
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Highly soluble drugs tend to release from preparations at high speeds, which make them need to be taken at frequent intervals. Additionally, some drugs need to be controlled to release in vivo at certain periods, so as to achieve therapeutic effects. Thus, the objective of this study is to design injectable microparticulate systems with controllable in vivo release profile. Biodegradable PLGA was used as the matrix material to fabricate microspheres using the traditional double emulsification-solvent evaporation method as well as improved techniques, with gel (5% gelatine or 25% F127) or LP powders as the inner phases. Their physicochemical properties were systemically investigated. Microspheres prepared by modified methods had an increase in drug loading (15.50, 16.72, 15.66%, respectively) and encapsulation efficiencies (73.46, 79.42, 74.40%, respectively) when compared with traditional methods (12.01 and 57.06%). The morphology of the particles was characterized by optical microscope (OM) and scanning electron microscopy (SEM), and the amorphous nature of the encapsulated drug was confirmed by differential scanning calorimetry (DSC) and X-ray diffraction (XRD) analysis. To evaluate their release behaviour, the in vitro degradation, in vitro release and in vivo pharmacodynamics were subsequently studied. Traditional microspheres prepared in this study with water as the inner phase had a relatively short release period within 16 d when compared with modified microspheres with 5% gelatine as the inner phase, which resulted in a smooth release profile and appropriate plasma LP concentrations over 21 d. Thus this type of modified microspheres can be better used in drugs requiring sustained release. The other two formulations containing 25% F127 and LP micropowders presented two-stage release profiles, resulting in fluctuant plasma LP concentrations which may be suitable for drugs requiring controlled release. All the results suggested that drug release rates from the microspheres prepared by various methods were mainly controlled by either the porosity inside the microspheres or the degradation of materials, which could, therefore, lead to different release behaviours. This results indicated great potential of the PLGA microsphere formulation as an injectable depot for controllable in vivo release profile via rational core phase design.
Core/shell microspheres fabricated by modified double emulsification-solvent evaporation methods, with various inner phases, to obtain high loading drugs system, as well as appropriate release behaviours. Accordingly, control in vivo release profile via rational core phase design.
Graphical Abstract

Introduction
Over the past few years, parenteral microspheres have been extensively utilized to deliver a variety of therapeutics, including those containing protein/peptides, chemotherapeutics and immunomodulatory factors in a controlled manner over periods of weeks to months [Citation1–8]. The majority of these microspheres are formed from poly (lactic-co-glycolic acid) (PLGA), owing to the excellent biodegradability, biocompatibility and controlled degradation speed [Citation9] that this matrix material provides.
Losartan potassium (LP) is a highly soluble drug. There have been many efforts to prepare novel formulations of LP tablets to improve its oral bioavailability [Citation10,Citation11], as it undergoes hepatic first-pass metabolism, which is a major obstacle limiting intrinsic absorption. Additionally, LP is required to be administrated 3–4 times daily in order to maintain plasma drug concentration, due to its short half-life (t1/2) of 1.5–2.5 h [Citation12]. Not only is the frequent administration burden on patients, but it can also cause various adverse effects, such as migraines, anaemia, gastrointestinal disorders, congestive heart failure, renal diseases and so on [Citation13]. There are also other occasions that LP should be controlled to release at certain time to reach therapeutic effects [Citation14]. Based on these, it is desirable to be able to deliver soluble drugs like LP in a sustained or controlled release dosage form, to enhance therapeutic efficacy.
To encapsulate hydrophilic drugs into microspheres, the water-in-oil-in-water (w/o/w) approach is typically used. This conventional double emulsion-technique is easy to fabricate and provides good results, including acceptable entrapment of drugs and appropriate drug release times when delivering macromolecular drugs [Citation15]. However, when encapsulating low molecular weight hydrophilic drugs, such as LP, microspheres prepared by the double emulsification-solvent evaporation method result in a low encapsulation efficiency [Citation16], likely as a result of the holes in the microspheres, which provide the possibility for drugs to diffuse outside to the outer phase. Using a saturated aqueous solution of drugs as the inner phase to prepare microspheres could decrease the internal diffusion, so as to improve the encapsulation efficiency [Citation17]. However, this approach has not been extensively investigated and few reports exist. Additionally, this strategy has been shown to be prone to an initial burst release [Citation18]. Efforts have been made to overcome these disadvantages [Citation19,Citation20], but encapsulation and release of hydrophilic therapeutics remain unsatisfactory through this method.
