
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The unique tumour microenvironment (TM) of pancreatic ductal adenocarcinoma (PDA) including highly desmoplastic ECM and low tumour perfusion supports a considerable barrier for effective delivery of nanomedicines. Effectively modulating PDA microenvironment to enhance tumour drug delivery represents a pinpoint in the field of PDA treatment. In this study, it was the first time that biomimetic nanoparticles, which were designed in the form of erythrocyte membrane-camouflaged PLGA nanoparticles (MNP), were utilized for PDA microenvironment modulation. Cyclopamine (CYC), an inhibitor of Hedgehog pathway that contributed a lot to desmoplastic ECM of PDA, was selected as the model drug and successfully encapsulated into MNP. Advantages of CYC-loaded MNP (CMNP) included favourable biocompatibility, long circulation time, and powerful TM modulation effect. CMNP could effectively deliver CYC to the tumour site, disrupt tumour ECM, increase functional vessels, and improve tumour perfusion significantly. The combination treatment with CMNP and PTX-loaded MNP (PMNP) successfully improved PTX delivery to tumour, resulting in remarkable tumour growth inhibition in vivo. Therefore, biomimetic nanoparticles provide a new strategy for modulating PDA TM and will have great potential to improve the therapeutic effects of nanomedicines for PDA patients.
Introduction
Pancreatic ductal adenocarcinoma (PDA), a common and lethal malignancy causing plenty of deaths every year, is characterized by an extensive desmoplastic reactional tumour microenvironment [Citation1] composed of a large number of tumour-associated fibroblasts (TAF) and abundant and dense extracellular matrix (ECM) [Citation2] which compromised effective nanomedicine delivery for PDA treatment. Abundant TAF and ECM could compress scarce tumour vessels to reduce tumour perfusion, resulting in little nanomedicine arrival at tumour site. In addition, the dense ECM could hinder effective penetration of nanomedicine inside tumour tissues to achieve a homogeneous distribution. Therefore, the tumour microenvironment characterized by abundant TAF and extensive ECM has seriously hindered the application of nanomedicine for the treatment of PDA, and it is urgent to establish a more rational and effective drug delivery strategy for PDA treatment.
As the Hedgehog (Hh) cellular signalling pathway plays an important role in desmoplastic ECM of PDA, it has become a potential therapeutic target in PDA treatment [Citation3]. Cyclopamine, a reliable extraction from Veratrum californicum that can inhibit the activity of Hh pathway by binding to the receptor Smoothend (SMO, a seven-transmenbrane protein), is a good candidate with high potential to inhibit ECM desmoplasia of PDA [Citation4]. Previous studies have confirmed that ECM disruption and tumour perfusion improvement in PDA mouse model by oral administration of free cyclopamine led to improved drug delivery to the tumour site [Citation4]. However, as a water-insoluble drug (∼5 µg/mL), the bioavailability of cyclopamine by oral administration was very low [Citation5]. In addition, delivery of free cyclopamine to PDA was always seriously compromised by the complex tumour microenvironment. To achieve effective tumour microenvironment modulation of PDA, a large dose of cyclopamine for oral administration was necessary and may lead to unpredictable toxicity to PDA patients, posing a great challenge for clinical translation. Therefore, finding a proper way to improve the bioavailability and simultaneously lower the dosage of cyclopamine is needed to be addressed.
Nanomedicine, being regarded as one of the most promising treatment to surpass traditional chemotherapy as a front-line cancer treatment [Citation6], has attracted extensive attention in recent decades. Different approaches including modifications on nanoparticle size, surface, shape, and flexibility have been explored to extend nanomedicine residence time in vivo [Citation7] to improve the tumour accumulation of nanomedicine via enhanced permeation and retention effect. Among them, PEGylation of nanomedicine has become a classical method to reduce nonspecific uptake of nanomedicine by mononuclear phagocyte system (MPS) and guarantee the long circulation time (12–24 h) and anti-tumour efficacy of nanomedicine to a certain degree. Inspired by the long circulation of natural red blood cells (RBCs) (100–120 days) [Citation8], biomimetic nanoparticles have recently been proposed by coating nanoparticles with RBC membrane (MNP) which endowed nanoparticles properties of RBC membrane such as less susceptible to uptake by MPS and much longer circulation time than traditional PEG decoration [Citation9]. With this long blood circulation property, these biomimetic nanoparticles have more chance to reach tumours and accumulate inside tumours, greatly benefiting drug delivery to tumours, especially PDA with poor perfusion and abundant ECM. Therefore, delivering cyclopamine with biomimetic nanoparticles might possess many advantages such as longer circulation time in vivo, higher bioavailability in tumours, capability of modulating the tumour microenvironment more effectively, and reduced toxicity of cyclopamine, which represented a rather safe and highly effective modulator of PDA microenvironment. To the best of our knowledge, there were few studies using biomimetic nanoparticles to deliver drugs to modulate PDA tumour microenvironment.
