
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Loss of dopamine-secreting neurons in the midbrain causes Parkinson’s disease. L-DOPA, the precursor to the neurotransmitters dopamine, crosses vast majority of physiological and biochemical barriers that dopamine cannot. But most levodopa is decarboxylated to dopamine before it reaches the brain. This causes to little therapeutic gain with strong peripheral side effects. Benserazide is an irreversible inhibitor of peripheral aromatic L-amino acid decarboxylase that prevents the breakdown of levodopa in the bloodstream. The challenges are to increase the therapeutic efficiency, the bioavailability and decreasing the unfavourable side effects of Levodopa drug. Biocompatible nano-sized drug carriers could address these challenges at molecular level. Thus calculations of drug loading ability of acid-functionalized CNT for the benserazide as a nanodug carrier complex for L-DOPA were performed. In this regard, evaluation of all adsorption features of the most stable conformer of benserazide molecule onto carboxylated carbon nanotube is critical. To determine the minimum energy conformer of benserazide, the molecular structure and conformational analysis of 512 possible conformers have been subjected to first principle quantum mechanical calculations. Our work established a novel and easy-to-make formulation of benserazide/carboxylated CNT conjugate with extremely high drug loading efficiency of Levodopa for Parkinson disease treatment.
Introduction
Preserving drugs from deactivation throughout the physiological state of body is necessary to improve the physicochemical properties and targeting of the pharmacological profile.
Provide drug levels within the therapeutic range, decrease in toxicity and side effects, decrease the dosage of drug, increase in efficacy of drugs and bioavailability are some of the advantages of targeting of drugs [Citation1]. Conformational dependent of drug bioactivity is a new physical insight in quantum pharmacology which affects the mode of drug interactions. In molecular medicine chemistry, the foremost step in drug design is to investigate the desirable drug-receptor interactions. Interfunctional group geometry distances in drug-receptor binding are based on the correct fit between the active sites of preferred energetic conformer of drug and receptor. Correct and stable geometry of drug prevent the physical steric repulsion and thus enhance the pharmacological activity of drug. It’s worth mentioning that the electrostatic repulsion of drug-receptor binding interactions directly correlates with the drug electron distribution. Conformational analyses of drugs help us to investigate the stereoelectronic properties of drugs which ultimately influence the biological activity.
Parkinson’s disease is thought to be caused by too little of dopamine in the brain [Citation2]. To help controlling movement, levodopa, precursor to the neurotransmitter dopamine, changes into dopamine in the brain. The compound benserazide (BZ) (DL-serine-2-[(2,3,4-trihydroxyphenyl)methyl] hydrazide) (C10H15N3O5) an irreversible inhibitor of peripheral aromatic L-amino acid decarboxylase (AADC), with L-Dopa (LD) is a combined therapy for age-related neurodegenerative disorder, Parkinson disease. Most LD is decarboxylated to dopamine (D) before it reaches the brain since that is unable to cross the brain–blood barrier. Drug inhibited dopamine decarboxylate allows dopamine to build up solely in brain instead. BZ at the recommended therapeutic dose not crossing the blood–brain barrier to any significant degree and combined therapy with levodopa reduces the amount of LD required for optimal therapeutic benefit and permits an earlier response to therapy and it is rapidly absorbed after oral administration. Benserazide prevents the breakdown of levodopa in the bloodstream and inhibits the aforementioned decarboxylation, so more levodopa can enter the brain and also reduce some of levodopa’s side effects [Citation3,Citation4].
Hydrogen bonding which exists in all states of matter play dominant role in biological phenomena and chemical reactions. The HB geometrical and activity influences are one of the most important problems in pharmacology thus the prediction of intramolecular hydrogen bonding (HB) properties are crucial. These strong interactions, which first was purposed by Benesi and Hildebrand as charge transfer bonds [Citation5], are formed by different parts of the same molecules or ions that lead to the creation of the so-called quasi-ring attached to the other rings, aromatic or heterocyclic systems. The non-covalent HBs interactions are significantly weaker than a covalent chemical bond. The proton donating and acceptor bonds, substituent, hybridization and solvent are the most important factors that influence the HBs [Citation6].
Drug delivery systems (DDS) develop platforms for smart drug release and real-time assessments of therapeutic. Nanomaterial with their intriguing properties has attracted great interests in active nanotherapeutic targeting [Citation7,Citation8]. Meanwhile, carbon nanotubes that are able to penetrate biological membranes and possess loading capability of bioactive agents offer excellent medical applications [Citation9–12].
Pristine carbon nanotubes (CNT) without any handing bonds are chemically inert [Citation13,Citation14]. Because of the hydrophobic nature which inhibits the dispersion in biological media, they are incompatible with all solvents and cannot be employed in DDS. Functionalized CNT with improved solubility and multiple attachment sides can be loaded with drugs. They can form supramolecular complexes with non-covalent interactions and covalent bonds and encapsulation of drugs inside the tube. They also have offered new applications in nanomedicine with cellular uptake of drugs which can be promoted with their capacity of cell penetration. CNTs can be modified by introducing carboxylic groups. The presence of the oxygen-containing groups in carboxylated carbon nanotube (c-CNT) with excellent biocompatibility can offer the probability of introducing the targeting and drugs at the same time [Citation15].
