
Abstract
Background
This study aims to via unveiling the novel mechanisms of KLF16 in regulating expression of genes involved in glioma.
Methods
KLF16 or KLF16-siRNA was transfected to U87MG cells by lentivirus. Colony formation assay was applied for detecting cell proliferation. MTT assay was adopted to assess cell viability. TUNEL assay was selected to evaluate cell apoptosis. Flow cytometry was used to determine cell cycle. Real-time PCR was performed to test mRNA expression. Western blot was used to detect protein level. Luciferase assay was applied to confirm the regulatory relationship between KLF16 and Mitochondrial transcription factor A (TFAM). Chromatin immunoprecipitation was adopted to test the protein binding site. The nude mouse transplantation tumour experiment was selected to test cancer cell proliferation in vivo.
Results
KLF16 was decreased in glioma cells and tissues. KLF16 obviously restrained U87MG cell proliferation both in vivo and in vitro. KLF16 transfection reduced mRNA and protein levels related to cell proliferation. KLF16 targeted a putative binding site near the transcription start sites (TSSs) of TFAM gene, thus suppressing glioma cell proliferation. KLF16-siRNA exhibited the opposite impact. KLF16 presented significant negative correlation with TFAM level in glioma patients.
Conclusions
KLF16 is a key regulator of glioma cell proliferation by directly targeting TFAM.
Introduction
Malignant gliomas are the most common and fatal tumours in the central nervous system. In spite of abundant improvements in surgical techniques and therapeutic strategies, the prognosis for patients with gliomas remains poor. Especially, the median survival is only 12–15 months for patients with glioblastomas (GBM), the most common and aggressive type of malignant gliomas, hugely attributed to the rapid tumour growth and infiltration of tumour cells throughout the brain [Citation1,Citation2]. Although oncogenic mutations and amplification, such as EGFR and RAS, and the loss of function of tumour suppressor genes, such PTEN and p53, have been involved in glioma progression, the molecular mechanisms of glioma malignancy remain poorly investigated. Thus far, no effective prognostic or therapeutic targets have been developed [Citation3–6]. Therefore, exploring functional tumour-associated genes may not only find novel mechanisms of glioma progression but may also allow for the identification of novel targets that could serve as possible targets for therapeutic intervention.
Krüppel-like factor (KLF) members, including KLFs 1–17, are zinc finger-containing transcription factors that play crucial roles in the regulation of differentiation and development as well as biological processes central to the development of malignancies [Citation7,Citation8]. Numerous evidence indicates a significant role for this family of transcription factors in carcinogenesis, acting as oncogenes and/or tumour suppressors under distinct cellular contexts [Citation7,Citation8]. Typically, KLF5 promotes proliferation of human breast, bladder and pancreatic cancer cells [Citation9–11]. KLF6 is frequently lost in many cancers by somatic mutation, loss of heterozygosity or promoter methylation [Citation12–14]. KLF4 suppresses tumour growth in multiple cancers, including gastric, colorectal, hepatocellular and lung carcinomas [Citation15–18], but promotes cell cycle progression in pancreatic cancer [Citation19]. Moreover, KLF9 regulates the transactivation of progesterone-responsive promoters and has been found to be downregulated in endometrial carcinoma and colorectal cancer [Citation20–23], suggesting that KLF9 downregulation might be linked to cancer development. Therefore, a better understanding of the expression pattern and biological role of KLF9 in glioma progression may provide clues for novel targets for therapeutic intervention.
The KLF family plays important roles in tumourigenesis by regulating oncogenic or tumour-suppressive genes through binding GC-rich DNA sequences in gene promoter regions [Citation24,Citation25]. Mitochondria are fundamental to metabolic homeostasis [Citation26]. Mitochondrial transcription factor A (TFAM), a high-mobility group (HMG) protein, plays a central role in mitochondrial DNA (mtDNA) replication, transcription and inheritance [Citation27]. Aberrant function of mitochondria has been reported to be associated with various metabolic disorders, such as cancer [Citation28]. Thus, it is possible that KLFs exert their functions through regulating the expression of TFAM.