In this study, cyclopamine-loaded biomitmetic nanoparticles (CMNP) were for the first time developed to modulate PDA tumour microenvironment (). Changes of tumour microenvironment after CMNP treatment including ECM, functional vessels density, and tumour perfusion were investigated by H&E staining, immunofluorescence staining and in vivo photoacoustic imaging, and compared with oral cyclopamine treatment. Additionally, the long circulation of MNP was verified and compared with PEGylated nanoparticles. The accumulation of MNP in tumours after CMNP treatment was investigated by in vivo imaging and biodistribution experiments. Finally, the therapeutic benefit of paclitaxel-loaded MNP (PNP) combined with CMNP treatment was assessed by pharmacodynamics experiments ().
Materials and methods
Materials
Carboxy-terminated 50:50 poly(DL-lactide-co-glycolide) (PLGA, 0.67 dl∕g) was purchased from LACTEL Absorbable Polymers (USA). Cylopamine was obtained from Stru Chem Co., Ltd (Wuxi, China). Paclitaxel was obtained from Dalian Meilun Biotech Co., Ltd (Dalian, China). Cremophor® EL (polyoxyethlene castor oil derivatives) was purchased from BASF (Germany). Fibronectin rabbit polyclonal primary antibody was purchased from Santa Cruz Biotechnology (USA), and CD31 goat polyclonal primary antibody was from R&D (USA). Cy™ 3-conjugated Affinipure donkey anti-goat secondary antibody and Alexa fluor® 647-conjugated donkey-rabbit secondary antibody were purchased from Jackson (USA). DyLight®488-labelled tomato lectin (Lycopersicon esculentum) was from Vector (USA). Hoechst 33342 was from Beyotime Biotechnology Co., Ltd. (Nantong, China). 1, 10-Dioctadecyl-3, 3, 3-tetramethylindo-tricarbocyanineiodide (DiR) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) were purchased from Biotium (Invitrogen, USA). Coumarin-6 was purchased from Sigma (St. Louis, MO, USA). All the other reagents were of analytical grade and were from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Deionized water was produced by a Millipore water purification system (Millipore, Bedford, MA).
Human-derived pancreatic cancer Capan-2 cell line and human umbilical vein endothelial cell (HUVEC) line were ordered from American Type Culture Collection (ATCC, MD, USA). Male Balb/c nude mice and Balb/c mice aged 6–8 weeks were bought from the Shanghai Slac Lab Animal Ltd. (Shanghai, China). All animal experiments were conducted in accordance with protocols evaluated and approved by the Experiment Animal Ethics Committee of Fudan University.
Nanoparticle preparation
Nanoparticles (NP) loaded with cyclopamine (CYC), paclitaxel (PTX), coumarin-6, DiD or DiR were prepared by an emulsion/solvent evaporation method as previously described [Citation10]. RBC ghosts were prepared as previously reported by Zhang et al. [Citation11]. Briefly, the whole blood was collected from the Balb/c mice and washed red blood cell were collected after few centrifugation. After hypotonic medium treatment, the resulting RBC ghosts were collected. Detailed methods are presented in Supplemental File.
For preparation of RBC membrane-coated NP, 8 mg of NP loaded with CYC, PTX, DiD, DiR, or coumarin-6 were mixed with 1 ml of RBC ghosts obtained above. After the mixture was sonicated in a capped glass vial at a frequency of 53 kHz and a power of 100 W for 2 min, RBC membrane-coated NP (MNP) solution was obtained [Citation12].
Characterization of MNP
The morphology of MNP was characterized using transmission emission microscopy (TEM) (H-600, Hitachi, Japan) after negative staining with 1% uranyl acetate. The size distribution and zeta potential of MNP were measured by dynamic light scattering (DLS) using a Malvern Nano ZS (Malvern Instruments, UK). MNP were stored in 1 × PBS at 4 °C for stability investigation, including particle size, and zeta potential monitoring every day for a week.
To determine the drug loading, PTX or CYC-loaded nanoparticles were dissolved in methanol and the concentration of PTX or CYC was measured by high performance liquid chromatography (HPLC) [Citation13]. To evaluate the in vitro release behaviour of CYC or PTX from CYC-loaded MNP (CMNP) or PTX-loaded MNP (PMNP), the mixture of PBS (0.01 M, pH 7.4) and methanol at a volume ratio of 1:3 was used as the release medium [Citation13]. The detailed methods are presented in Supplemental File.