To improve the ability of these microsphere systems to reach high encapsulation efficiencies and attain well-controlled release rates, gels are being considered in this work, to impede the hydrophilic drugs from diffusing to the external phase, and thus obtain appropriate release behaviours. Gels, like gelatine and Pluronic 407 (F127) have previously gained attention due to their thermo-reversible characteristics. Gelatine is a biopolymer with a solution-to-gel phase transition in response to temperature decreasing from high to low [Citation21], while F127 exists as liquid state at low temperature, but becomes a gel at higher temperatures [Citation22]. It is therefore possible to adjust the temperature of the outer water phase to transform the in-situ gels from liquid to gel, thereby fixing drugs into the inner phase and increasing entrapment rates [Citation23]. As well, the gel solvent should effectively control the release of the drugs, promoting sustained release and minimizing burst effects [Citation24,Citation25]. The preparation of hydrophilic drug-loaded microspheres by a solid-in-oil-in-water (s/o/w) emulsion method has recently been reported, particularly in regards to proteins [Citation26,Citation27], as proteins in a solid-state can retain activities in organic solvents, rather than denaturing in their soluble state [Citation28]. Certainly, this method shows promise as an improvement over the classical w/o/w method to improve encapsulation efficiency and modify release behaviour [Citation29].
The goal of this study is to investigate improved double emulsion techniques to create biocompatible PLGA microspheres for the encapsulation of a high rate of soluble drugs, resulting in controllable in vivo release profiles. Different inner phases, such as water, gelatine, F127 and LP micropowders were used, in order to investigate their effects on the characteristics of the resultant microspheres. As there have been few reports about controlling in vivo release profile via rational formulation design, this study should provide novel preparations for soluble drugs to reach appropriate release behaviours for different purpose.
Materials and methods
Materials
LP was obtained as a gift from Beijing Twinluck Pharmaceutical Co., Ltd. (Beijing, China). PLGA (lactide:glycolide 75:25, 20k) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). Polyvinyl alcohol (PVA-217SB) was a kind gift from Kuraray Co. Ltd. (Osaka, Japan). Gelatine was obtained from Yuwang Group (Shandong, China). Pluronic 407 (F127) was supplied from Sigma-Aldrich (St Louis, MO). All other chemicals were of analytical or chromatographic grade.
Methods
Preparation of PLGA microspheres (LP-MSs)
LP-MSs were prepared using a double emulsification-solvent evaporation method [Citation30,Citation31] with PLGA as the matrix material, according to the compositions listed in .
Table 1. Physical characteristics of microsphere formulations with different inner phases.
Briefly, to prepare water-in-oil-in-water(w/o/w) PLGA microspheres, LP was dissolved into 50 μl water, Gel 1 (5% gelatine solution) and Gel 2 (25% F 127), respectively, to be utilized as the inner phase. Based on previous reports, gelatine and F127 have the most suitable vicidity when encapsulating or releasing drugs at the concentrations of 5 and 25%, respectively, and thus these concentrations were used here [Citation32–35]. The 5% gelatine was kept in a water bath heating at 37 °C and the 25% F127 was used immediately after removal from the fridge at 4 °C to maintain its liquid state. At the same time, PLGA was dissolved in 1 ml dichloromethane (DCM), after which the entire water phase was added to the organic phase, homogenizing at 12,000 rpm for 30 s with a high-speed disperser (ULTRA-TURRAX® T18 digital, IKA Werke GmbH & Co., Staufen, Germany). The resulting water-in-oil emulsion (at this point at room temperature and thus the gelatine and F127 had turned to their gel state) was emulsified in 40 ml aqueous PVA solution (1%, w/v), followed by homogenization for 1 min at the speed of 8000 rpm. The final emulsion was then dispersed into 100 ml deionized water. The organic solvent was then removed by a rotary evaporator at 37 °C, and the LP-MSs were collected by filtration, washed with purified water, and freeze-dried in a FD-1 freeze dryer (LABFREEZ Instruments Co., Ltd., Changsha, China) without addition of cryoprotectant ().
Figure 1. The schematic diagram of LP-MSs.
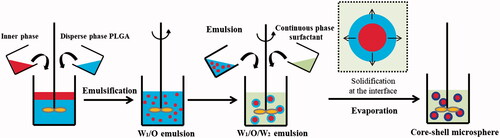
To prepare solid-in-oil-in-water (s/o/w) PLGA microspheres, the procedure was as above with the modification of using micropowders of LP as the inner phase.
Characterization of LP-MSs
The particle size and particle size distribution of the LP-MSs were determined with a Laser Particle Size Analyser (BT-9300S, Bettersize Co., Ltd., Dandong, China) before and after freeze drying.
The LP-MSs were observed under a BA300 Pol. optical microscope (Motic Ins., Xiamen, China) to confirm whether crystallization occurred. The morphology of the microspheres was investigated using an S-3400 scanning electron microscope (Hitachi High Technologies, Kyoto, Japan) after sputter coating with gold.