The major goal of this study is to explore a novel drug delivery as a guest-(c-CNT) conjugate through the modification of c-CNT by benserazide to improve targeting Levodopa therapy of Parkinson. We have investigated the potential applications of c-CNT for loading and interaction of benserazide using ab initio DFT simulation.
The study is categorized under the following heads:
Finding optimized geometry of benserazide, c-CNT and the complexes formed by the non-covalent interaction of benserazide with c-CNT.
Calculation of adsorption energies and possible hydrogen bindings for the result complexes.
Investigation of the inter–intra interactions in terms of AIM and NBO analysis results.
Modulation in electronic properties of c-CNT due to the effect of the adsorption of benserazide in terms of HOMO-LUMO energy gap, the global reactivity descriptors and density of states (DOS).
Computational details
Theoretical calculations were performed using Gaussian 09 program package (Gaussian, Inc., Wallingford, CT) [Citation16]. Geometry optimization was performed at density functional theory level using different basis sets. Beckes three-parameter hybrid exchange functional [Citation17] (B3) combined with both the Lee-Yang-Parr gradient-corrected correlation functional (LYP) were utilized in DFT calculations. All the atoms are allowed to relax during geometry optimization. Positive values for wavenumbers verify the stability of the geometries. A natural bond orbital (NBO) analysis [Citation18] using natural population analysis (NPA) of optimized structures was performed to understand the orbital interactions and charge transfer investigations. The characteristics of the bond critical point (BCP) and ring critical point (RCP) which analysed with the electron density at the critical point (ρ) and its Laplacian (∇2ρ) were performed within Bader’s atoms in molecules theory (AIM) using the AIM2000 code [Citation19] to study the nature of intramolecular HB.
All possible attachments of the functional groups of the benserazide molecule onto to the carboxylated site of the SWCNT were investigated to find the most preferred mode of adsorption.
The adsorption energy of BZ onto c-CNT is calculated using the following equation:
(1)
(1)
where EBZ/c-CNT is the total energy of c-CNT with BZ molecule and EBZ and Ec-CNT are the total energy of BZ molecule and c-CNT, respectively.
The chemical reactivity and site selectivity of the complexes were explained by quantum molecular DFT based descriptors like global hardness (η), chemical potential (μ) and electrophilicity index (ω).
In an N-electron system chemical potential and chemical hardness are given as [Citation20]:
(2)
(2)
(3)
(3)
where μ and η are defined as the first and second derivatives of the total energy with respect to the external potential, respectively.
According to Parr, the electrophilicity index defined as [Citation21]:
(4)
(4)
Quantum theory of atoms in molecules is applied to analyse the total electron density at the critical points (HC) in terms of the potential electron density (VC) and the kinetic electron density (GC) at the bond critical points (BCPs) which expressed in the following relation [Citation22]:
(5)
(5)
The following relationships were assigned to derive the HB by Abramov [Citation23]:
(6)
(6)
(7)
(7)
Results and discussion
Quantum chemical studies of benserazide
Conformational properties of BZ
Quantum mechanics calculations enable us to express the geometry of molecules as a function of energy and by minimizing this energy function, the optimized geometries can be determined. In this study, we assumed the benserazide drug as a flexible molecule and without any conformational constraint, 512 different conformers were found to explore the most stable and most probable nano conformer correspond to energy minima with lowest steric and electrostatic repulsion which would contribute in pharmacokinetic with levodopa, the Parkinson disease drug, which in body converts to dopamine. After all, the intermolecular hydrogen bonding in benserazide molecule, which is the main interaction stabilizing the nano conformers affect the biological activity, were investigated.
On the basis of the conformer’s standard theoretical definition, BZ with 33 atoms has 512 conformers. Based on hydrogen bonded to N–N bond, we group all these conformers into two classes: cis and trans with 256 and 256 conformers, respectively. Based on functional groups OH and NH2 groups these conformers can be classified into four cases: TT (trans H&H and trans OH&NH2), CC (cis H&H and cis OH&NH2), TC (trans H&H and cis OH&NH2), CT (cis H&H and trans OH&NH2), that each one consists of 128 conformers. These conformers optimized at HF/6–311 G (d, p) and B3LYP/6–311++G (d, p) level. Our theoretical calculations on BZ show that its TT conformers are more stable than the others. Probing all hydrogen-bonded and non-hydrogen-bonded systems in all conformers by mean energy calculations illustrate the following stability orders for BZ conformers.
TT > CT > TC > CC
As shown in most recent reports, the DFT/B3LYP type calculations have been successful in predicting various molecular properties than the other ab initio (such as HF and MP2) type. The title conformers show two homonuclear intramolecular HBs, i.e. O–H···.O and N–H···.N and two heteronuclear HBs, i.e. O–H···N and N–H···O. The most important changes concern the distance of the hydrogen bridge atom, the extension of the O–H and N–H bond lengths and the shortening of the O(H)···O and N(H)···N distances.