In this study, we found that KLF16 was robustly reduced in glioma tissues and cell lines. Silencing KLF16 promoted, while overexpressing KLF16 impaired, glioma cells proliferation both in vitro and in vivo. Furthermore, we identified that KLF16 transcriptionally repressed TFAM expression by interacting with its promoter. Taken together, our findings uncover a novel mechanism for KLF function in tumourigenesis and may suggest new targets for clinical intervention against human cancers.
Methods
Cell culture and hypoxia condition
Tissues and cells
Glioma tissues were obtained from the Department of Neurosurgery, the Second Affiliated Hospital, Fujian Medical University, Quanzhou, China. Normal brain tissues were obtained from individuals who died in traffic accidents and confirmed to be free of any pre-existing pathologically detectable conditions. Approval from the Institutional Research Ethics Committee and donor consent were obtained.
Normal human astrocyte (NHA) cells (ScienCell, Carlsbad, CA) were cultured under the recommended conditions. Glioma cell lines U87MG, U118MG, U251MG, SNB19, LN18 and LN444 were routinely maintained in DMEM medium (Invitrogen, Carlsbad, CA) supplemented with 10% foetal bovine serum (HyClone, Logan, UT).
MTT assay
The U87MG cells were seeded at 96-well plate at 2 × 103cells/well for cultivation. Cells were then transfected with by KLF16 or KLF16 siRNA as described above for 24 h. During the last 4 h of each day of culture, cells were treated added with MTT (at 50 mg per well (Sigma, St. Louis, MO). The formazan was dissolved by dimethyl sulfoxide (DMSO) and the absorbance was measured tested at 450 nm using a microplate reader (Bio-tek, Winooski, VT).
Cell cycle analysis
U87MG cells were collected and washed by phosphate buffered saline (PBS) for three times. The cells were fixed in 1 ml 70% precooled ethanol at 4 °C overnight. Next, the cells were washed with PBS and incubated in 100 mg/l RNase at 37 °C for 30 min. After stained by 50 mg/l PI at 4 °C avoid of light for 30 min, the cells were detected tested on flow cytometry at 488 nm. The primary result was analysed by FlowJo software (Tree Star, Inc., Ashland, OR) to acquire the hypodiploid peak, so as to calculate the cell ratio in G0/G1 phase, S phase and G2/M phase, respectively. All the experiments were repeated for three times.
Real-time PCR
Total RNA was extracted by the kit obtained from Takara, Tokyo, Japan. RNA was eluted in 50 ml of RNase-free water and preserved at −70 °C. To analyse gene expression, the real-time PCR mixture system containing primers, cDNA templates and SYBR Green qPCR Master Mix were reacted by real-time PCR according to standard methods. Fold changes in gene expression were calculated upon 2−△△Ct method with GAPDH used as an internal control.
Western blot
U87MG cells were lysed with loading lysis buffer that was diluted from 5 × loading lysis buffer (2.5 ml Tris–HCl at 0.5 mol/l, 0.39 g dithiothreitol, 0.5 g sodiumdodecyl sulphate, 0.025 g bromophenol blue and 2.5 ml glycerin). Equal amount of protein was transferred to the polyvinylidene fluoride (PVDF) membrane. Next, the membrane was blocked by skim milk and further incubated with primary antibodies at 4 °C for 1 h. Then the membranes were incubated in horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 60 min. The binding signals were visualized by enhanced chemiluminescence (Thermo Fisher Scientific, Rugby, UK).
Cell transfection
Cells were transfected using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) according to the manual. For the gene expression analyses, cells were seeded in 6-well plates and transfected with 1 μg plasmid DNA per well; 50 ng KLF16 or KFL16 siRNA, 300 ng pDUO-hMD-2/CD14 adjusted with pcDNA3.1 to 1 μg.The total plasmid load was adjusted to 1 μg plasmid DNA with pcDNA3.1.
