin to enhance therapeutic efficacy and reduce its toxicity.
Synthesis of folate-conjugated pluronic (PF-FA)
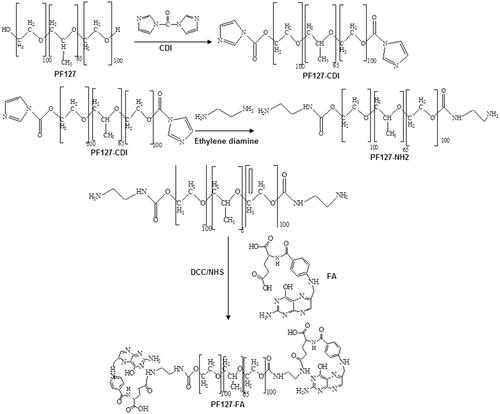
PF-FA was synthesized using the various molar ratios of FA to PF to ensure that at least one or more of the two hydroxyl groups of PF were conjugated with the carboxylic acid groups of FA. For synthesis of PF-FA, PF was first activated by CDI to form an imidazole carbamate intermediate (PF-CDI). Subsequently, PF-CDI reacted with ethylenediamine to form amino terminated PF-NH2. FA was reacted with DCC and NHS for activation of folate, from which carboxylic group reacts with PF-NH2 to achieve PF-FA conjugates. The synthetic scheme of PF-FA conjugate is shown in .
Figure 1. Synthetics scheme of folate-conjugated pluronic127 (PF127-FA).
Characterization of PF-FA conjugates
Fourier transform infrared spectroscopy
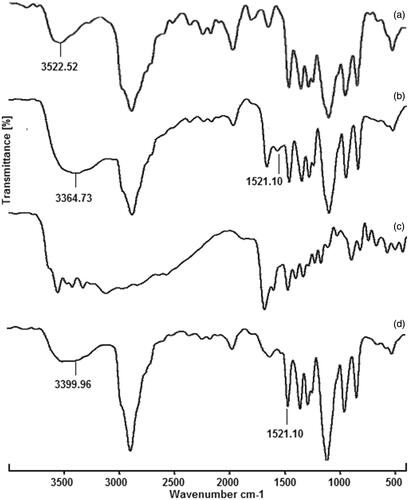
The FT-IR spectra of PF, PF-NH2, FA and PF-FA are shown in . PF showed characteristic hydroxyl group peak at 3522.5 cm−1 whereas PF127-NH2 showed a new absorption peak at 3364 and 1521 cm−1, corresponding to the amine group (N–H, broad) and carbonyl group (C = O) peak, respectively [Citation35]. In FA, the peaks appeared in between 3600 and 3000 cm−1 were of hydroxyl (–OH) stretching and N–H stretching vibrations. The C = O stretching vibration of carboxyl group appeared at 1692 cm−1, while the peak at 1640 cm−1 assigned to the C = O bond stretching vibration of –CO–NH group. The peak appeared at 1605 cm−1 was bending mode of N–H vibration and at 1412 cm−1 corresponds to the –OH deformation band of phenyl skeleton. The characteristic absorption peak of phenyl ring appeared at 1483 cm−1. In PF-FA, the absorption peaks occurring at 3399 cm−1 (N–H, stretch) and 1521 cm−1 (C = O, sharp) are associated with the amide group (–CO–NH) in the PF-FA structure. FTIR spectra were consistent with the desired polymer product [Citation36].
Figure 2. Fourier transform infrared spectra of (a) pluronic127 (PF127), (b) PF127-NH2 (c) folic acid (FA) and folate-conjugated pluronic127 (PF127-FA).
Proton NMR
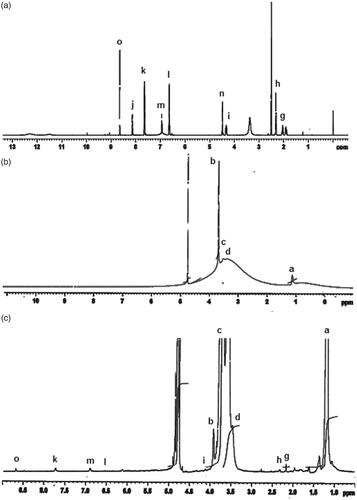
The appearance of characteristics peaks in the 1 H NMR spectrum of PF-FA conjugation confirms that FA is successfully incorporated with PF (). The NMR spectrum of FA exhibits typical peaks at 1.93 ppm (β-CH2 of glutamic acid), 2.07 ppm (γ-CH2 of glutamic acid), 4.51 ppm (methylene proton), 6.66 and 6.98 ppm (aromatic protons), 8.17 ppm (aliphatic amide proton) and 8.67 ppm (pteridine proton). The peaks at 2.50 and 3.30 ppm should be ignored as they are corresponding to DMSO and H2O peaks, respectively [Citation36]. The NMR spectrum of PF shows a broad peak at 3.47–3.84 ppm attributed to –CH2CHO and –CH2CH2O–segments of PPO and PEO and peak that are characteristic of –CH3 groups of PPO appears at 1.1 ppm. The spectrum of PF-FA contains both signals originating from FA and PF, but relatively weaker intensity for the signal of FA which may be due to the small molecular weight of FA (441 Da) compared to that of PF (12,500 Da). The degree of FA substitution onto PF was determined to be ∼0.09 mol%.