Differential scanning calorimeter (DSC) curves were measured using DSC1 equipment (Mettler-Toledo AG, Küsnacht, Switzerland) equipped with a refrigerated cooling system, and the data was processed using ORANGE version 8 software (Orange County Health Care Agency, Santa Ana, CA). Samples, including preparations or active pharmaceutical ingredient (API), were hermetically sealed in aluminium pans, and then heated from 30 to 300 °C at a rate of 10 °C/min under a nitrogen atmosphere.
X-ray powder diffraction (XRPD) patterns were measured on a X-ray powder diffractometer (Shimadzu, Japan) from 5 to 60° at a step rate of 5°/min.
Drug loading (DL) and drug entrapment (EE)
Approximately 15 mg of LP-MSs were weighed and transferred into a 50 ml volumetric flask, and then 2 ml DCM was added to damage the microspheres. The samples were sonicated for 5 min until the microspheres were completely dissolved. Deionized water was used to dilute to the full volume, and the sample was well-shaken to ensure all drugs were extracted from the water. The samples were left to settle for 10 min, and then the upper aqueous solution was analysed using UV–Vis spectrophotometry (UV5100, Ningbo Wayee Textile Co., Ltd., Ningbo, China) at 278 nm to determine the concentration of LP (gelatine and F127 have little absorbance at 278 nm, thus it is appropriate to measure at this wavelength). The DL and EE were then calculated as below.
In vitro release
In vitro release studies of LP from the PLGA microspheres were carried out in phosphate buffered saline (PBS, pH 6.8). To assure sink conditions, approximately 15 mg of microspheres was suspended in 5 ml PBS and incubated at 37 ± 0.5 °C in a shaking bath (ZHWY 110X30, Zhicheng Instrument Co., Shanghai, China) which was vibrating at 100 cycles/min. At pre-determined time intervals, samples were centrifuged at 5000 rpm for 5 min to precipitate the microspheres, which allowed the withdrawal of all the release media, and then replaced with the same volume of fresh media. The same volume of fresh media was replaced each time, and the amount of drug released from the LP-MSs was determined by UV–Vis spectrophotometry at 278 nm.
Gel permeation chromatography (GPC)
Investigating how the materials degrade sheds light on the mechanism of drug release, and thus the decrease in molecular weight (Mw) of the microspheres during release was analysed by Gel Permeation Chromatography (GPC) (Waters). A certain amount of LP-MSs was dissolved in tetrahydrofuran (THF), the solution was filtered through 0.45 μm membrane filters, and then 100 µl samples were injected for GPC analysis. The data was calculated by Empower GPC software (Waters Corporation, Milford, MA).
In vitro degradation
The prepared PLGA microspheres were dried and imaged by SEM. The same batch preparation of LP-MSs was then used for the in vitro release experiments. For drug release tests, at pre-determined time intervals, the microspheres were collected after centrifugation, residual salt washed off with water and then freeze-dried. The obtained powders were imaged by SEM once more and analysed via gel permeation chromatography (GPC).
In vivo pharmacodynamics study
To further investigate the drug absorption in vivo, male Sprague–Dawley rats weighing from 180 to 200 g were randomly divided into five groups (n = 5), with group A as control group, and group B–E as experimental groups. In our pre-experiment, due to excessive absorption, effective blood concentration could not be detected in a short time (4 h) after intramuscular injection of LP solution. On the other hand, due to the fact that most of the LP formulations on current markets are oral tablets, in order to visually compare the differences between the preparations we fabricated and those on the market, we took oral LP tablets on the market as control group. In addition, to highlight the discrepancy of release, we administrated each group for single dose. Since, the LP tablets have a release period for only one day, while our prepared agents could release drugs for nearly a month, we gave dose of control group the amount of a day, and that of experimental groups the amount of a month. In brief, an oral dose of 10 mg/kg of LP solution was intragastrically administrated to animals in group A, and groups B–E were injected intramuscularly with LP-MSs of formulations 1–4 (equivalent to 90 mg LP/kg). After drug administration, blood samples were collected at predetermined time intervals of 1, 2, 4, 8, 12, 24, 48 (D3), 72 (D4), 96 (D5), 120 (D6), 144 (D7), 168 (D8), 216 (D10), 264 (D12), 312 (D14), 360 (D16), 480 (D21), 648 (D28) and 816 h (D35). The samples were centrifuged at 6000 rpm for 10 min, and the supernatant was frozen at −20 °C until analysis. The LC-MS/MS method used was reported from the previous study [Citation36,Citation37] (The analysis were chromatographed on a C18 column by using an 85:15, v/v mixture of methanol and 0.1% v/v formic acid as the mobile phase at a flow rate of 1.0 ml/min. The mass transition [M-H] ions used for detection were m/z 421.0 –> 127.0).