Cell uptake
Cell uptake of MNP was determined on Canpan-2 cells. Briefly, cells were seeded into a 96-well plate at the density of 104 cells per well, and incubated at 37 °C for 24 h to reach 80% confluence. After treatment with CMNP (cyclopamine concentration of 5 µM) or PBS for 6 h, cells were incubated with Coumarin-6-labelled MNP (Cou6-MNP) for 30 min, and then were observed under a fluorescence microscope (Leica DMI 4000B, Germany).
Cytotoxicity assay
HUVECs were used to determine the cell growth inhibition ability of CMNP and PMNP and Capan-2 cells were used to determine the tumour cell killing effect of CMNP and PMNP. Briefly, approximately 104 cells per well were seeded into a 96-well plate, and incubated at 37 °C for 24 h to reach 80% confluence. After being washed twice with PBS, the cells were treated with varying concentrations of CMNP, PMNP or MNP at 37 °C for 24 h. The cell viability was then tested by MTT assay.
Tumour microenvironment modulation by CMNP treatment
To establish the PDA tumour-bearing mouse models, 5 × 106 Capan-2 cells suspended in 100 μL of PBS were subcutaneously implanted into the right hind flank region of nude mice. Tumour diameter was determined using a caliper. When tumour diameter reached approximately 3 mm, animals were randomly divided into three groups including control group, CYC group, and CMNP group, and treated with different CYC formulations for 18 days. Mice in the control group received an intravenous injection of 100 μL of PBS once a day for 18 days. Mice in the CYC group were orally administrated with 100 μL of cyclopamine solution (CYC concentration of 10 mg/ml) at the CYC dose of 50 mg/kg once a day for 18 days. Cyclopamine was dissolved in the mixture of ethanol, Cremophor EL, and deionized water with the cyclopamine concentration of 10 mg/mL. Mice in the CMNP group were injected with 100 μL of CMNP solution containing 0.1 mg of cyclopamine once a day for 18 days.
To investigate the ECM modulation effect of CMNP, after CMNP treatment ended, mouse models were sacrificed, and perfused with 4% paraformaldehyde, and frozen tumour slices with the thickness of 20 µm were prepared for immunofluorescence staining of fibronectin [Citation14]. Briefly, tumour slices were successively immune-stained with fibronectin rabbit polyclonal primary antibody (1:100) at 4 °C overnight, Alexa fluor® 647-conjugated donkey-rabbit secondary antibody (1:100) at room temperature for 1 h, and Hoechst 33342 (1 µg/ml) at room temperature for 5 min. Afterward, the slices were mounted in Dako fluorescence mounting medium and observed under confocal microscopy (ZEISS, 710, LSM, Oberkochen, Germany).
To investigate the ECM modulation effect of CMNP on tumour blood perfusion, in vivo blood perfusion at tumour site was demonstrated by photoacoustic imaging of oxyhaemoglobin signal. After CMNP treatment ended, mouse models were anesthetized with isoflurane at a concentration of 1.5–2%, and placed on the working plate for photoacoustic scanning using a Vevo LAZR system (FUJIFLM VisualSonics, Toronto, Canada) with a linear array transducer (LZ400, 30 Hz centre frequency). The data were analyzed by Vevo LAB software.
Lectin-labelling experiment was also performed to investigate the ECM modulation effect of CMNP on tumour blood perfusion. After CMNP treatment ended, mouse models were sacrificed at 1 h after an intravenous injection of Dylight 488-labled lectin (5 mg/kg) and perfused with 4% paraformaldehyde. Tumours were harvested for frozen slice preparation and immunostaining. Briefly, tumour slices were successively stained with CD31 primary goat polyclone antibody (1:100) at 4 °C overnight, CyTM 3-conjugated donkey anti-goat IgG secondary antibody (1:100) at room temperature for 1 h, and Hoechst 33342 (1 µg/ml) at room temperature for 5 min. Finally, the slices were mounted in Dako fluorescence mounting medium and observed under the confocal microscopy (ZEISS, 710, LSM, Germany).