Anchorage-independent growth ability assay
A total of 2 × 103 cells were trypsinized and suspended in 2 ml of complete medium plus 0.3% agar (Sigma, St. Louis, MO). The agar-cell mixture was plated on top of a bottom layer with 1% complete medium agar mixture. After 10 d, viable colonies larger than 0.05 mm were counted.
Xenograft tumour model
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of the Second Affiliated Hospital, Fujian Medical University. Briefly, BALB/c-nu mice (5–6 weeks, 18–20 g, n = 5) were inoculated subcutaneously with U87MG/vector cells (2 × 106) in the left dorsal flank and with U87MG/KLF16 cells (2 × 106) in the right dorsal flank per mouse. Tumour volumes were determined every 3 d. Length, width and thickness measurements were obtained with callipers and tumour volumes were calculated. Tumour volume was calculated using the equation (L × W2)/2. On day 40, the mice were euthanized and imaged, and the tumours were excised and weighed. Serial 6.0 μm sections were cut and subjected to immunohistochemical staining using an anti-Ki67 antibody (Dako, Glostrup, Denmark). Proliferation index was quantified by counting the proportion of Ki67-positive cells.
Luciferase assay
Cells (2 × 104) were seeded in triplicate in 48-well plates and cultured for 24 h. One hundred nanograms of TFAM promoter luciferase plasmid plus 5 ng of pRL-TK Renilla plasmid (Promega, Madison, WI) were transfected into the indicated cells using Lipofectamine 2000 reagent (Invitrogen Co., Carlsbad, CA) according to the manufacturer’s recommendations. Luciferase and Renilla signals were measured 36 h after transfection using the Dual Luciferase Reporter Assay Kit (Promega, Madison, WI) according to the manufacturer’s protocol.
Statistical analysis
All data analyses were carried out by SPSS version 15.0 (SPSS Inc., Chicago, IL) software. The data were depicted as mean ± SD and compared by t-test or one-way ANOVA. A p < .05 was considered as statistical significance.
Results
KLF16 is downregulated in glioma cell lines and tissues
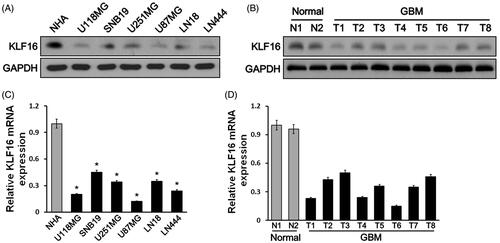
Western blot analysis showed that KLF16 protein expression levels were repressed in six human glioma cell lines compared with NHA cells (). KLF16 protein levels were obviously lower in GBM tumours with the two normal brain tissues (). Furthermore, real-time PCR analysis revealed that KLF16 was transcriptionally downregulated in glioma cell lines and tissues ()).
Figure 1. KLF16 expression is reduced in glioma tissues and cell lines. (A and B) Western blotting analysis of KLF16 expression in normal human astrocyte (NHA) cells and 6 glioma cell lines (A) and in two normal brain tissues and eight human glioma tissues (B). GAPDH served as the loading control. (C and D) Real-time PCR analysis of KLF16 expression in NHA and 6 glioma cell lines (C) and in two normal brain tissues and eight human glioma tissues (D). Transcript levels were normalized to GAPDH expression. *p < .05 vs. NHA cells.
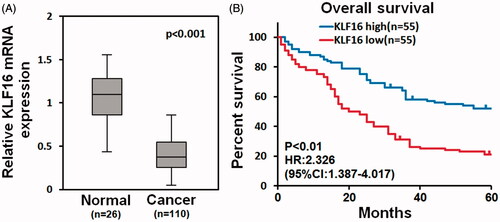
KLF16 is associated with better prognosis of glioma patients
Analysis of TCGA mRNA array data showed that KLF16 levels were significantly downregulated in human GBM tissues (n = 110) compared with that in normal brain tissues (n = 26, ). Furthermore, survival analysis demonstrated that glioma patients with low KLF16 level exhibited poor prognosis (HR =2.328, 95% CI =1.387–4.017, ). Taken together, these results suggest that KLF16 is downregulated in human gliomas and related to better prognosis.