Figure 3. +H NMR spectra of (a) folic acid (FA), (b) pluronic127 (PF127) and (c) folate-conjugated pluronic127 (PF127-FA).
UV spectrometric analysis
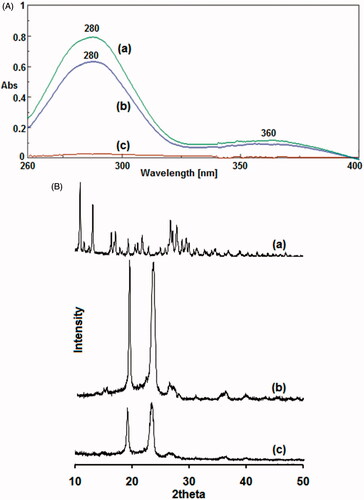
Preliminary determination was acquired using UV–VIS spectra in which PF does not show any absorbance peaks from 260 to 400 nm. FA showed its characteristic peaks at 280 and 360 nm. In PF-FA conjugates, the characteristic UV absorbance peaks around 360 and 280 nm appeared in the spectra indicating the presence of FA in the conjugate [Citation37] (). Degree of conjugation of FA onto PF was ∼2.69% as determined from UV.
Figure 4. A. UV–VIS absorbance spectra of (a) folic acid (FA), (b) folate-conjugated pluronic127 (PF127-FA) and (c) pluronic127 (PF127). B. Powder X-ray diffraction patterns of (a) folic acid (FA), (b) pluronic127 (PF127) and (c) folate-conjugated pluronic127 (PF127-FA).
Powder X-ray diffraction spectroscopy
PF-FA conjugates form partly amorphous rather than crystal structure as observed from powder X-ray diffraction pattern (). The diffractogram of FA showed characteristics diffraction peaks at 2Φ values of 10.79, 10.99, 12.25, 16.25, 16.97, 19.21, 20.35, 20.63, 21.61, 22.81, 26.63, 27.73, 28.05 and 29.35° indicating crystalline nature. PF showed sharp diffraction peaks at 19.17 and 23.33° indicating crystalline nature. PF-FA showed different diffraction peaks of PF at 2Φ values of 19.05 and 23.17° with less intensity and not observed FA peaks indicating amorphous form.
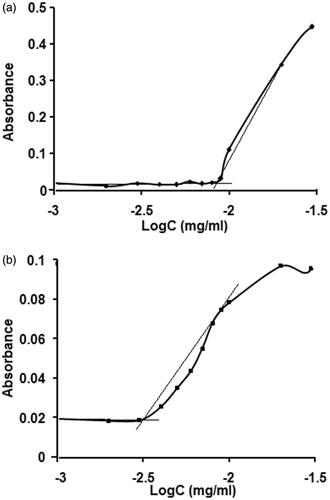
Critical micelle concentration
CMC is an important parameter for the stability of drug-loaded micelles, both in vitro and in vivo. In this study, iodine was used as a hydrophobic probe to monitor the formation of PF and PF–FA micelles. As a small hydrophobic molecule, iodine prefers to enter the hydrophobic microenvironment of copolymers causing the conversion of I3 to I2 from the excess KI in the solution which maintains the saturated aqueous concentration of I2. At the low concentration, PF-FA molecules are well dispersed in the aqueous medium. As the concentration increased, the free energy of the system rises because of unfavourable interaction between hydrophobic domain and at the surrounding water molecule. At a specific concentration, termed the CMC, amphiphilic molecules with the appropriate geometry, orients themselves in such a way that the hydrophobic segments are isolated from the aqueous environment, achieving a state of minimum energy that leads to the formation of colloidal assemblies termed micelles. The CMC of PF-FA was determined to be 0.03 mg/ml which was much lower than that of PF (0.09 mg/ml) (). The obvious decrease in CMC suggested that the PF-FA micelles would provide good stability for the drug in the suspension and make them relatively insensitive to dilution and gives them a longer circulation time compared to surfactant micelles in-vivo [Citation21].