All the data were expressed as mean ± standard deviation. Comparison between groups was performed using the Student t-test.
Statistical analysis
Statistical analysis was carried out using a paired Student t-test to evaluate systemic significant differences among different approaches. The significance was accepted at p < .05. *p < .05 and **p < .01 represented general difference and high difference, respectively.
Results
Physicochemical properties of LP-MSs
The main physicochemical properties of the different LP-MSs, including drug loading, encapsulation efficiency and mean particle size, are shown in . No significant differences in mean particle size were observed for formulations F-1 and F-2, while F-3 and F-4 had larger sizes. There were obvious increases in drug loading and encapsulation efficiency for the formulations using improved inner phases, compared to the microspheres prepared by traditional technique with water as the inner solvent.
The micrographs () observed by optical microscope show that there were needle-sharp crystals in F-1 and massive crystals in F-4, but these were not be seen in formulations of F-2 and F-3. As well, shapes were spherical in formulations F-1, F-2 and F-3, while they were irregular and uneven in F-4, with obvious lumps inside and outside the microspheres.
Figure 2. Micrographs of different inner phase of liquid (1), Gel 1 (2), Gel 2 (3) and solid (4).

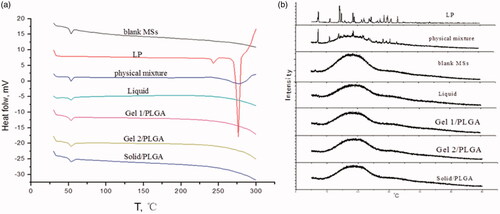
Losartan potassium in its natural state has two different crystal forms, with melting points of Tm =235.47 °C (Form I) and Tm =273.37 °C (Form II), respectively. The DSC thermograms and XRPD patterns () show that there were crystal peaks present in LP and the physical mixture, while they were not be seen in the prepared formulations, which illustrates that LP embedded in microspheres exist as an amorphous state.
Figure 3. DSC thermograms (a) and XRPD patterns (b) of blank microspheres. LP: physical mixture and LP-MSs.
In vitro degradation of the LP-MSs
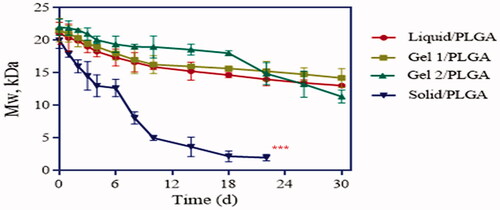
The decrease in Mw for the different MSs is shown in the GPC traces (), which show that there was a rapid degradation for F-4 (prepared by the s-o-w method), while the three other formulations had sustained decrease rates. The profiles of the microspheres with inner phases of water and 5% gelatine were similar, having a slow degradation rate before day 15, followed by maintaining even levels after. F-3, prepared with 25% F127 as the inner phase, decreased slowly in the first 18 days, but then began to decrease with a relatively fast rate in the next few days.
Figure 4. Degradation of different LP-loaded microspheres after exposure to PBS (pH 6.8) at 37 °C (n = 3) (***labels significant differences).
Morphological changes of the microspheres are shown in SEM images () and were quite varied. F-1 and F-2 still had spherical structures, with porosities inside the microspheres, while large cracks were observed through microspheres of F-3. The results indicated that in these formulations, the drug diffused through holes, with little PLGA degradation. However, a completely different morphology with irregular and uneven structures for F-4 showed that the drug was released by another mechanism which we would discuss later.
Figure 5. SEM images of different inner phase of liquid (1), Gel 1 (2), Gel 2 (3) and solid (4) before and after exposure to PBS (pH 6.8).

In vitro drug release
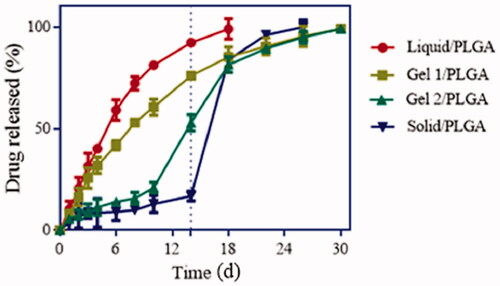
The real-time release profiles of the microsphere formulations are shown in . F-1, prepared with water as the inner phase, was found to release the drug with an initial burst phase and failed to extend the release after day 18. F-2, with the inner phase of 5% gelatine, could release the drug over 30 d, but still had an accelerated release rate when compared with the two other formulations for F-3 and F-4. However, these two formulations still had a relatively well-proportioned release. F-3 and F-4, with 25% F127 liquor and LP micropowders as the inner phases, showed two-stage in vitro release profiles, with prolonged periods with little burst rates in the early stages and burst drug release rates afterwards. These four release profiles indicate that microspheres prepared with different inner phases may have different release mechanisms, which will be discussed further.