TT group
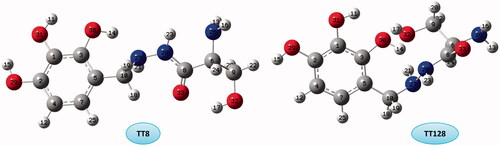
TT group included 128 conformers that they are more stable than the other conformers. The results of theoretical calculations on the stability orders of TT conformers at all levels of theory illustrate that TT8 is the most stable and TT128 is the less stable (). O–H···O and O–H···N HBs are seen in both conformers but TT8 shows N–H···N and TT128 shows N–H···O HBs.
Figure 1. The optimized structure and numbering of the most stable and unstable of TT conformers.
CC group
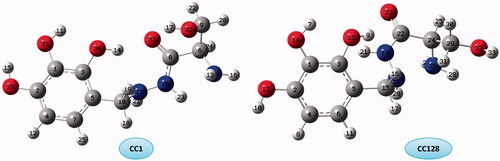
CC group has 128 conformers that they are less stable than the other conformers. Theoretical calculations of CC conformers at all levels of theory shows that CC1 and CC128 are the most stable and the less stable conformers, respectively (). O–H···O and O–H···N HBs are seen in both conformers but only CC1 shows N–H···N HB.
Figure 2. The optimized structure and numbering of the most stable and unstable of CC conformers.
TC group
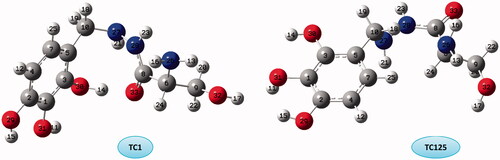
The comparison of the relative energies of the different TC conformers shows that the most stable conformer is TC1 with O–H···O and N–H···N HBs and TC125 is the most unstable one without any intramolecular HB ().
Figure 3. The optimized structure and numbering of the most stable and unstable of TC conformers.
CT group
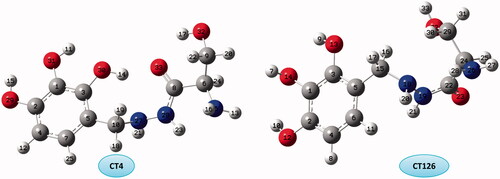
CT4 conformer with all of HBs types except N–H···O is the most stable conformer and CT126 with only N–H···N HB is the unstable conformer in this group ().
Figure 4. The optimized structure and numbering of the most stable and unstable of CT conformers.
Our theoretical calculations on BZ show that CT4 and CT26 are the most stable and unstable conformers than the others, respectively.
QTAIM: relation between geometrical and topological parameters
We apply density functional theory (DFT) and QTAIM theory to analyse the properties of the intramolecular HBs interactions of the most stable and unstable systems described.
Analysis of bond critical point (BCP) properties
To get deeper insight into the characteristics of intramolecular HBs, wide spectrum of HBs within the species have been analysed [Citation24–27]. The optimized structures of title compounds are shown in with numbering of the atoms. The optimized structure parameters of studied molecules are listed in in accordance with the atom numbering given in . shows O–H bond lengths of the most stable conformer (CT4), are detected in the range of 0.98–0.99 Ǻ, the H···O contacts are of about 1.98–2.13 Ǻ and H···N contacts are about 2.17–2.35 Ǻ. In the case of N–H bond the electron density at the corresponding BCP amounts to 0.3138 Å and for H···N interaction such electron density amounts to 0.024 Å. Besides, the corresponding Laplacian values of the O···H and N···H interaction contacts are positive. The positive Laplacian is the topological evidence of the noncovalency of closed-shell interactions [Citation28–30]. A very well-known relationship between the proton-donating bond length and the proton-acceptor distance often found for studied conformers. It indicates that the greater the elongation of hydrogen donor bond is the shorter the hydrogen acceptor contact is which means the stronger is the interaction. The electron density at the corresponding bond length reflects the strength of any pair interactions: this density is greater for shorter distances. presents the molecular graph of the most stable conformer.
Figure 5. The molecular graphs of CT4 obtained from the B3LYP/6–31 G(d,p) wave function.
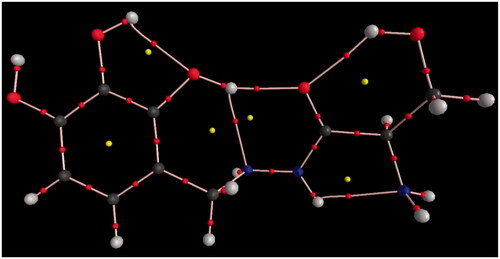
Table 1. The geometrical parameters (bond distance in Å) and topological parameters (in a.u.) of in more stable conformers.