Pharmacokinetics of MNP
For pharmacokinetics studies, PEGylated nanoparticles (PEG-NP) sized 100 nm were prepared as previously described [Citation11] and used as control. Male Balb/c mice aged 6–8 weeks were intravenously injected with 0.5 mg of DiD-labelled MNP (DiD-MNP), NP, or PEG-NP in 150 μL of sucrose solution. 30 μL of blood were collected by cheek pouch puncture at varying time points (1 min, 5 min, 10 min, 30 min, 1 h, 3 h, 8 h, 24 h and 48 h) after nanoparticle injection. Blood samples were diluted with PBS in 96-well plates and the fluorescence was further determined by a microplate reader (TECAN M1000, Männedorf, Switzerland) [Citation11]. The nanoparticle concentration in the blood was expressed as the percentage of injected dose per millilitre (% ID/mL). The pharmacokinetics parameters including area under the concentration − time curve (AUC0−t), mean residence time (MRT0−t), elimination rate constant (k), clearance (Cl), and the half-life (t1/2) were calculated by DAS 3.0 pharmacokinetics software.
In vivo imaging and biodistribution of MNP after CMNP treatment
After the CMNP treatment ended, PDA-bearing mouse models were intravenously injected with 100 μL of DiR-labelled MNP (DiR-MNP) at a DiR dose of 0.5 mg/kg. Twenty-four hours after nanoparticle injection, in vivo fluorescence imaging of mouse models was carried out using the In vivo IVIS spectrum imaging system (PerkinElmer, MA, USA). The mice were then sacrificed and subjected to heart perfusion with 4% paraformaldehyde. Tumour tissues and other major organs (heart, liver, spleen, lung, kidney, and brain) were collected and imaged using the same imaging system, and the corresponding fluorescence signals ex vivo were recorded.
Anti-tumour efficacy
PDA-bearing mouse models were constructed as described above. When the tumour diameter reached approximately 3 mm, mouse models were randomly divided into six groups: blank group, CMNP group, PMNP group, CMNP + PTX group, CYC + PMNP group, and CMNP + PMNP group. The day of grouping was regarded as the 1st day. For the blank group, mice did not receive any treatment. For the CMNP group, mice were intravenously injected with CMNP at a CYC dose of 5 mg/kg every day for 18 days. For the PMNP group, mice were intravenously injected with PMNP every three days at a PTX dose of 6 mg/kg from the 14th day to the 26th day. For the CMNP + PTX group, mice were not only treated with CMNP as the CMNP group but also treated with free PTX (Taxol®) at a PTX dose of 6 mg/mL every three day from the 14th day to the 26th day through tail vein injection. For the CYC + PMNP group, mice were orally administrated with CYC solution at the CYC dose of 50 mg/kg once a day for 18 days as described above and then received PMNP injection every three day at a PTX dose of 6 mg/kg from the 14th day to the 26th day. For the CMNP + PMNP group, mice were not only treated with CMNP as the CMNP group but also treated with PMNP as the PMNP group.
During the pharmacodynamics experiment, the tumour volume of mice from different groups (n = 5) was monitored every three days for 28 days. Tumour diameter was determined using a caliper, and tumour volume (V) was calculated using the following equation: , where a and b represented the maximum diameter and the minimum diameter, respectively. When tumour size of a mouse reached above 1 cm, the mouse was defined dead and mouse survival curve was plotted to assess the tumour growth inhibition effect of different treatments. On the 28th day, the mice from different groups (n = 5) were sacrificed and perfused with 4% paraformaldehyde. The tumours were collected for weight recording and photography. Tumour growth inhibition rates (TGIR) based on tumour size (TGIRV) and tumour weight (TGIRW) were calculated by the following formulas, respectively: TGIRV = (Vc–Vt)/Vc, TGIRW = (Wc–Wt)/Wc. In these formulas, Vc and Vt indicated tumour volume in the control group and that in the treatment group, respectively; Wc and Wt indicated tumour weight in the control group and that in the treatment group, respectively. At the end of the study, tumours from each group (n = 5) were excised and sliced for cell apoptosis detection by terminal dUTP-mediated nick-end-labelling (TUNEL) test and histology analysis by hematoxylin and eosin (H&E) staining.
In vivo systematic toxicity
During the pharmacodynamics experiment, the body weights of mice from different groups (n = 5) were measured by digital caliper measurement and digital weighing machine every three days for 28 days. On the 28th day, the mice from different groups (n = 5) were sacrificed, and major organs including heart, liver, spleen, lung, and kidney were collected, fixed in 4% paraformaldehyde, and sliced for histology analysis by hematoxylin and eosin (H&E) staining. Tissue inflammation or necrosis was observed under an inverted fluorescence microscope (LEICA DMI 4000 B).