Figure 6. In vitro drug release profiles of LP-MSs with different inner phases, at 37 °C in PBS (pH 6.8) (n = 3).
In vivo pharmacodynamics studies
Sustained and appropriate plasma LP concentrations were expected for the LP-MSs. In this study, mean concentration-time curve and pharmacokinetic parameters of LP solution and LP-loaded MSs after oral and intramuscular administrations to rats are shown in and . Compared to the controlled group, the microspheres had far longer t1/2 and mean retention time (MRT). The results indicated that microspheres could better reach the goals of prolong release, which could also be confirmed by the Clearance (CL). But tmax of F-1 and F-2 were just 10.67 and 4 h, far shorter than that of F-3 and F-4 (232.09 and 344.21 h, respectively), which was mainly because of the burst release of F-1 and F-2. Like the in vitro release study, four formulations all showed a certain degree of increase in Cmax, but the rate of increase was more significantly for F-1. AUC0–t (ug·h/l) for F-1, 2, 3 and 4 were 22,761.67, 30,313.00, 45,105.17 and 56,203.17, respectively, which were all dozens of times more than that of LP solution (1255.33).
Figure 7. Plasma LP concentrations in rats received controlled group and experiment group of preparations with different inner phases, respectively (n = 5).
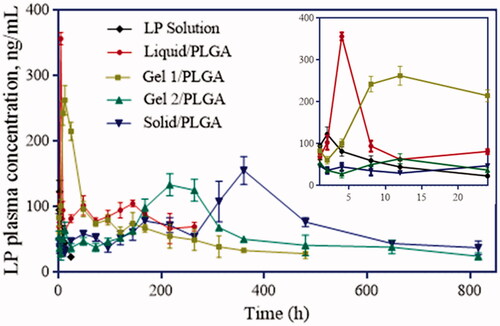
Table 2. Pharmacokinetic evaluation parameters of drug in SD rats after oral and intramuscular administrations of LP solution and LP-loaded MSs.
From the in vivo release test (), plasma LP concentrations of the control group decreased rapidly to below 14 ng/ml at approximately 24 h after oral administration of LP solution. However, all the formulations prepared in this study had sustained plasma LP concentrations. Burst release within 12 h for F-1 (water) and F-2 (5% gelatine) and as clear from the release profiles. However, F-2 was apparent as more promising formulation, as it demonstrated more sustained release in vivo when compared to all the formulations. F-1 had a relatively short release time (12 days) and higher burst release rate, and thus F-2 could be considered a more superior formulation. As for F-3 (25% F127) and F-4 (LP powders), they had low daily dose at former few days followed by increase doses after D8 (F-3) and D14 (F-4). This special release property of the two formulations provided possibility for them to be used for controlled or delayed release.
Discussion
It was evident that the series of prepared microspheres had different physicochemical properties as a result of the manufacturing changes, which included the disperse system and preparation methods ( and ). Due to the differences of the disperse systems and drug states in the inner phase, it is possible that the rate of dispersion removal during the microspheres preparation process may be different. For microspheres fabricated by traditional w/o/w technique, with osmotic pressure between the inside and outside phases and no viscous material inside the microspheres, water may carry a certain amount of drugs to the outer layer or surface when it is removed through holes in the microspheres. For the other two inner phases of F-2 and F-3, gelatine polymer and F127 could increase the viscidity of inner phase, so as to more tightly bind the drugs, hindering outside water from diffusing into the inner core and removing the encapsulated drug [Citation38,Citation39], so accordingly decreased burst release for F-2 and F-3 (), as well as increased drug loading and encapsulation efficiency. However, as gelatine existed as a liquid state at the release temperature of 37 °C, while F127 was gel under such conditions (), the viscidity of the inner phase for F-3 was higher, resulting in a relatively decelerating drug release in this formulation. As for the method of s/o/w, we hypothesized that the LP powders plugged up the holes which water diffused through, so although a little would be dissolved, the majority of drugs inside the core were isolated from the outer phase, increasing drug loading and drug entrapment, resulting in slower drug release rates in early stages. Certainly, due to the strong hydrophilicity of LP, the drug barrier was temporary, and thus together with the degradation of PLGA, burst release was soon observed. In addition, the whole LP powders instead of dispersing evenly in solution were directly wrapped into PLGA, which could easily result in irregular morphologies (), and powders could clearly be seen inside the microspheres in the micrographs, which were not observed in the other formulations.
Figure 8. Liquid and gel states of Gel1 and Gel 2 at different temperatures, including the process of preparation and in vivo administration.