Figure 2. KLF16 is associated with better prognosis of glioma patients. (A) Analysis of TCGA mRNA array data revealed that KLF16 levels were significantly downregulated in human primary GBMs (n = 110) compared with normal brain tissues (n = 26). (B) Survival analysis of glioma patients with high and low KLF16 expression level.
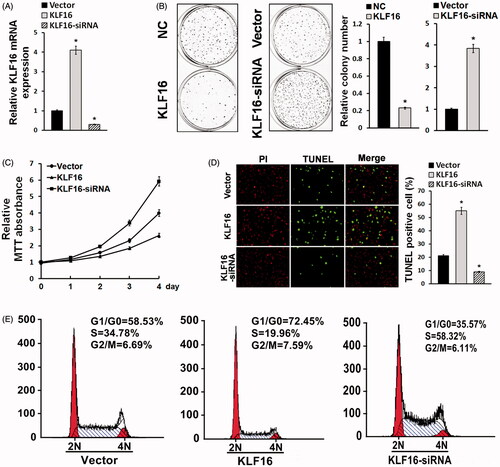
KLF16-regulated glioma cell proliferation
Since KLF16 was downregulated in glioma cells compared with normal control, we proposed that KLF16 was involved in regulating glioma cell proliferation. U87MG cells were transfected by KLF16 plasmid or siRNA. Real-time PCR showed that KLF16 mRNA was significantly overexpressed in U87MG cells after KLF16 transfection and declined after KLF16 siRNA transfection (). Colony formation assay and MTT assay revealed that KLF16 upregulation obviously suppressed U87MG cell proliferation, while KLF16 siRNA markedly enhanced glioma cell growth ()). TUNEL assay exhibited that KLF16 significantly facilitated glioma cell apoptosis, whereas KLF16 siRNA transfection obviously restrained the apoptosis of U87MG cells (). Flow cytometry demonstrated that KLF16 transfection apparently arrested cell cycle, whereas KLF16 siRNA markedly accelerated cell cycle of U87MG compared with control ().
Figure 3. KLF16-regulated glioma cell proliferation. (A) Real-time PCR analysis of KLF16 expression in U87MG cells infected with KLF16 plasmid or KLF16 siRNA. mRNA expression levels were normalized to GAPDH. (B) Colony formation assay demonstrated the influence of KLF16 on U87MG cell clone formation ability. (C) MTT assays revealed that KLF16 downregulation induced proliferation of U87MG cells. (D) Representative micrographs and quantification of TUNEL incorporation in KLF16 overexpression and silencing cells. (E) Flow cytometric analysis of cell cycle in KLF16 overexpression and silencing glioma cells. *p < .05.
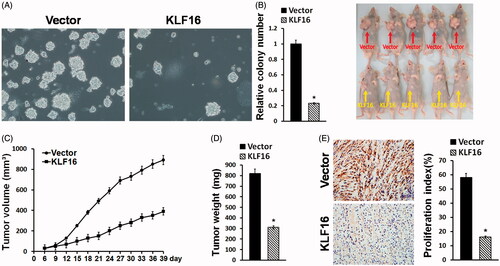
KLF16 overexpression inhibited glioma cell tumourigenicity
The tumour-suppressive role of KLF16 overexpression in the tumourigenicity of glioma cells was further examined in vitro and in vivo. As shown in , KLF16 overexpression dramatically reduced, while silencing of KLF16 increased, the anchorage-independent growth of glioma cells. Importantly, the tumours formed by KLF16-overexpressing U87MG cells were substantially smaller, in both size and weight, than the control tumours ()). In addition, KLF16-overexpressing tumours displayed a lower proliferation index, as indicated by the percentage of Ki67-positive cells (). Therefore, our results suggest that KLF16 overexpression inhibits glioma cell tumourigenicity.