Statistical analysis
Statistical differences were evaluated with unpaired Student's t-test for two groups' comparison and one-way ANOVA followed by Bonferroni analysis for multiple group comparison. Data were expressed as mean ± standard deviation (SD), and p values < .05 was considered statistically different.
Results
Characterization of MNP
In this study, the emulsion method without any surfactant was used to prepare PLGA nanoparticles RBC membrane was cloaked on these nanoparticles. The RBC membrane coating on the nanoparticles was verified by transmission electron microscopy. As shown in , a unilamellar membrane with the thickness of approximately 15 nm could be clearly observed around the nanoparticle. DLS revealed that the size of NP (68.8 ± 5.8 nm) was increased by 12 nm after RBC membrane coating (80.4 ± 11.3 nm), which agreed well with TEM results (). The zeta potential of NP was −45.1 ± 1.9 mV. After RBC membrane coating on NP, the zeta potential of MNP was increased to −29.5 ± 4.1 mV, which was similar to that of RBC membrane vesicles (−29.1 ± 4.5 mV), indicating the successful RBC membrane coating. After PTX loading in MNP, their size (86.0 ± 31.7 nm) and zeta potential (-25.1 ± 4.7 mV) were not significantly changed. However, CYC encapsulation in MNP slightly decreased their size (66.1 ± 17.2 nm) but did not significantly changed their zeta potential (−26.1 ± 3.2 mV) (). This size decrease might result from the strong drug–polymer interaction between CYC and PLGA, which still needs further investigation. To evaluate the stability of CMNP and PMNP in PBS at 4 °C, the size and zeta potential were monitored once a day for 6 days, as unstable nanoparticles tended to aggregate leading to size increase and zeta potential change. It was shown that both of the size and zeta potential of CMNP and PMNP stayed steady within 6 days (), demonstrating excellent colloidal stability in vitro. The drug-loading capacity of cyclopamine and PTX by MNP were 4.30 ± 0.59% and 4.50 ± 0.84%, respectively. In vitro drug release demonstrated that the release of cyclopamine or paclitaxel from CMNP or PMNP was significantly slower compared with that of free drug (). Only 26.2 ± 6.0% of cyclopamine or 30.2 ± 5.9% of paclitaxel was released from CMNP or PMNP within the first 2 h. By contrast, 98.2 ± 5.5% of cyclopamine or 85.7 ± 0.6% of paclitaxel was released from free drug within the first 2 h.
Figure 1. Schematic graph of MNP preparation and MNP delivery in stroma-rich PDA with and without CMNP treatment. NP were loaded with cyclopamine or paclitaxel and coated with red blood cell membrane. Without CMNP treatment, the tumour vessels are compressed by surrounding ECM components, leading to poor blood perfusion and limited MNP delivery. In contrast, after CMNP treatment, ECM components such as fibronectin were disrupted and tumour vessels were decompressed, resulting in enhanced blood perfusion and more effective delivery of PMNP. Thus, chemotherapy effect could be improved and the tumour growth was inhibited.
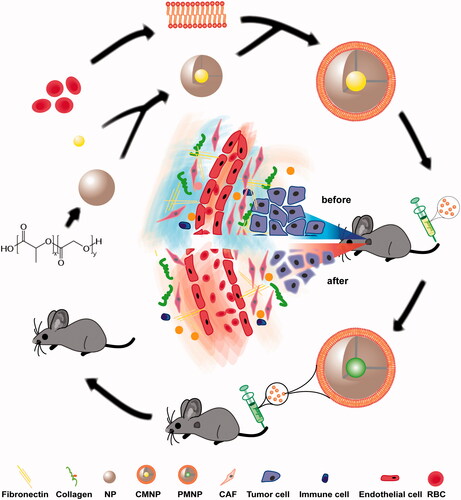
Figure 2. Characterizations of MNP (n = 4). (A) TEM image of MNP following 1% uranyl acetate staining. (B) Zeta potential and size of NP, RBC vesicle (MV), MNP, CMNP and PMNP. (C) Size monitoring of NP, CMNP and PMNP in 1 × PBS at 4 °C for 6 days. (D) Zeta potential monitoring of CMNP and PMNP at 4 °C for 6 days. (E) Drug release curves of cyclopamine from CMNP and free cyclopamine at 37 °C for 4 days. (F) Drug release curves of paclitaxel from PMNP and free PTX at 37 °C for 4 days.