Figure 5. Plot of the absorbance intensity of I2 as a function of (a) pluronic127 (PF127) and (b) folate-conjugated pluronic127 (PF127-FA) concentrations.
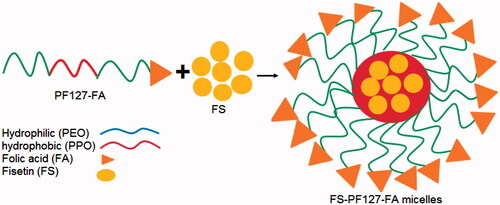
Fisetin-loaded PF-FA micelles
In micelles, inner hydrophobic core which involves hydrophobic–hydrophobic interaction between nonpolar head groups and the outer shell surface with hydrophilic groups. The micelles formed by PF-FA are viewed as a hydrophobic (PPO) core surrounded by a larger shell made from the hydrophilic (PEO) chain with their water of hydration (). The drug is immersed in hydrophobic core of the micelles by hydrophobic interactions and hydrogen bonds.
Figure 6. Schematic representation of FS-PF-FA micelles (fisetin loaded folate-conjugated pluronic127 micelles).
Characterization of FS-PF and FS-PF-FA micelles
Micelles size
Micelles size has a direct effect on the stability, drug release, biodistribution and cellular uptake. The micelles size measured by the laser diffraction technique is shown in . The mean size of FS-PF was 89.3 ± 4.1 nm whereas the mean size of FS-PF-FA was found to be increased to 103.2 ± 6.1 nm. The increased size of ligand conjugated micelles is in agreement with the literature. Clearly, this implies that FA contributes to interactions that favour the micelle growth. FA is composed of three primary structures 2-amino-4-hydroxy-6-methyl-pteridin, para-aminobenzoic acid and L-glutamic acid. Since it comprised of hydrophilic carboxyl and amino groups, it can interact via hydrogen bonding. Therefore, we speculate that attractive interactions between FA moieties in the micelle may lead to micellar growth and a broader size distribution. Sizes of both formulations are above 50 nm and below 200 nm which are suitable for efficient drug delivery via parenteral route [Citation38]. This is a point of distinctive advantage since nanoparticles bigger than 10 nm could escape renal clearance and smaller than 200 nm can escape phagocytosis by macrophages, have prolonged circulation half-lives and are more favourable for passive targeting to solid tumour via EPR effect.
Figure 7. A. Particle size distribution and B. Zeta potential of (a) fisetin-loaded pluronic127 micelles (FS-PF) and fisetin-loaded folate-conjugated pluronic127 micelles (FS-PF-FA).
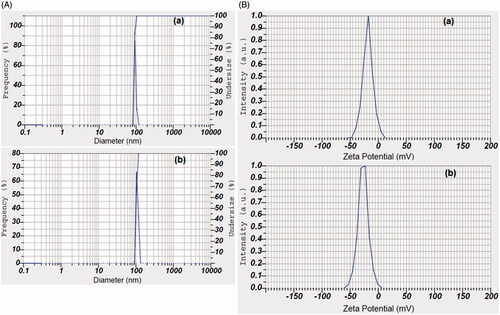
Zeta potential
Surface charges of the particles are reflected by zeta potential which plays a significant role in both colloidal stability and interaction with physiological body cells. The FS-PF showed negative zeta potential (−18.3 ± 0.65 mV) () which may be due to terminal polyethylene oxide (PEO) group of PF present on surface of micelle [Citation38], whereas FS-PF127-FA micelles showed higher negative zeta potential (−26.7 ± 0.44 mV) which may be due to presence of carboxylate groups of FA at the periphery [Citation39]. The high magnitude of negative zeta potential of micelles escarps from reticular endothelial system, have longer blood circulation time and reach to cancer cell through enhanced permeation retention effect (EPR).
Encapsulation efficiency
The encapsulation efficiency of FS-PF and FS-PF-FA were of 84.12 ± 2.15 and 82.50 ± 1.78%, respectively. Micelles achieved high encapsulation efficiency which may be due to bulky nonpolar head group of the PF molecules that form a strong hydrophobic core which interacts with FS and permits more encapsulation. Introduction of FA had negligible impact on encapsulation efficiency in micelles. PF block copolymer has much higher solubilization capacities and more selective towards aromatic and heterocyclic compounds than towards aliphatic molecules compared with conventional low molecular weight surfactants [Citation40]. Since FS molecules contain both aromatic and heterocyclic groups.