Figure 4. KLF16 overexpression inhibits glioma cell tumourigenicity. (A) Representative micrographs and quantification of colonies produced in an anchorage independent growth assay. Colonies larger than 0.05 mm in diameter were scored. Scale bars: 50 μm. (B) A xenograft model in nude mice. The indicated cells were injected into the dorsal flank of the mice. Tumour-bearing mice were euthanized and imaged on day 40. (C) Tumour volumes were measured on the indicated days. (D) Mean tumour weights. (E) Proliferation index was determined by counting the proportion of Ki67-positive cells. *p < .05.
KLF16-inhibited glioma cell proliferation via TFAM downregulation
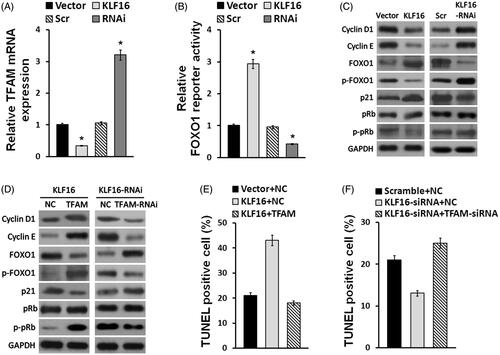
TFAM is a key oncogene that plays important role in a wide variety of cancers, including glioma [Citation29]. Interestingly, real-time PCR indicated that TFAM expression was downregulated in KLF16-elevated cells and upregulated in KLF16-silenced cells (). In addition, a luciferase reporter assay showed that KLF16 overexpression significantly increased, while silencing KLF9 decreased, the luciferase activity linked to FOXO1 (). Subsequently, we found that KLF16 overexpression increased p21 and FOXO1 expressions, but decreased cyclin D1 and cyclin E expressions, leading to the reduced phosphorylation of Rb and FOXO1 proteins. Conversely, KLF16 silencing had an opposite effect on p21, cyclin D1, cyclin E, p-FOXO1 and p-Rb expressions (). We next examined whether TFAM repression contributes to KLF16-mediated cell apoptosis. As shown in ), elevation of TFAM abrogated the effect of KLF16 overexpression, whereas inhibition of TFAM impaired the effect of KLF16 silencing, on p21, cyclin D1, cyclin E, p-FOXO1 and p-Rb expressions and the percentage of TUNEL positive cells. These results suggest that TFAM is a functionally relevant effector of KLF16 in regulating glioma cell proliferation.
Figure 5. KLF16 inhibits glioma cell proliferation via TFAM downregulation. (A) Real-time PCR analysis of TFAM expression in KLF16-overexpressing and KLF16-silenced cells compared with controls. Transcript levels were normalized to GAPDH expression. (B) Luciferase assay of the indicated cells transfected with pGL3-FOXO1. (C and D) Western blotting analysis of cyclin D1, cyclin E, p21, p-FOXO1, FOXO1, p-Rb and Rb expression in the indicated cells. GAPDH served as the loading control. (E) Percentage of TUNEL incorporating cells in KLF16-overexpressing, KLF16-overexpressing plus TFAM overexpressing and control cells. (F) Percentage of TUNEL positive cells in KLF16-silenced, KLF16-silenced plus TFAM inhibited and control cells. Each bar represents the mean ± SD of three independent experiments. *p < .05.