The in vitro degradation of the LP-MSs was investigated via GPC and SEM, and the in vitro release study also indicated different release mechanisms for the different formulations. For F-4 (prepared by s/o/w method) a drastic decrease of Mw over time was observed through the release period, as shown in . The surface of this formulation could be observed to transform from smooth to rough and uneven (). Both these two phenomenon indicated an obvious difference to the other formulations. Despite the fast Mw decrease rate, the microspheres still had a low cumulative release rate of LP during the first 14 d, which was less than 20% (showed in ). At this stage, the molecular weight dispersity was broad [Citation40], which indicated that the initial erosion happened to the outer layer, with little inner drug being exposed, and thus presented a low profile before day 14. Subsequently, as the degradation increased, more encapsulated drug was then exposed to the dissolution medium, resulting in an acceleration of drug release during the later periods. There were similar degradation and release profiles for F-1 and F-2 (with water and 5% gelatine as the inner phase). Both the formulations had slow Mw decrease rates and well-proportioned drug release rates. The SEM images () demonstrated that F-1 and F-2 still maintained a spherical shape after the release tests, although porous structures could be observed in the microspheres. This is explained by the process of exposure to the dissolution medium leading to outside water diffusing into the microspheres through holes, and then the drugs dissolving out from the microspheres. The GPC and SEM images further support that the drug release mechanism was mainly the diffusion through holes embedded in the microspheres. Additionally, the release of F-3 was slower and longer than F-2 (30 and 18 d, respectively), due to the viscous inner phase, which could retain the drug for a longer period. Furthermore, the slowest decrease rate of F-3 (prepared with 25% F127 as the inner phase) indicated that it had a different release mechanism as is discussed as follows. It could clearly be seen that both the degradation and release profiles had two stages ( and ). At the first stage, the Mw decreased very slowly, which indicates drug release through diffusion through holes. However, due to the gel state of F127 at these conditions, a large number of drugs were fixed within the microspheres, resulting in the slowest release. Another obvious characteristic is shown in that there were large cracks in the microspheres, which was supposedly the reason for the second-stage drug release. As water diffused into the microspheres, F127 gradually absorbed the water, resulting in swelling. Eventually, the microsphere matrix could not withstand the large volume of gel, and they began to break up. The broken microspheres were then exposed to more dissolution medium and thus had an accelerated erosion speed. On the other hand, without the coating of PLGA, drugs within F127 were then released from F127 directly to the dissolution medium accordingly cause the rapid second stage release. Theoretically, gelatine polymer has the same swelling property that can also absorb the water to be swelling. But at the release situation at 37 °C, gelatine polymer presented liquid state while F127 gel, thus resulting in slight or even not obvious microspheres cracked in F-2.
From the in vitro degradation and release behaviours, F-2 was chosen to be a promising formulation to realize appropriately sustained release of LP, and this was consistent with the in vivo pharmacodynamics studies. Comparison with oral administration in shows that the four formulations prepared could all have sustained release rates. AUC0∼t (ug·h/l) of F-3 and F-4 were higher than F-1 and F-2. What’s more, tmax of F-3 and F-4 were longer, but their release behaviours were different from F-1 and F-2. They had lower plasma LP concentration at first and higher at the second stage (). This property provides possibility for them to be used for delayed release of drugs requiring to release at certain period. In contrast, F-1 and F-2 had relatively shorter tmax and higher Cmax, with the reason that they released drugs with a fast speed at beginning, as could be seen in the in vivo pharmacodynamics profiles. However, overall both the in vitro and in vivo release behaviours, F-1 and F-2 could maintain proper plasma concentrations, which could better achieve the purpose of sustained release. Since the sustained period of F-1 was somehow shorter and the burst release was higher, F-2 was finally chosen to be the most promising formulation for sustained release. As for F-3 and F-4, they have efficient drug concentration at certain time, which means they can be used in purpose of controlled release.
However, there are still some deficiencies should be improved in these formulations. For F-2, it still had a certain rate of burst release, maybe resulting in too high or even toxic dose at former days. Thus some reasonable improvement should be done to reduce the burst release, such as adding additives into the inner phase to increase its viscosity. For F-3 and F-4, if they are supposed to be delayed-release system, former plasma concentration should be reduced to limit their initial in vivo effects. We hypothesis this can be realized by adding mixed proportions of organic phase containing two or more degradable polymers. And all these possible improvements will be investigated in our following researchers.