Analysis of ring critical point (RCP) properties
The characteristics of corresponding RCP are also analysed for investigated conformers. It was found that the ring critical point properties may be often treated as the hydrogen bond strength measure. Here the pseudo-ring containing N–H···N, O–H···N and O–H···O intramolecular HBs is created and hence also the RCP exists. The characteristics of RCPs of studied conformers are given in . It is worth mentioning that the greater electron density at RCP of intramolecular HB corresponds to the stronger interaction.
The intramolecular hydrogen bond energy
In this investigation, the approximate values of the intramolecular HB energies were estimated by the Espinosa and Molins method [Citation31]. The excellent relationship obtained between the H-bond energy and the electron density at the H···O and H···N bond critical point. In the most stable investigated conformer, CT4, the values of O–H···O energies are −19.97, −28.4 and −39.11 and the O–H···N and N–H···N energies are −12.73 and −23.97 (kcalmol−1), respectively, which reveal the relationship between the high H-bond energies and the stability. The ratio of |V|/G criterion that was proposed by Espinosa in which V is the potential and G is the kinetic electron energy densities at BCP were calculated for title conformers. On the basis of this ratio, for closed shell interaction, |V|/G and for shared shell interaction, |V|/G〉2. The intermediate falls between these two limits. These parameters were calculated for the most stable and lowest stable conformers. Hydrogen-bonded complexes fall in the closed-shell or intermediate region. Three Eigenvalues of the Laplacian, λ1, λ2 and λ3 were calculated for investigated conformers and the ratio of |λ1|/λ3 which is less than 0.25 for closed-shell interaction and greater than 1 for shared-shell interactions obtained [Citation24]. Results are listed in .
Table 2. Topological parameters (in a.u.) and intramolecular hydrogen bond energies (in kJ/mol) of stable and unstable conformers of benserazide.
NBO analysis
NBO analysis has been performed on the BZ conformers at B3LYP/6–311++G(d, p) level of theory. Delocalization of electron density from occupied Lewis-type (donor) NBOs to properly unoccupied non-Lewis type (acceptor) NBOs within molecule investigate with NBO analysis. Strongest stable interactions take place between effective donor and effective acceptors. The second-order perturbation interaction energy E(2) expresses the more intensive interaction between bonding and antibonding orbitals and qualify the extent of hydrogen bonding. As can be seen in , in CT4 conformer the most important and strong interaction between LP(2) O33 and σ* H14-O30 with the stabilization energy of 12.32 kcalmol−1 has been observed. Lone pair (LP1) nitrogen atom participates in LP(1)→σ* H14-O30 with the 22.46 kcalmol−1 energy value in TT8 conformer. In CC1 conformer the energy value of LP(2) O33→σ* H14-O30 is 15.36 kcalmol−1 and the energy values of LP(1)→σ* H14-O30 and LP(2) O33→σ* H14-O30 interactions in TC1 conformer are 10.4 and 23.74 kcalmol−1, respectively, indicating that these interactions produce a great stabilization in the title conformers.
Table 3. The NBO analysis of stable and unstable conformers of benserazide (includes the second order perturbation energy of some important orbital interactions).
HOMO-LUMO analysis
Molecular electrical transport properties can be determined by the energy gap between highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) frontier molecular orbitals. The chemical stability DFT based descriptors such as stability (ΔE), chemical hardness (η), chemical softness (S), electronegativity (χ), Ionization Potential (IP) and electron affinity (A) were obtained by using the MO energy values at the same level (see ). As can be seen in , the relatively high value of ΔEHOMO-LUMO indicates that the title conformers present high chemical stability and low reactivity. CT4 conformer with the smallest HOMO-LUMO gap is the softest molecule and is the most polarizable one with the largest dipole moment value thus it has the most reactivity with L-Dopa to play the decarboxylase inhibiting role.
Table 4. Total energy, dipole moment, and MO analyses of stable and unstable conformers of benserazide.
Vibrational analysis
Frequency calculated at the same level of theory, B3LYP/6–311++G (d, p), for all 512 conformers revealed no imaginary frequencies, indicating that an optimal geometry at these levels of approximation was found for the title conformers. The harmonic stretching vibrational frequencies of O···H and N···H for the most stable conformations of BZ, i.e. CT4, TT8, CC1 and TC1 are listed in . The vibrational bands assignments have been made by GaussView molecular visualization program. As can be seen from , strong bonds at 3619.74, 3323.33, 3611.39 and 3527.35 cm−1 assigned as O–H stretching and the values of 3592.74, 3489.48 and 3648.71 cm−1 assigned as N–H stretching in CT4, the most stable conformer. The discrepancy observed between the calculated frequencies could be explained due to the fact that intramolecular HB formation blue shifts the O–H and N–H of stretching frequencies. An increase of the corresponding intensity of O–H and N–H absorption band indicates the intramolecular HB formation [Citation32,Citation33].
Table 5. The harmonic stretching vibrational frequencies of O…H and N…H for the most stable conformations of benserazide.