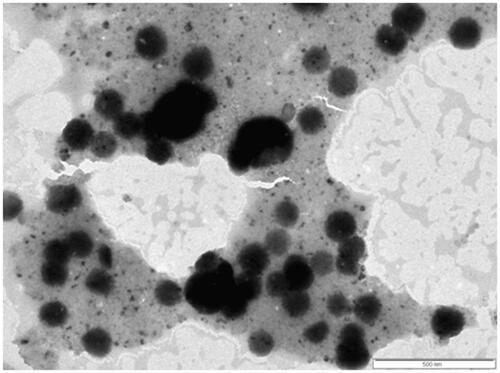
Surface morphology
Surface morphology of the FS-PF127-FA was assessed using transmission electron microscope (TEM) from which it can be seen that the micelles have smooth surfaces and are spherical in shape (). The micelles shown in the TEM are little bigger in size than that tested by laser diffraction technique, which is due to low melting point of pluronic (55 °C). The micelles undergo the melting and expansion to certain extent under the high energy electron beam in TEM which makes them seem bigger in TEM image than in laser diffraction test [Citation41].
Figure 8. Transmission electron microscopy image of FS-PF-FA (fisetin-loaded folate-conjugated pluronic127 micelles).
In-vitro drug release
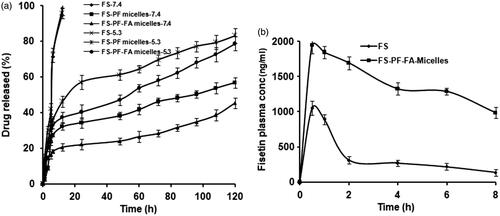
The fate of a drug from a drug carrier depends on various experimental factors, such as pH, degradation rate, particle size, interaction between drug and the surface and behaviour of polymer in solvent. FS-PF-FA micelles were exposed to pH 7.4 (corresponding to physiological pH) and 5.3 (to mimic the slightly acidic environment in the MCF-7 breast cancer cells) conditions at 37 °C to measure the release of FS in vitro and evaluate the feasibility of using FS-PF-FA micelles to target MCF-7 breast cancer cells. The FS release from micelles is shown in . FS-PF/FS-FA-PF micelles showed similar release patterns differing only slightly in terms of burst and cumulative drug release. The release of free FS was found to be rapid and reached 100% cumulative almost in less than 12 h. In case of micelles, an initial burst release was observed for first 12 h (30–40%), after which the release of FS from micelles increased steadily up to 120 h. It was observed that at pH 5.3, about 80% of FS was released over a 5-d period, whereas only about 50% FS was released in the same period at pH 7.4 which could be due to higher partitioning of FS at acidic pH [Citation42]. This could be helpful and add to the advantage of selective and targeted uptake in cancer cells where the pH is slightly acidic compared to normal physiological pH could be due to the enhanced solubility of FS at acidic pH resulting in a higher drug release [Citation43]. The pH-dependent release may help to improve efficiency of FS-PF-FA micelles as efficient delivery system, where normal cells are not affected. The acidic environment in the endosome may trigger the rapid release of FS from the FS-PF-FA micelles, thereby greatly enhancing the cell cytotoxicity.
Figure 9. Cumulative (a) FS release from FS-PF micelles and FS-PF-FA micelles at 37 °C in phosphate buffer solution (pH 5.3 and 7.4). The data points are average of at least three experiments. Error bars represent the range over which the value was observed. (b) The mean plasma concentration-time profiles of FS after i.p administration of FS solution and FS-PF-FA micelles in rat. Each point refers to mean ± SD (n = 3).
Pharmacokinetics study in rats
The influence of the micelles formulation on FS bioavailability was examined in rats. Upon intravenous injection of a classical nanoparticle, the particles interact with the apolipoproteins and are captured by the reticulo-endothelial system leading to their rapid elimination from the blood compartment [Citation44]. To avoid this followed i.p. route of administration for the bioavailability studies [Citation16]. Plasma levels of FS from FS-PF-FA micelles were compared with free FS (). FS-PF-FA micelles yielded higher FS plasma concentrations as compared to free FS. The relevant pharmacokinetic parameters derived by non-compartmental analysis are listed in . The FS-PF-FA micelles showed 1.8-, 2-, 6.3- and 2.2-fold increase in Cmax, t1/2, AUC and MRT (p < .05), respectively. There was no significant difference in the time to reach peak concentration (Tmax) of the FS-PF-FA micelles and free FS.
Table 1. Mean non-compartment pharmacokinetic parameters of FS after single i.p dose (15 mg/kg) administration of FS-PF-FA micelles and free FS in rat plasma.