KLF16 directly represses TFAM expression
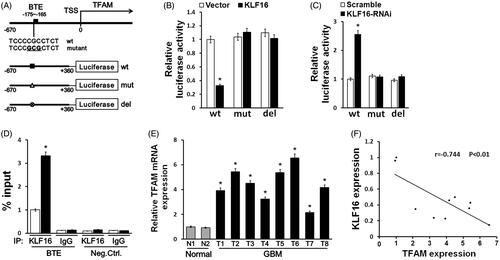
Since KLF16 is known to function as a transcription repressor by binding to GC-rich basic transcription element (BTE) in gene promoter regions [Citation24,Citation30], we further investigated whether KLF16 represses TFAM expression by interacting with its promoter. Analysis by the TRANSFAC programme predicted a BTE in the TFAM promoter region (). The luciferase reporter assay showed that KLF16 overexpression significantly decreased, while silencing KLF16 increased, the luciferase activity driven by the BTE in the TFAM promoter ()). In addition, the luciferase activities of the TFAM promoter that contained mutated or deleted BTE did not show obvious alterations in response to either KLF16 overexpression or knockdown ()). Moreover, a chromatin immunoprecipitation (ChIP) assay showed high binding affinity of endogenous KLF16 to the BTE region in TFAM promoter (). Therefore, our results suggest that KLF9 suppresses TFAM expression by directly interacting with the TFAM promoter. Finally, we examined whether the KLF16-induced TFAM repression identified in our study is clinically relevant. As shown in , we found that TFAM was robustly upregulated in glioma tissues compared with normal brain tissues. Statistical analyses revealed that TFAM levels inversely correlated with the KLF16 expression levels (r = −0.744, p < .01; ). Taken together, our results suggest that KLF16 downregulation promotes TFAM expression, leading to uncontrolled cell proliferation in gliomas.
Figure 6. KLF16 directly represses TFAM expression and is clinically relevant. (A) Schematic illustration of the predicted BTE site and the cloned luciferase reporter containing the TFAM promoter. BTE was mutated or deleted to verify that the site is essential for KLF16 regulation. (B) The effect of KLF16 on the transactivation of the TFAM promoter with or without BTE site. (C) The effect of KLF16-siRNA on the transactivation of the TFAM promoter with or without BTE site. (D) Regions of the promoter physically associated with KLF16 were analysed using the ChIP assay. (E) Real-time PCR analysis of TFAM expression in 2 normal brain tissues and 8 human glioma tissues. (F) Reduced KLF16 expression significantly correlated with TFAM overexpression in glioma samples. Each bar represents the mean ± SD of three independent experiments. *p < .05.
Discussion
In recent decades, KLF s emerged as important regulators in carcinogenesis, acting as oncogenes and/or tumour suppressors under distinct cellular contexts [Citation7,Citation8]. Of note, KLF16 was recently found to be associated with neurite outgrowth in retinal ganglion cell [Citation31], suggesting a possible tumour suppressor role of KLF16 in cancer. However, the role and mechanism of KLF16 in glioma progression remain unclear. In this report, we present evidence that the zinc finger-containing transcription factor KLF16 has an important tumour-suppressive role in glioma. First, KLF16 expression was downregulated in both in vitro cultured cell lines and in clinical samples. Second, silencing KLF16 promoted, while overexpressing KLF16 impaired cell proliferation and anchorage-independent growth of glioma cells. Third, re-constitution of KLF16 inhibited glioma tumour growth in a xenograft mouse model. Fourth, KLF16 directly repressed TFAM expression, leading to downregulation of p-Rb, p-FOXO1, cyclin E, and cyclin D1 and upregulation of p21. Furthermore, KLF16 downregulation is clinically relevant to the upregulation of TFAM expression in glioma tissues. Thus, our results not only suggest that KLF16 functions as a tumour suppressor in glioma but also support the notion that dysregulation of KLFs is involved inhuman cancers.