Quantum chemical studies of carboxylated carbon nanotube
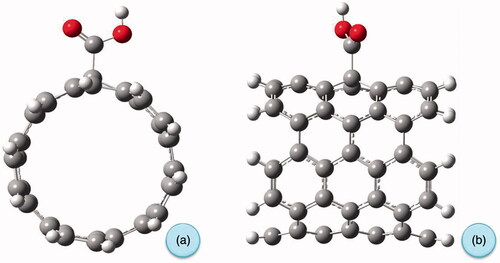
Covalent functionalization by generating carboxylic acid groups on the surface of the CNT is one of the widely used strategies to increase water miscibility, biocompatibility and decrease cytotoxicity. Drug molecules can be coupled to the surface of carboxylated CNTs for the medicinal applications. The carboxylated armchair (5,5) single-walled carbon nanotube-containing 70 carbon atoms, 20 hydrogen atoms with a length of 9.23 Ǻ and a diameter of 7.63Ǻ, functionalized with carboxylic acid group which replaced in the middle of the SWCNT fragment, were used as a model in this investigation. Both ends of the SWCNT are saturated with hydrogen atoms. Full geometry optimization was computed at B3LYP method with 6–311 G(d) basis set. shows the optimized geometry of the studied c-CNT from front and side views. For pristine SWCNT, the average C–C bond length of 1.42Ǻ was obtained and a C–C bond length of 1.55 Ǻ forming sp3-type hybridization results between the carbon nanotube and COOH group. The difference between HOMO and LUMO are of 2.16 eV for the pristine SWCNT and of 2.09 eV for the c-CNT, respectively. It can be observed that after the functionalization, the HOMO and LUMO energy gap decreases. This lowering of HOMO-LUMO band gap influences the biological activity and reactivity of the c-CNT in drug carrier systems.
Figure 6. Optimized geometry of carboxylated carbon nanotube (a) from front and (b) from side view.
Structure and electronic properties of nano drug carrier formed by coupling c-CNT with benserazide
Molecular conformation
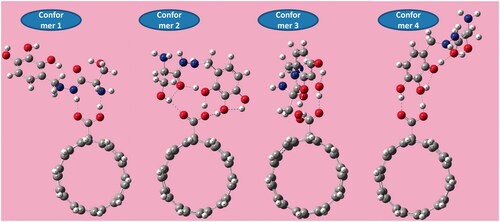
To explore the potential of c-CNTs as transfer vectors and evaluating the stability of BZ-c-CNT complexes, all four possible molecular configurations of adsorbed BZ onto c-CNT were investigated. Full optimized geometry of all possible complexes of BZ adsorbed on c-CNT was performed at DFT/B3LYP/6–311 G(d). represents the optimized geometric structure of the considered systems. The adsorption energy for the energetically favourable complex and the equilibrium distances are 94.5 kJ/mol, 1.69 and 1.55 Å, respectively.
Figure 7. Non-covalent adsorption features of benserazide onto carboxylated carbon nanotube.
After full optimization of the adsorption of BZ onto c-CNT, the interactions are manifested through the hydrogen binding (HB) of amine and oxygen atoms of BZ. To get deeper insight into characteristics of intramolecular HBs, QTAIM is applied to analyse the topological parameters. shows the molecular graphs obtained from AIM approach. The binding energies (calculated by EquationEquation (1)(1)
(1) ) and the relevant interatomic distances for all configurations studied are shown in . These binding energy values reveal a chemisorption process of the BZ over the c-CNT surface.
Figure 8. Molecular graphs of the conformers calculated at B3LYP/6–311 G(d) level using AIM approach.
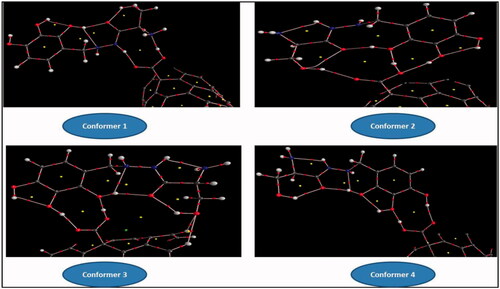
Table 6. Adsorption energy, Eads in kJ/mol, optimum distance of interaction, in Ǻ, electron density (ρBCP), Laplacian (∇2ρBCP), electron kinetic energy density (GBCP), electron potential energy density (VBCP), total electron energy density (HBCP) and hydrogen bond energy (EHB) at the bond critical point (BCP) of all configurations calculated at B3LYP/6–311 G(d) level.
Intermolecular hydrogen bond energies
The strength is one of the most important features of the intermolecular HB leads to structural modification. The stability of conformers is mainly due to the strong homonuclear intermolecular HBs of the hydroxyl groups of the ring of BZ with the c-CNT system. To predict the properties of HB energies, EquationEquations 4–6 were implemented. The electron densities and positive values of Laplacian are consistent with electrostatic character of HBs. Electron densities at corresponding HBs, Laplacian and energetic topological parameters are collected in . Analysing the sign of Laplacian indicates that the internuclear region is diminution of electron charge. It also verifies the closed-shell interactions. The strength of the HBs is reflected by the greater electron densities. Results also indicate that for strong HBs, ∇2ρ(rCP)〉0 and HBCP〈0 which is exactly related to Hammond–Leffler postulate [Citation34,Citation35].