FS-PF-FA micelles displayed higher Cmax and long terminal half-life. This would lead to long residence time with slow plasma elimination and consequent remarkable improvement in in-vivo anticancer effect. As compared with free FS, FS-PF-FA micelle showed lower clearance due to less metabolism through glucuronidation; because of micelle structure protects the entrapped drug from degradation or metabolism. The enhanced FS bioavailability is probably due to faster absorption and the slow drug release from micelles causing prolonged residence time in blood. Intraperitoneal administration also favours the lymphatic distribution of drugs, especially anticancer drugs which must access the lymph nodes that frequently harbour metastases [Citation45]. Similar results were reported for the hydrophobic anticancer paclitaxel where i.p. administration led to a significant improvement in its bioavailability [Citation46].
In-vivo toxicity assessment in rats
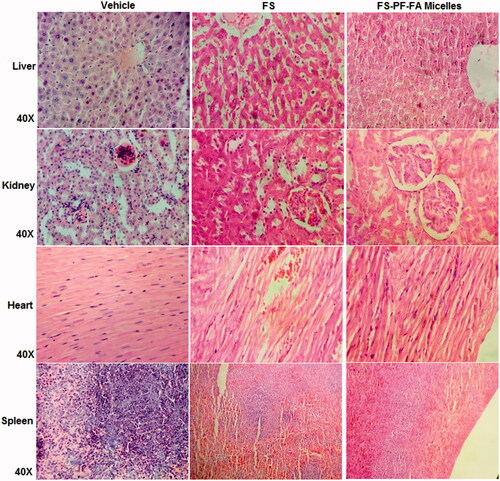
FS-PF-FA micelles reduced systemic toxicity as compared to free FS which was reflected in tissue toxicity study by macroscopic examination on vital organs. As shown in , the free FS caused abnormal tissue toxicity such as congested sinusoids, moderate degeneration of hepatocytes, perivascular cuffing, hepatocellular necrosis and focal infiltration of inflammatory cells in liver; myocardial necrosis, infiltration of inflammatory cells and myocardial degeneration, congested blood vessels, minimal haemorrhages and mild myocardial necrosis in heart; congested blood vessels, severe necrosis of tubules and glomeruli, lymphocytic infiltration and congestion of blood vessels, congested glomeruli, moderate tubular degeneration in kidney; markedly reduced white pulp and presence of megakaryocyt, reduced size of follicle, minimal haemorrhages in spleen, whereas FS-PF-FA micelles treatment did not cause any significant tissue toxicity on vital organs. The reason behind the reduced systemic toxicity for FS-PF-FA micelles formulations could be attributed to the slow release of the FS from the micelles over prolonged period of time in comparison to the free FS which is released in 8 h duration as evidenced in pharmacokinetics study. Also, the survival data further supports the reduction in the systemic toxicity with the FS-PF-FA micelles with 10% increased life span (ILS) in comparison to free FS. The results demonstrated no signs of normal tissue toxicity in FS-PF-FA micelles treated group indicating its nontoxic nature. This study clearly substantiates the safety of FS-PF-FA micelles supporting the need for further clinical exploration.
Figure 10. Tissue toxicity assessment of FS and FS-PF-FA micelles (fisetin loaded folate-conjugated pluronic127 micelles) in rats. The histopathologic examination of tissue sections of liver, kidney, heart and spleen was made after rats were sacrificed and staining with H and E stain; magnification 40X.
In-vitro anticancer study
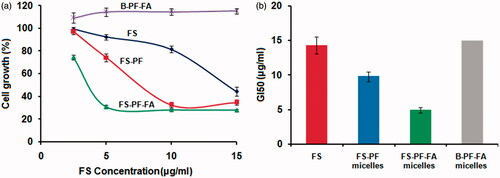
The in-vitro anticancer activity of FS, FS-PF, FS-PF-FA and blank FS-PF-FA micelles were investigated against folate overexpressed human breast cancer MCF-7 cells using in vitro SRB assay ().The advantages of the PF-FA micelles formulation of FS was quantified by the GI50 value which is defined as the drug concentration needed to inhibit 50% of the incubated cells growth in a designed time period [Citation47]. From , it is observed that the GI50 for FS is 14.3 ± 1.2 µg/ml while it was greatly decreased to 9.8 ± 0.78 µg/ml, i.e. a 31.46% decrease for the FS-PF. Furthermore, the GI50 value for FS-PF-FA was 4.9 ± 0.4 µg/ml, i.e. a 65.737% decrease compared to FS and 50% decrease compare to FS-PF (). The enhanced anticancer activity of FS-PF micelles is attributed to the enhanced solubility of FS in micelles solution, increased stability of FS inside the micelles core, high drug transportation by passive targeting to cancer cells, the controlled drug release and P-gp inhibition properties of PF [Citation48]. Further, improved anticancer activity of FS-PF-FA demonstrated active targeting effects to the cancer cells which may suggest that the FA-conjugated micelles were endocytosed via a folate receptor-mediated mechanism [Citation49].