Cell uptake and cytotoxicity assay
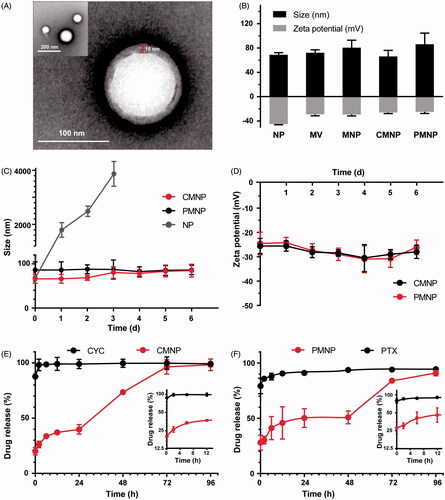
To evaluate the cell uptake capability of MNP, coumarin-6 was used to label the MNP and Canpan-2 cells after MNP treatment were imaged by a fluorescence microscope. As shown in , cell uptake of MNP with or without CMNP pretreatment showed no significant difference, indicating CMNP pretreatment did not affect MNP uptake by Canpan-2 cells.
Figure 3. Cell uptake of MNP and cell viability after CMNP or PMNP treatment (n = 4). (A) Fluorescence images of MNP-treated Canpan-2 cells with or without CMNP pretreatment for 6 h. (B) Cell viabilities of Capan-2 cells after incubation with various concentrations of CMNP (cyclopamine concentration varied from 0, 1, 5, and 20 µM) at 37 °C for 24 h. (C) Cell viabilities of Capan-2 cells after incubation with various concentrations of PMNP (PTX concentration varied from 0, 0.25, 1, 10 and 20 µg/mL) 37 °C for 24 h. *p < .05, ***p < .001, compared with the control group (PTX concentration equals to 0 µg/mL). (D) Cell viabilities of HUVEC cells after incubation with various concentrations of CMNP (cyclopamine concentration varied from 0, 1, 5, and 20 µM) at 37 °C for 24 h. (E) Cell viabilities of HUVEC cells after incubation with various concentrations of PMNP (PTX concentration varied from 0, 0.25, 1, 10 and 20 µg/mL) 37 °C for 24 h.
Cytotoxicity of CMNP and PMNP were determined by MTT analysis against Capan-2 cells ()) and HUVEC cells ()). It was shown both CMNP and PMNP had little influence upon the cell viability of HUVEC cells. However, CMNP demonstrated little cytotoxicity to Canpan-2 cells and PMNP decreased the Capan-2 cell viability as the concentration of PTX increased.
Effect of CMNP on tumour microenvironment
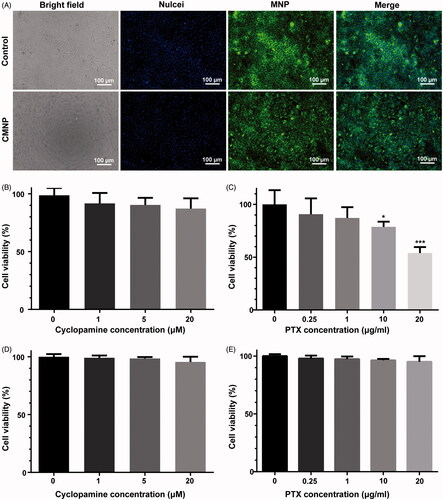
To explore the effect of CMNP on tumour microenvironment, Capan-2 tumour xenografts were utilized as PDA models and tumour ECM morphology and tumour blood perfusion were investigated after CMNP treatments for 18 days. Fibronectins highly expressed in Capan-2 tumours were used as the marker of ECM [Citation14,Citation15]. H & E staining of tumour slices from Capan-2 xenografts showed the ECM was highly desmoplastic with abundant and compact fibronectins bundles, which divided the tumour parenchyma into separate compartments in the control group without drug treatment. As a contrast, fibronectin bundles were fractured and loosely distributed after treatment with oral CYC or CMNP (). According to the fluorescence intensity of fibronectin per field in six slices from each group determined by the confocal microscopy, the fibronectin expression in Capan-2 xenografts of the CMNP group and the CYC was 37.8 ± 15.8%, 45.7 ± 6.0% of that of the control group, respectively, indicating CMNP and CYC treatment significantly disrupted ECM in tumours.
Figure 4. ECM disruptions by CMNP treatment evaluated by (A) H & E staining and (B) immunofluorescence staining (n = 6). After treatment with PBS, CYC or CMNP for 18 days, Capan-2 tumour xenografts were obtained for H&E staining and immunofluorescence staining.