Frontier molecular orbital analysis
The HOMO and LUMO isosurfaces of BZ and c-CNT are given in . For c-CNT, the HOMO is delocalized throughout the nanotube sidewall and carboxyl group whereas LUMO is delocalized along the C-C bonds parallel to the carbon nanotube axis. In BZ molecule with the presence of the aromatic ring, hydroxyl groups and –CONHNH2 bond, HOMO is delocalized on the aromatic ring and hydroxyl groups while the LUMO plot is delocalized on aliphatic part with contribution on the electronegative nitrogen and hydrogen atoms. The side view of HOMO in the adsorption of BZ onto c-CNT and the corresponding LUMO and the energy gap is shown in . It can be seen that the adsorption of BZ molecule does not bring major perturbation in HOMO electronic charge distribution of c-CNT. The absence of charge distribution on BZ molecule illustrates the unperturbed conformational framework.
Figure 9. Frontier molecular orbitals (HOMO-LUMO) plots with corresponding energy gap of (a) carboxylated carbon nanotube and (b) benserazide molecule.
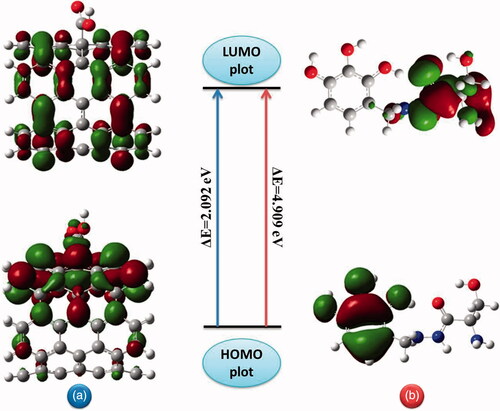
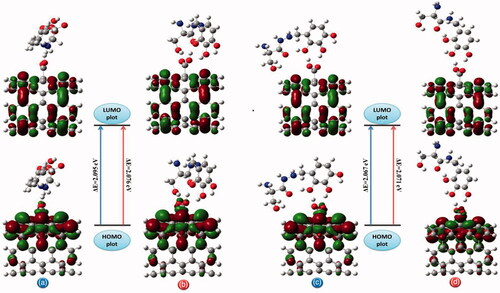
Figure 10. The pictorial illustration of HOMO and LUMO frontier molecular orbitals with corresponding energy gap for different possible complexes calculated by B3LYP method with 6–311 G(d) basis set; (a) conformer 1, (b) conformer 2, (c) conformer 3 and (d) conformer 4.
DFT-based reactivity and stability descriptors
Quantum molecular DFT based descriptors for Bz/c-CNT and its constituents are calculated and compared in . Gas phase geometry of BZ has a HOMO-LUMO energy gap of 4.91 eV. On the other hand, c-CNT found to be more reactive than the (5,5) SWCNT counterpart. Although in BZ adsorbed onto c-CNT the change in chemical hardness values are not very dramatic but the most stable conformer shows the highest stability. The chemical potential of conformer 3 is higher suggesting it to be more reactive. Lowering the electrophilicity index of mentioned conformer leads to behaves as a better nucleophile. These results support with the calculated values of ionization potential and electron affinity values.
Table 7. Total energy (Hartree) and global reactivity descriptors (all units in eV) for BZ/c-CNT drug carrier conformers and its constituents.
Molecular electrostatic potential (MPE) surface analysis
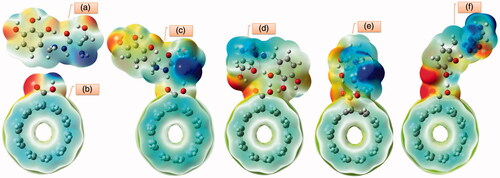
Electrostatic potential maps enable us to visualize the size, shape and charge distributions. Different colours used to represent the values of surface electrostatic potentials. The orange colour represents the most negative and the blue colour corresponds to the most positive site [Citation36,Citation37]. MPE surfaces for investigated BZ, c-CNT and investigated conformers over the optimized electronic structures using DFT B3LYP method with 6–311 G(d) basis set are plotted in . The carboxylic site of c-CNT provides the most electrophilic and nucleophilic region in the nanotube. The bluish green colours are related to less positive region. From the MPE of BZ is evident that the negative charge covers the C=O and O–H groups and the positive region is over the hydrogen of amine groups. Different colours represent and visualize different electrostatic potentials at the surface. The region of attractive potential appears in red and those of repulsion potential appear in blue thus the N–H regions with positive electrostatic potential, participate in nucleophilic reactions. Oxygen atoms of hydroxyl groups with higher electronegative and higher electron density participate in electrophilic attacks. The values of electrostatic potentials are responsible for binding of BZ to the receptor binding sites of L-DOPA.
Figure 11. MPE plots of (a) Benserazide, (b) c-CNT, (c) conformer 1, (d) conformer 2, (e) conformer 3 and (f) conformer 4 calculated at B3LYP/6–311 G(d).