Figure 11. (a) Microphotograph of MCF-7 cancer cell (b), (c), (d) and (e) FS, FS-PF micelles, FS-PF-FA micelles and blank PF-FA micelles treated MCF-7 cancer cell.
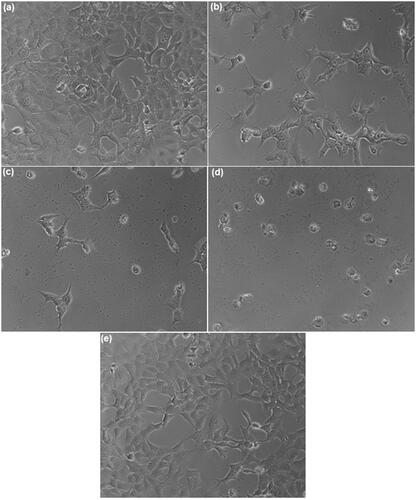
Figure 12. (a) The antiproliferative effect at different concentration in MCF-7 cells was determined using the SRB assay and (b) GI50 value of FS, FS-PF micelles, FS-PF-FA micelles and blank PF-FA micelles on MCF-7 cancer cell. Each point represents an average ± SD (n = 3).
Conclusions
In this study, FS loaded FA-conjugated PF micelles (FS-PF-FA micelles) were successfully prepared. The FS-PF-FA micelles displayed smaller in size, smooth surfaces with spherical in shape, good encapsulation efficiency, pH-dependent FS release with no sign of tissue toxicity. Further, the FS-PF-FA micelles demonstrated active targeting effect on folate overexpressed human breast cancer MCF-7 cells and displayed 6-fold enhanced intraperitoneal bioavailability as compared to FS solution which may bring about reduction in dose as well as cost and increase patient compliance. Our results indicate that FS-PF-FA micelles may be promising vehicles for delivering FS to cancer cells that overexpress this receptor. Further studies will focus on the in-vivo antitumor efficiency and the optimization of the drug dose to obtain the most effective dose with less side effects during the treatment.
Acknowledgements
The authors sincerely acknowledge Tata Memorial Center, Advanced Centre for Treatment Research and Education in Cancer (ACTREC), Mumbai, India for the in vitro anticancer study. They also wish to acknowledge Department of Physics and chemistry, Pune University, Pune, India, for availing SEM and 1 H NMR facility.
Disclosure statement
The authors declare that they have no conflicts of interest to disclose.
Related Research Data
References
- Akhter MH, Rizwanullah M, Ahmad J, et al. Nanocarriers in advanced drug targeting: setting novel paradigm in cancer therapeutics. Artif Cells Nanomeds Biotechnol. 2017 [Aug 22]; [12 p.]. DOI:https://doi.org/10.1080/21691401.2017.1366333
- Arai Y, Watanabe S, Kimira M, et al. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J Nutr. 2000;130:2243–5220.
- Inkielewicz-Stepniak I, Radomski MW, Wozniak M. Fisetin prevents fluoride and dexamethasone-induced oxidative damage in osteoblast and hippocampal cells. Food Chem Toxicol. 2012;50:583–589.
- Cho Y, Chung JH, Do HJ, et al. Effect of fisetin supplementation on hepatic lipogenesis and glucose metabolism in Sprague-Dawley rats fed on a high fat diet. Food Chem. 2013;139:720–727.
- Ju YK, Young KJ, Won J, et al. Fisetin induces apoptosis in Huh-7 cells via down regulation of BIRC8 and Bcl2L2. Food Chem Toxicol. 2010;48:2259–2264.
- Raygude KS, Kandhare AD, Ghosh P, et al. Anticonvulsant effect of fisetin by modulation of endogenous biomarkers. Biomed Prevent Nutr. 2012;2:215–222.
- Touil YS, Seguin J, Scherman D, et al. Improved antiangiogenic and antitumour activity of the combination of the natural flavonoid fisetin and cyclophosphamide in Lewis lung carcinoma-bearing mice. Cancer Chemother Pharmacol. 2011;68:445–455.