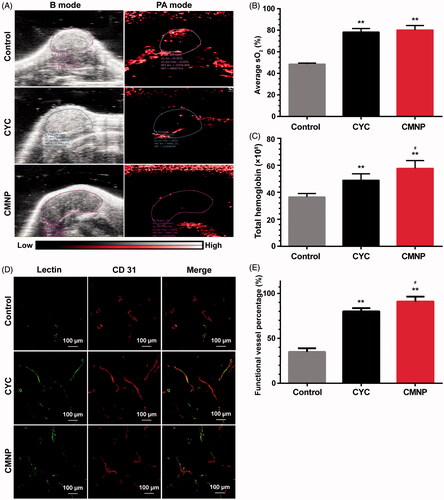
Photoacoustic imaging of oxyhaemoglobin signal at tumour site ()) revealed total haemoglobin signal in the CMNP group and the CYC group were significantly stronger than that of the control group, indicating significant tumour perfusion improvement after CMNP or CYC treatment. In addition, the haemoglobin signal in the CMNP group was significantly higher than that of the CYC group even the cyclopamine dose of in the CMNP group was only 10% of that of the CYC group. The blood oxygen saturation in the CMNP group (80.2 ± 7.2%) and the CYC group (78.3 ± 5.8%) was significantly higher than that of the control group (48.5 ± 1.8%), which was probably because of the tumour perfusion improvement. No significant differences were found in the blood oxygen saturation between the CMNP group and the CYC group. Lectin-labelling experiment revealed the percentage of DyLight® 488-lectin positive vessels (well-perfused) in all vessels (CD31 positive) was significantly increased from 34.7 ± 7.8% in the control group to 90.2 ± 9.3% in the CMNP group and 79.4 ± 7.3% in the CYC treatment group (). Furthermore, the percentage of well-perfused vessels in all vessels of the CMNP group was significantly higher than that of the CYC group. All these results demonstrated CMNP treatment significantly increased tumour perfusion. Factually, oral cyclopamine treatment could achieve significant ECM disruption and tumour perfusion improvement. Compared with oral cyclopamine treatment, only 10% of the cyclopamine dose was used in the CMNP group but achieved similar or better ECM disruption and tumour perfusion improvement. These results might be due to the long circulation nature of MNP and the increased bioavailability of cyclopamine in tumours.
Figure 5. Tumour perfusion improved by CMNP treatment (n = 6). (A) In vivo photoacoustic imaging of Capan-2 tumour xenografts after treatment with PBS, CYC or CMNP for 18 days and (B) the corresponding average oxygen saturation and (C) total haemoglobin level in tumour regions. (D) The fluorescence imaging of tumour slices after lectin 488 labelling and (E) the percentage of functional vessels (lectin 488 positive) in tumour vessels after CMNP treatment. **p < .01 compared with the control group, #p < .05 compared with the CYC group.
Pharmacokinetics of MNP
To study the in vivo pharmacokinetics of MNP, a fluorescence tracker DID was used to label nanoparticles [Citation11]. The fluorescence intensity of DID in the blood samples obtained at preset time points after nanoparticle injection was determined and the pharmacokinetics curves were plotted. As shown in , MNP had a significantly longer circulation time (t1/2 = 12.36 h) than bare nanoparticles (1.51 h) and PEG-NP (t1/2 = 8.67 h), and the blood clearance of MNP was only 3.6% of that of NP, indicating RBC membrane coating could significantly extend the in vivo circulation of nanoparticles (), which consisted well to previous study [Citation11].
Figure 6. Pharmacokinetics of MNP and enhanced tumour delivery of MNP by CMNP treatment (n = 6). (A) Pharmacokinetics curves of NP, MNP and PEG-NP. (B) t1/2 of NP, MNP and PEG-NP in blood. **p < .01, compared with the NP group. ##p < .01, compared with the PEG-NP group. (C) In vivo fluorescence imaging of mouse models pretreated with CMNP at 24 h after MNP injection and (D) the corresponding fluorescence intensity of tumours determined by In vivo IVIS spectrum imaging system. The circle indicated the tumour. *p < .05, compared with the control group. (E) Ex vivo fluorescence imaging of major organs and tumours harvested at 24 h after MNP injection. (F) Biodistribution of MNP in major organs and tumours at 24 h after MNP injection. ***p < .001 compared the CYC group or the CMNP group.
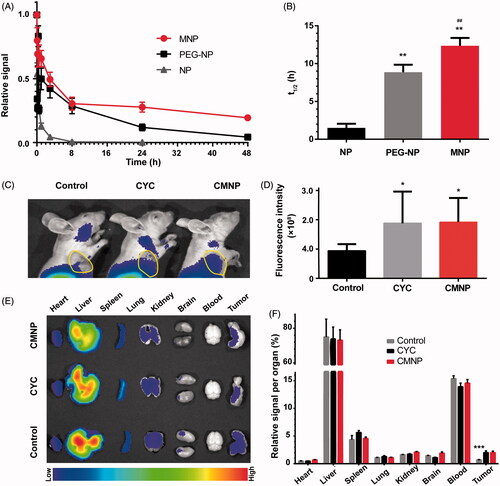
Table 1. Pharmacokinetics parameters of PEG-NP and MNP.