Density of states
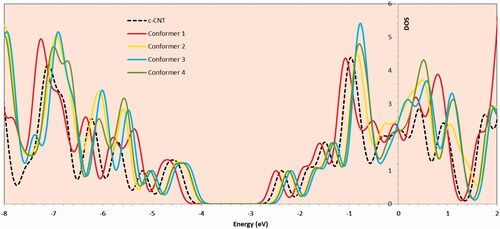
The total DOS of the conformers were predicted to elucidate the electronic properties and the influence of BZ adsorbed onto c-CNT. shows the DOS for the adsorption systems of BZ/c-CNT. By comparison, the maximum diminution of bong gap could be seen in the DOS diagram of conformers 3. As shown in this Figure, a slightly left move happens to the DOS around the Fermi Level.
Figure 12. Total DOS of the c-CNT and Bz/c-CNT adsorption conformers.
Conclusion
Effective neurodegradative diseases therapy is the most important challenge in the field of pharmaceutical technology. Since most of the brain-directed pharmaceuticals are incapable of crossing the blood–brain barrier (BBB), only a minor number of them have reached the market. Anatomic structure of astrocyte, pericytes and endothelial cells and the presence of tight junctions of the BBB instead of large fenestration between cells restrict the passage of the drugs to the central nervous system by efflux. Only small molecules, such as water, some gases and some lipid-soluble compounds can penetrate through BBB by passive transcellular diffusion due to the existence of high electrical resistance (1500–2000 Ωcm2) between the endothelial cells. It should be stated that the size, 3D structure, surface properties and charge state of the molecules affect the transport dynamics of the BBB. Zeta potential is also an important parameter that affects the passage through the BBB. Nanoparticles with high Zeta potential (high positive charge) cause toxicity so, moderated or negative zeta potential nanoparticles could be able to cross the BBB in brain delivery systems nanoparticles. Chemical modification of drugs and prodrugs, local delivery by neurosurgery and nanoparticles-mediated delivery are efficiently released drug strategies into the brain. Nano drug delivery offers advantages of non-invasive, low cost, biodegradability, long-term stability and high controllability to load and efficient targeting. Nanoparticles can be modulated in terms of shape, size, hydrophobicity, coating chemistry and surface charge-controlling that these factors can enhance the BBB penetration ability of nanocarriers. To evaluate the drug delivery potential of c-CNT-BZ for LD in Parkinson disease, all possible stable conformers of BZ have been studied. Our results demonstrate that within 512 possible BZ conformers, CT4 with 3 five-membered and 3 six-membered ring type strong hydrogen bond could be the best candidate conformer in MADOPAR (Levodopa + benserazide) anti-Parkinson drug. It was confirmed that the adsorption efficiency of chemically modified SWCNT with carboxylic acid group through hydrogen bonding and large surface area make c-CNT a good candidate in drug carrier process of benserazide. This new complex overcomes the limitations of other carbon nanotube-based systems. Carboxylic acid groups introduced onto the walls of the single-walled carbon nanotubes could facilitate further functionalization with benserazide molecule as a nano drug carrier, loading Levodopa and increase dispersibility of CNTs in water and become less toxic, more effective and a safe targeting treatment of Parkinson disease.
Acknowledgements
The authors wish to thank Graduate University of Advanced Technology, Kerman, Iran, for their support.
Disclosure statement
All authors reported no disclosures.
References
- Vashist SK, Zheng D, Pastorin G, et al. Delivery of drugs and biomolecules using carbon nanotubes. Carbon. 2011;49:4077–4097.
- Pardeshi CV, Belgamwar VS. Ropinirole-dextran sulfate nanoplex for nasal administration against Parkinson’s disease: in silico molecular modeling and in vitro–ex vivo evaluation. Artif Cells Nanomed Biotechnol. 2017;45:635–648.
- Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009;72(21):S1–S136.
- Singh D, Kapahi H, Rashid M, et al. Recent prospective of surface engineered nanoparticles in the management of neurodegenerative disorders. Artif Cells Nanomed Biotechnol. 2016;44:780–791.
- Benesi HA, Hildebrand J. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J Am Chem Soc. 1949;71:2703–2707.
- Raissi H, Yoosefian M. Substituent effect on the reaction mechanism of proton transfer in formamide. Int J Quantum Chem. 2012;112:2378–2381.
- Bajwa N, Kumar Mehra N, Jain K, et al. Targeted anticancer drug delivery through anthracycline antibiotic bearing functionalized quantum dots. Artif Cells Nanomed Biotechnol. 2016;44(7):1774–1782.
- Kaur S, Mehra NK, Jain K, et al. Development and evaluation of targeting ligand-anchored CNTs as prospective targeted drug delivery system. Artif Cells Nanomed Biotechnol. 2017;45(2)242–250.
- Zhang Y, Bai Y, Yan B. Functionalized carbon nanotubes for potential medicinal applications. Drug Discov Today. 2010;15:428–435.