- Tripathi R, Samadder T, Gupta S, et al. Anticancer activity of a combination of cisplatin and fisetin in embryonal carcinoma cells and xenograft tumors. Mol Cancer Ther. 2011;10:255–268.
- Yu SH, Yang PM, Peng CW, et al. Securin depletion sensitizes human colon cancer cells to fisetin-induced apoptosis. Cancer Lett. 2011;300:94–104.
- Adhami VM, Syed DN, Khan N, et al. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol. 2012;84:1277–1281.
- Chen Y, Wu Q, Song L, et al. Polymeric micelles encapsulating fisetin improve therapeutic effect in colon cancer. ACS Appl Mater Interfaces. 2014;14:534–542.
- Chien CS, Shen KH, Huang JS, et al. Antimetastatic potential of fisetin involves inactivation of the PI3K/Akt and JNK signaling pathways with downregulation of MMP-2/9 expressions in prostate cancer PC-3 cells. Mol Cell Biochem. 2010;333:169–180.
- Khan N, Asim M, Afaq F, et al. A novel dietary flavonoid fisetin inhibits androgen receptor signaling and tumor growth in athymic nude mice. Cancer Res. 2008;68:8555–8563.
- Mignet N, Seguin J, Ramos Romano M, et al. Development of a liposomal formulation of the natural flavonoid fisetin. Int J Pharm. 2012;423:69–76.
- Touil YS, Auzeil N, Boulinguez F, et al. Fisetin disposition and metabolism in mice: identification of geraldol as an active metabolite. Biochem Pharmacol. 2011;82:1731–1739.
- Seguin J, Brulle J, Boyer R, et al. Liposomal encapsulation of the natural flavonoid fisetin improves bioavailability and antitumor efficacy. Int J Pharm. 2013;444:146–154.
- Ragelle H, Crauste-Manciet S, Seguin J, et al. Nanoemulsion formulation of fisetin improves bioavailability and antitumour activity in mice. Int J Pharm. 2012;427:452–459.
- Crauste-Manciet S, Larquet E, Khawand K, et al. Lipidic spherulites: formulation optimisation by paired optical and cryoelectron microscopy. Eur J Pharm Biopharm. 2013;85:1088–1094.
- Capek I. Degradation of kinetically-stable o/w emulsions. Adv Colloid Interface Sci. 2004;107:125–155.
- Bothiraja C, Yojana BD, Pawar AP, et al. Fisetin-loaded nanocochleates: formulation, characterisation, in vitro anticancer testing, bioavailability and biodistribution study. Expert Opin Drug Deliv. 2014;11:17–29.
- Pawar A, Patel R, Arulmozhi S, et al. D-α-Tocopheryl polyethylene glycol 1000 succinate conjugated folic acid nanomicelles: towards enhanced bioavailability, stability, safety, prolonged drug release and synergized anticancer effect of plumbagin. RSC Adv. 2016;6:78106–18121.
- Jadhav P, Bothiraja C, Pawar A. Resveratrol-piperine loaded mixed micelles: formulation, characterization, bioavailability, safety and in vitro anticancer activity. RSC Adv. 2016;6:112795–112805.
- Yasir M, Sara UVS, Chauhan I, et al. Solid lipid nanoparticles for nose to brain delivery of donepezil: formulation, optimization by Box–Behnken design, in vitro and in vivo evaluation. Artif Cells Nanomeds Biotechnol. 2017 [Aug 26]; [14 p.]. DOI:https://doi.org/10.1080/21691401.2017.1394872
- Rajpoot K, Jain SK. Colorectal cancer-targeted delivery of oxaliplatin via folic acid-grafted solid lipid nanoparticles: preparation, optimization, and in vitro evaluation. Artif Cells Nanomeds Biotechnol. 2017 [Aug 29]; [12 p.]. DOI:https://doi.org/10.1080/21691401.2017.1366338
- Chiani M, Norouzian D, Shokrgozar MA, et al. Folic acid conjugated nanoliposomes as promising carriers for targeted delivery of bleomycin. Artif Cells Nanomeds Biotechnol. 2017 [Jun 23]; [7 p.]. DOI:https://doi.org/10.1080/21691401.2017.1337029
- Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153:198–205.
- Eliezar J, Scaranoa W, Boaseb NR, et al. In vivo evaluation of folate decorated crosslinked micelles for the delivery of platinum anticancer drugs. Biomacromolecules. 2015;16:515–523.
- Saxena V, Hussain MD. Poloxamer 407/TPGS mixed micelles for delivery of gambogic acid to breast and multidrug-resistant cancer. Int J Nanomedicne. 2012;7:713–721.