In recent decades, KLFs emerged as important regulators in carcinogenesis, acting as oncogenes and/or tumour suppressors under distinct cellular contexts [Citation7,Citation8]. Of note, KLF16 was recently found to inhibit retinal ganglion cell (RGC) neurite outgrowth and enhance RGC growth cone collapse in response to exogenous ephrinA5 ligands [Citation32], suggesting a possible tumour suppressor role of KLF16 in cancer. However, the role and mechanism of KLF16 in glioma progression remain unclear. In this report, we present evidence that the zinc finger-containing transcription factor KLF16 has an important tumour-suppressive role in glioma. First, KLF16 expression was downregulated in both in vitro cultured cell lines and in clinical samples. Second, silencing KLF16 promoted, while overexpressing KLF16 impaired cell proliferation and anchorage-independent growth of glioma cells. Third, re-constitution of KLF16 inhibited glioma tumour growth in a xenograft mouse model. Fourth, KLF16 directly repressed TFAM expression, leading to downregulation of p-Rb, cyclin E, p-FOXO1, and cyclin D1, and upregulation of p21. Furthermore, KLF16 downregulation is clinically relevant to the upregulation of TFAM expression in glioma tissues. Thus, our results not only suggest that KLF16 functions as a tumour suppressor in glioma but also support the notion that dysregulation of KLF s is involved inhuman cancers.
TFAM, a transcription factor for mtDNA, is required for mtDNA replication and transcription. TFAM has been reported to participate in the regulation of cell survival, proliferation and migration. In injured rat carotid artery, vascular smooth muscle cell proliferation is dependent on the upregulation of TFAM expression [Citation30]. It has been demonstrated that TFAM regulates p21 (WAF1/CIP1), a critical regulator of cell cycle progression, as knockdown of TFAM expression was found to induce p21-dependent G1 cell cycle arrest [Citation33]. Moreover, TFAM is involved in the development and progression of malignant tumours. Frequent truncating mutation of TFAM has been shown to induce mtDNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer [Citation34]. In addition, TFAM is found to be elevated in glioma, indicating its potential oncogene role in cancer.
In conclusion, we observed that KLF16 is downregulated in glioma. It can suppress TFAM, which plays an oncogene role in cancer. Aberrant overexpression of KLF16 in glioma causes restrained TFAM expression inU87MG, allowing cells to arrest cell proliferation and cell cycle. Our results provide new insight into glioma and a potential choice for cancer therapy, as KLF16 activity clearly affects the critical mechanism in glioma progress.
Disclosure statement
The authors declare no competing financial interests.
Additional information
Funding
References
- Mischel PS, Cloughesy TF. Targeted molecular therapy of GBM. Brain Pathol. 2003;13:52–61.
- Feng H, Hu B, Jarzynka MJ, et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci USA. 2012;109:3018–3023.
- Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333.
- Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068.
- Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–626.
- Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013;331:139–146.
- Tetreault MP, Yang Y, Katz JP. Kruppel-like factors in cancer. Nat Rev Cancer. 2013;13:701–713.
- McConnell BB, Yang VW. Mammalian Krüppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381.
- Chen C, Benjamin MS, Sun X, et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. Int J Cancer. 2006;118:1346–1355.
- Mori A, Moser C, Lang SA, et al. Up-regulation of Kruppel-like factor 5 in pancreatic cancer is promoted by interleukin-1beta signaling and hypoxia-inducible factor-1alpha. Mol Cancer Res. 2009;7:1390–1398.
- Takagi K, Miki Y, Onodera Y, et al. Kruppel-like factor 5 in human breast carcinoma: a potent prognostic factor induced by androgens. Endocr Relat Cancer. 2012;19:741–750.
- Kremer-Tal S, Reeves HL, Narla G, et al. Frequent inactivation of the tumor suppressor Kruppel-like factor 6 (KLF6) in hepatocellular carcinoma. Hepatology. 2004;40:1047–1052.
- Narla G, Heath KE, Reeves HL, et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science. 2001;294:2563–2566.
- Reeves HL, Narla G, Ogunbiyi O, et al. Kruppel-like factor 6 (KLF6) is a tumor-suppressor gene frequently inactivated in colorectal cancer. Gastroenterology. 2004;126:1090–1103.
- Li Q, Gao Y, Jia Z, et al. Dysregulated Kruppel-like factor 4 and vitamin D receptor signaling contribute to progression of hepatocellular carcinoma. Gastroenterology. 2012;143:799–810.