- Sharma S, Mehra NK, Jain K, et al. Effect of functionalization on drug delivery potential of carbon nanotubes. Artif Cells Nanomed Biotechnol. 2016;44:1851–1860.
- Zong W, Chen J, Han W, et al. Preparation of PVA/amino multi-walled carbon nanotubes nanocomposite microspheres for endotoxin adsorption. Artif Cells Nanomed Biotechnol. 2018;46(1):185–191.
- Farvadi F, Tamaddon A, Sobhani Z, et al. Polyionic complex of single-walled carbon nanotubes and PEG-grafted-hyperbranched polyethyleneimine (PEG-PEI-SWNT) for an improved doxorubicin loading and delivery: development and in vitro characterization. Artif Cells Nanomed Biotechnol. 2017;45:855–863.
- Yoosefian M, Raissi H, Mola A. The hybrid of Pd and SWCNT (Pd loaded on SWCNT) as an efficient sensor for the formaldehyde molecule detection: a DFT study. Sensor Actuat B Chem. 2015;212:55–62.
- Yoosefian M, Barzgari Z, Yoosefian J. Ab initio study of Pd-decorated single-walled carbon nanotube with C-vacancy as CO sensor. Struct Chem. 2014;25:9–19.
- Chen Z, Pierre D, He H, et al. Adsorption behavior of epirubicin hydrochloride on carboxylated carbon nanotubes. Int J Pharm. 2011;405:153–161.
- Frisch M, Trucks G, Schlegel H, et al. Gaussian 09, Revision B. 01. Wallingford (CT): Gaussian, Inc; 2009.
- Becke AD. Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648.
- Glendening E, Reed A, Carpenter J, et al. NBO, version 3.1. Pittsburg (CA): Gaussian Inc.; 2003.
- Biegler K, Schonbohm J, Derdan R, et al. AIM2000, version 1, Bielefeld, Germany; 2000.
- Koopmans T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines atoms. Physica. 1934;1:104–113.
- Parr RG, Szentpaly L, Liu S. Electrophilicity index. J Am Chem Soc. 1999;121:1922–1924.
- Rozas I, Alkorta I, Elguero J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J Am Chem Soc. 2000;122:11154–11161.
- Abramov YA. On the possibility of kinetic energy density evaluation from the experimental electron-density distribution. Acta Crystallogr A Found Crystallogr. 1997;53:264–272.
- Koch U, Popelier P. Characterization of CHO hydrogen bonds on the basis of the charge density. J Phys Chem. 1995;99:9747–9754.
- Raissi H, Yoosefian M, Mollania F, et al. The effect of substitution on structure, intramolecular hydrogen bonding strength, electron density and resonance in 3-amino 2-iminomethyl acryl aldehyde. J Theor Comput Chem. 2012;11:925–939.
- Raissi H, Yoosefian M, Hajizadeh A, et al. Theoretical description of substituent effects in 2, 4-Pentanedione: AIM, NBO, and NMR study. BCSJ. 2012;85:87–92.
- Yoosefian M, Chermahini ZJ, Raissi H, et al. A theoretical study on the structure of 2-amino-1, 3, 4-thiadiazole and its 5-substituted derivatives in the gas phase, water, THF and DMSO solutions. J Mol Liq. 2015;203:137–142.
- Yoosefian M, Etminan N. Pd-doped single-walled carbon nanotube as a nanobiosensor for histidine amino acid, a DFT study. RSC Adv. 2015;5:31172–31178.
- Etminan N, Yoosefian M, Raissi H, et al. Solvent effects on the stability and the electronic properties of histidine/Pd-doped single-walled carbon nanotube biosensor. J Mol Liq. 2016;214:313–318.
- Yoosefian M, Etminan N. Density functional theory (DFT) study of a new novel bionanosensor hybrid; tryptophan/Pd doped single walled carbon nanotube. Physica E LowDimens Syst Nanostruct. 2016;81:116–121.
- Espinosa E, Molins E, Lecomte C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem Phys Lett.1998;285:170–173.
- Yoosefian M, Mola A. Solvent effects on binding energy, stability order and hydrogen bonding of guanine–cytosine base pair. J Mol Liq. 2015;209:526–530.
- Raissi H, Jalbout A, Fazli M, et al. Intramolecular hydrogen bonding in derivatives of 3‐amino‐propenethial. Int J Quantum Chem. 2009;109:1497–1504.
- Hammond GS. A correlation of reaction rates. J Am Chem Soc. 1955;77:334–338.
- Leffler JE. Parameters for the description of transition states. Science. 1953;117:340–341.
- Moro S, Bacilieri M, Ferrari C, et al. Autocorrelation of molecular electrostatic potential surface properties combined with partial least squares analysis as alternative attractive tool to generate ligand-based 3D-QSARs. Curr Drug Discov Technol. 2005;2:13–21.
- Luque FJ, López JM, Orozco M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the provision of solvent effects”. In: Theoretical chemistry accounts. Berlin: Springer; 2000. p. 343–345.