- Bothiraja C, Kapare HS, Pawar AP, et al. Development of plumbagin-loaded phospholipid-Tween® 80 mixed micelles: formulation, optimization, effect on breast cancer cells and human blood/serum compatibility testing. Ther Deliv. 2013;4:1247–1259.
- Bothiraja C, Pawar AP, Shaikh KS, et al. Eudragit EPO based nanoparticles suspension of andrographolide: in vitro and in vivo. Nanosci Nanotechnol Lett. 2009;10:156–164.
- Bothiraja C, Pawar AP. Improved bioavailability of orally administered andrographolide from pH-sensitive nanoparticles. Eur J Drug Metab Pharmacokinet. 2011;35:123–129.
- Bothiraja C, Pawar AP. Andrographolide, a novel bioactive phytoconstituent encapsulated in sustained release biodegradable nanoparticles. Int J Nanotechnol. 2011;8:764–778.
- Bothiraja C, Thorat UH, Pawar AP, et al. Chitosan coated layered clay montmorillonite nanocomposites modulate oral delivery of paclitaxel in colonic cancer. Mater Sci Technol. 2014; 29:B120–B126.
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780.
- Kwak M, Musser AJ, Lee J, et al. DNA-functionalised blend micelles: mix and fix polymeric hybrid nanostructures. Chem Commun. 2010;46:4935–4937.
- Lin JJ, Chen JS, Huang SJ, et al. Folic acid-Pluronic F127 magnetic nanoparticle clusters for combined targeting, diagnosis, and therapy applications. Biomaterials. 2009;30:5114–5124.
- Butt AM, Mohd Amin MC, Katas H. Synergistic effect of pH-responsive folate-functionalized poloxamer 407-TPGS-mixed micelles on targeted delivery of anticancer drugs. Int J Nanomedicine. 2015;10:1321–1334.
- Suthiwangcharoen N, Nagarajan R. Controlled design and construction of multifunctional nanoparticles by molecular self-assembly. RSC Adv. 2014;4:10076–10089.
- Varshosaz J, Hassanzadeh F, Sadeghi Aliabadi H, et al. Synthesis and characterization of folate-targeted dextran/retinoic acid micelles for doxorubicin delivery in acute leukemia. Bio Med Res Int. 2014;2:1–14.
- Niu J, Wang A, Ke Z, et al. Glucose transporter and folic acid receptor-mediated Pluronic P105 polymeric micelles loaded with doxorubicin for brain tumor treating. J Drug Target. 2014;22:712–723.
- Zhang D, Tao L, Zhao H, et al. A functional drug delivery platform for targeting and imaging cancer cells based on Pluronic F127. J Biomater Sci Polym Ed. 2015;26:468–482.
- Dalmark M, Storm HH. A Fickian diffusion transport process with features of transport catalysis. Doxorubicin transport in human red blood cells. J Gen Physiol. 1981;78:349–364.
- Zhao X, Liu P. Reduction-responsive core-shell-corona micelles based on triblock copolymer: novel synthetic strategy, characterization, and application as tumor microenvironment-responsive drug delivery system. ACS Appl Mater Interfaces. 2014;7:166–174.
- Afsharzadeh M, Hashemi M, Mokhtarrzadeh A, et al. Recent advances in codelivery systems based on polymeric nanoparticle for cancer treatment. Artif Cells Nanomeds Biotechnol. 2017 [Sep 28]; [16 p.]. DOI:https://doi.org/10.1080/21691401.2017.1376675
- Nishioka Y, Yoshino H. Lymphatic targeting with nanoparticulate system. Adv Drug Deliv Rev. 2001;47:55–64.
- Soma D, Kitayama J, Ishigami H, et al. Different tissue distribution of paclitaxel with intravenous and intraperitoneal administration. J Surg Res. 2009;155:142–146.
- Muthu MS, Kulkarni SA, Liu Y, et al. Development of docetaxel-loaded vitamin E TPGS micelles: formulation optimization, effects on brain cancer cells and biodistribution in rats. Nanomedicine. 2012;7:353–364.
- Pindiprolu SKSS, Krishnamurthy PT, Chintamaneni PK, et al. Nanocarrier based approaches for targeting breast cancer stem cells. Artif Cells Nanomeds Biotechnol. 2017 [Aug 21]; [14 p.]. DOI:https://doi.org/10.1080/21691401.2017.1366337
- Harei A, Osouli M, Bayat F, et al. Nanomedicine approaches for sirolimus delivery: a review of pharmaceutical properties and preclinical studies. Artif Cells Nanomeds Biotechnol. 2017 [Nov 29]; [14 p.]. DOI:https://doi.org/10.1080/21691401.2017.1408123