Conclusions
This work developed improved methods of double emulsification-solvent evaporation for preparing losartan potassium-loaded-microspheres, with water, 5% gelatine, 25% F127 or LP powders as the inner phases. The obtained pharmaceutical systems showed appropriate physicochemical properties, but different release behaviours among the formulations. Because of the drug diffusion mechanism through holes, microspheres with water and 5% gelatine as the inner phases had smoother and more stable release profiles, but the latter released more slowly (up to 21 d) due to the viscidity of the inner phase. Thus this type of modified microspheres could be better used in drugs requiring sustained release. Another mechanism existed in microspheres with LP powders as the inner phase was mainly the degradation of matrix materials. As the erosion of PLGA was a two-stage process, the release speed of this formulation was slow at first, and then increased with burst release over time, resulting in less daily dose in the former 8 d and peak plasma concentration at D14. A similar profile was shown in the microspheres with 25% F127 as the inner phase, but the mechanism was obviously different, with initially the same pore diffusion mechanism, but due to the internal gel state, the drug release was very slow. At the later stages, the microspheres cracked as a result of the swelling of F127, which resulted in the direct exposure of the whole gel-containing drugs, and thus presented with rapid release at this point. The special controlled release behaviours of F-3 and F-4 suggested that they could be designed for drugs requiring delayed release. In addition, in vivo experiments showed that all the pharmaceutical systems we have prepared were superior to LP solution in achieving long-term release. All these formulations with various release behaviours make it possible to control in vivo release profile by rational core phase design.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Zhai P, Chen XB, Schreyer DJ. PLGA/alginate composite microspheres for hydrophilic protein delivery. Mater Sci Eng C Mater Biol Appl. 2015;56:251–259.
- Wei Y, Wang Y, Zhang H, et al. A novel strategy for the preparation of porous microspheres and its application in peptide drug loading. J Colloid Interface Sci. 2016;478:46–53.
- Gu B, Wang Y, Burgess DJ. In vitro and in vivo performance of dexamethasone loaded PLGA microspheres prepared using polymer blends. Int J Pharm. 2015;496:534–540.
- Falconi M, Focaroli S, Teti G. Novel PLA microspheres with hydrophilic and bioadhesive surfaces for the controlled delivery of fenretinide. J Microencapsul. 2014;31:41–48.
- Shen J, Lee K, Choi S, et al. A reproducible accelerated in vitro release testing method for PLGA microspheres. Int J Pharm. 2016;498:274–282.
- Allahyari M, Mohabati R, Amiri S, et al. Synergistic effect of rSAG1 and rGRA2 antigens formulated in PLGA microspheres in eliciting immune protection against Toxoplasma gondii. Exp Parasitol. 2016;170:236–246.
- Doolaanea AA, Ismail AFH, Mansor NI, et al. Effect of surfactants on plasmid DNA stability and release from Poly (D,L-lactide-co-glycolide) microspheres. Trop J Pharm Res. 2015;14:1769.
- Gupta RK, Alroy J, Alonso MJ, et al. Chronic local tissue reactions, long-term immunogenicity and immunologic priming of mice and guinea pigs to tetanus toxoid encapsulated in biodegradable polymer microspheres composed of poly lactide-co-glycolide polymers. Vaccine. 1997;15:1716–1723.
- Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 2012; 64:72–82.
- Osamura T, Takeuchi Y, Onodera R, et al. Prediction of effects of punch shapes on tableting failure by using a multi-functional single-punch tablet press. Asian J Pharm Sci. 2017;12:412.
- Vashisth I, Ahad A, Aqil M, et al. Investigating the potential of essential oils as penetration enhancer for transdermal losartan delivery: effectiveness and mechanism of action. Asian J Pharm Sci. 2014;9:260–267.
- Chopra S, Patil GV, Motwani SK. Release modulating hydrophilic matrix systems of losartan potassium: optimization of formulation using statistical experimental design. Eur J Pharm Biopharm. 2007;66:73–82.
- Burnier M, Wuerzner G. Pharmacokinetic evaluation of losartan. Exp Opin Drug Metab Toxicol. 2011;7:643–649.
- Sereno GA, Loeches BD, Varas FMR, et al. A pharmaceutical controlled release composition of losartan. EP20110720522. 2011 May 20. Laboratorios Liconsa SA.
- Tamnak S, Mirhosseini H, Tan CP, et al. Encapsulation properties, release behavior and physicochemical characteristics of water-in-oil-in-water (W/O/W) emulsion stabilized with pectin–pea protein isolate conjugate and Tween 80. Food Hydrocoll. 2016;61:599–608.
- Li M, Rouaud O, Poncelet D. Microencapsulation by solvent evaporation: state of the art for process engineering approaches. Int J Pharm. 2008;363:26–39.
- Mandal TK, Tenjarla S. Preparation of biodegradable microcapsules of zidovudine using solvent evaporation: effect of the modification of aqueous phase. Int J Pharm. 1996;137:187–197.