- Zhang N, Zhang J, Shuai L, et al. Kruppel-like factor 4 negatively regulates beta-catenin expression and inhibits the proliferation, invasion and metastasis of gastric cancer. Int J Oncol. 2012;40:2038–2048.
- Yu T, Chen X, Zhang W, et al. Regulation of the potential marker for intestinal cells, Bmi1, by beta-catenin and the zinc finger protein KLF4: implications for colon cancer. J Biol Chem. 2012;287:3760–3768.
- Hu W, Hofstetter WL, Li H, et al. Putative tumor-suppressive function of Kruppel-like factor 4 in primary lung carcinoma. Clin Cancer Res. 2009;15:5688–5695.
- Wei D, Wang L, Kanai M, et al. KLF4alpha up-regulation promotes cell cycle progression and reduces survival time of patients with pancreatic cancer. Gastroenterology. 2010;139:2135–2145.
- Simmen RC, Pabona JM, Velarde MC, et al. The emerging role of Krüppel-like factors in endocrine-responsive cancers of female reproductive tissues. J Endocrinol. 2010;204:223–231.
- Kang L, Lu B, Xu J, et al. Downregulation of Krüppel-like factor 9 in human colorectal cancer. Pathol Int. 2008;58:334–338.
- Zhang XL, Zhang D, Michel FJ, et al. Selective interactions of Kruppel-like factor 9/basic transcription element-binding protein with progesterone receptor isoforms A and B determine transcriptional activity of progesterone-responsive genes in endometrial epithelial cells. J Biol Chem. 2003;278:21474–21482.
- Velarde MC, Zeng Z, McQuown JR, et al. Kruppel-like factor 9 is a negative regulator of ligand-dependent estrogen receptor alpha signaling in Ishikawa endometrial adenocarcinoma cells. Mol Endocrinol. 2007;21:2988–3001.
- Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol Cell Biol. 1993;13:2776–2786.
- Shields JM, Yang VW. Identification of the DNA sequence that interacts with the gut-enriched Krüppel-like factor. Nucleic Acids Res. 1998;26:796–802.
- Warda M, Kim HK, Kim N, et al. A matter of life, death and diseases: mitochondria from a proteomic perspective. Expert Rev Proteomics. 2013;10:97–111.
- Uchiumi T, Kang D. The role of TFAM-associated proteins in mitochondrial RNA metabolism. Biochim Biophys Acta. 2012;1820:565–570.
- Cheng Z, Ristow M. Mitochondria and metabolic homeostasis. Antioxid Redox Signal. 2013;19:240–242.
- Lee H, Park J, Tran Q, et al. Mitochondrial transcription factor A (TFAM) is upregulated in glioma. Mol Med Rep. 2017;15:3781–3786.
- Yoshida T, Azuma H, Aihara K, et al. Vascular smooth muscle cell proliferation is dependent upon upregulation of mitochondrial transcription factor A (mtTFA) expression in injured rat carotid artery. Atherosclerosis. 2005;178:39–47.
- Wang J, Galvao J, Beach KM, et al. Novel roles and mechanism for Krüppel-like factor 16 (KLF16) regulation of neurite outgrowth and ephrin receptor A5 (EphA5) expression in retinal ganglion cells. J Biol Chem. 2016;291:21422.
- Wang J, Galvao J, Beach KM, et al. Novel roles and mechanism for Krüppel-like factor 16 (KLF16) regulation of neurite outgrowth and Ephrin receptor A5 (EphA5) expression in retinal ganglion cells. J Biol Chem. 2016;291:18084–18095.
- Kim AJ, Jee HJ, Song N, et al. p21(WAF¹/C¹P¹) deficiency induces mitochondrial dysfunction in HCT116 colon cancer cells. Biochem Biophys Res Commun. 2013;430:653–658.
- Guo J, Zheng L, Liu W, et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011;71:2978–2987.