- Park JH, Ye M, Park K. Biodegradable polymers for microencapsulation of drugs. Molecules. 2005;10:146–161.
- Gaignaux A, Réeff J, Vriese CD, et al. Evaluation of the degradation of clonidine-loaded PLGA microspheres. J Microencapsul. 2013;30:681–691.
- Freytag T, Dashevsky A, Tillman L, et al. Improvement of the encapsulation efficiency of oligonucleotide-containing biodegradable microspheres. J Control Release. 2000;69:197–207.
- Goudoulas TB, Germann N. Phase transition kinetics and rheology of gelatin-alginate mixtures. Food Hydrocoll. 2017;66:49–60.
- Akash MS, Rehman K. Recent progress in biomedical applications of Pluronic (PF127): pharmaceutical perspectives. J Control Release. 2015;209:120–138.
- Zhang M, Yang B, Liu W, et al. Influence of hydroxypropyl methylcellulose, methylcellulose, gelatin, poloxamer 407 and poloxamer 188 on the formation and stability of soybean oil-in-water emulsions. Asian J Pharm Sci. 2017;12:521–531.
- Kim DY, Kwon DY, Kwon JS, et al. Synergistic anti-tumor activity through combinational intratumoral injection of an in-situ injectable drug depot. Biomaterials. 2016;85:232–245.
- Deng L, Kang X, Liu Y, et al. Effects of surfactants on the formation of gelatin nanofibres for controlled release of curcumin. Food Chem. 2017;231:70–77.
- Morita T, Horikiri Y, Yamahara H, et al. Formation and isolation of spherical fine protein microparticles through lyophilization of Protein-Poly(ethylene Glycol) aqueous mixture. Pharm Res. 2000;17:1367–1373.
- Paillard-Giteau A, Tran VT, Thomas O, et al. Effect of various additives and polymers on lysozyme release from PLGA microspheres prepared by an s/o/w emulsion technique. Eur J Pharm Biopharm. 2010;75:128–136.
- Sah H. Protein behavior at the water/methylene chloride interface. J Pharm Sci. 1999;88:1320–1325.
- Zhang Y, Lin J, Zhong Q. S/O/W emulsions prepared with sugar beet pectin to enhance the viability of probiotic Lactobacillus salivarius NRRL B-30514. Food Hydrocoll. 2016;52:804–810.
- Mosafer J, Teymouri M. Comparative study of superparamagnetic iron oxide/doxorubicin co-loaded poly (lactic-co-glycolic acid) nanospheres prepared by different emulsion solvent evaporation methods. Artif Cells. 2017;9:1–10.
- Yang H, Hao Y, Liu Q, et al. Preparation and in vitro study of hydrochloric norvancomycin encapsulated poly (d,l-lactide-co-glycolide, PLGA) microspheres for potential use in osteomyelitis. Artif Cells Nanomed Biotechnol. 2016;45:1326–1330.
- Coopes IH. Helix formation and formaldehyde crosslinking in gelatin solutions. J Polym Sci A-1 Polym Chem. 1970;8:1793–1811.
- El-Kamel AH. In vitro and in vivo evaluation of Pluronic F127-based ocular delivery system for timolol maleate. Int J Pharm. 2002;241:47–55.
- Koob T J, Hernandez D J. Mechanical and thermal properties of novel polymerized NDGA-gelatin hydrogels. Biomaterials. 2003;24:1285–1292.
- Ye F, Yaghmur A, Jensen H, et al. Real-time UV imaging of drug diffusion and release from Pluronic F127 hydrogels. Eur J Pharm Sci. 2011;43:236.
- Arun B, Narendar D, Veerabrahma K. Development of olmesartan medoxomil lipid-based nanoparticles and nanosuspension: preparation, characterization and comparative pharmacokinetic evaluation. Artif Cells Nanomed Biotechnol. 2017;46:126–137.
- Karra VK, Pilli NR, Inamadugu JK, et al. Simultaneous determination of losartan, losartan acid and amlodipine in human plasma by LC-MS/MS and its application to a human pharmacokinetic study. Pharm Methods. 2012;3:18–25.
- Wang P, Wang Q, Ren T, et al. Effects of pluronic F127-PEG multi-gel-core on the release profile and pharmacodynamics of Exenatide loaded in PLGA microspheres. Colloid Surf B Biointerfaces. 2016;147:360–367.
- Park KS, Kim C, Nam JO, et al. Synthesis and characterization of thermosensitive gelatin hydrogel microspheres in a microfluidic system. Macromol Res. 2016;24:529–536.
- Cai Q, Shi G, Bei J, et al. Enzymatic degradation behavior and mechanism of poly(lactide-co-glycolide) foams by trypsin. Biomaterials. 2003;24:629–638.