In vivo imaging and biodistribution of MNP after CMNP treatment
To investigate the biodistribution of MNP after CMNP treatment, a near-infrared tracker, DiR was used to label MNP for in vivo fluorescence imaging of model mice after nanoparticle injection. Results revealed that the fluorescence intensity of MNP at the tumour site in the CMNP group and the CYC group 24 h post MNP injection was significantly higher than that in the control group (). Ex vivo imaging of tumour xenografts obtained at 24 h after nanoparticle administration also displayed the fluorescence signal of tumours in the CMNP group and the CYC group was approximately 2.86-fold and 2.83-fold higher than that in other groups, respectively (). In addition, there were no significant differences in the fluorescence signal of other major organs between these groups (), indicating the specific effect of cyclopamine on tumour tissues and minimal effect on normal organs.
Anti-tumour efficacy
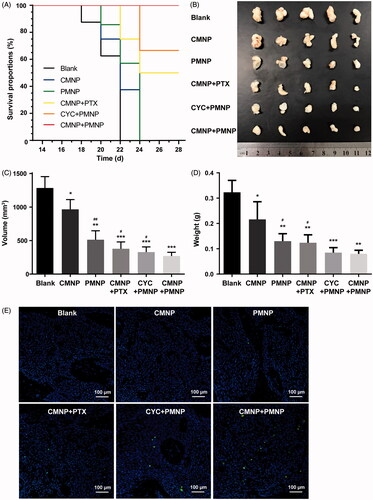
As shown in ), anti-tumour experiment results demonstrated the tumour volume from different treatment groups was significantly smaller than that from the blank group. Moreover, the tumour volume or tumour weight for the CMNP + PMNP group was significantly lower than any other treatment group. Compared with the blank group, the TIGRv of the PMNP group, the CMNP + PTX group, the CYC + PMNP group, and the CMNP + PMNP group were 60.1%, 70.6%, 74.5%, and 79.0%, respectively. The TIGRw of the PMNP group, the CMNP + PTX group, the CYC + PMNP group, and the CMNP + PMNP group were 59.7%, 61.7%, 73.8%, and 75.3%, respectively. Among all the treatment groups, the CMNP + PMNP group showed the strongest tumour growth inhibition. In addition, more extensive tumour apoptosis was found in tumours from the CMNP + PMNP group compared with other groups (). These results altogether indicated that PMNP combined with CMNP treatment achieved the best chemotherapeutic effect, probably resulting from the efficient tumour microenvironment modulation by CMNP treatment and thus improved delivery of PMNP to tumours based on the long circulation nature of MNP.
Figure 7. Antitumour efficacy of PMNP combined with CMNP (n = 5). (A) Survival curve of mouse models after different treatments recorded from 14th day to 28th day. (B) Bight images of tumour xenografts obtained at the study end point. (C) Tumour volume and (D) tumour weight of tumour xenografts at the study end point. *p < .05, **p < .01, ***p < .001, compared the Blank group. #p < .05, ##p < .01, compared with the CMNP + PMNP group. (E) Tumour cell apoptosis assay by dUTP-mediated nick-end-labelling (TUNEL).
In vivo systematic toxicity
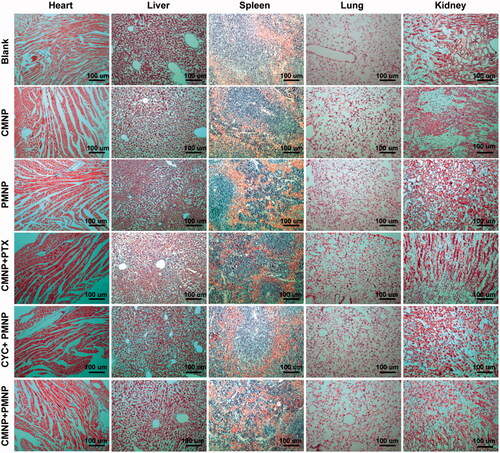
To evaluate the safety of CMNP and PMNP in vivo, body weight and living state such as the daily uptake of food and water of animal models in all treatment groups were monitored during the whole experiment. Results showed there were no significant changes among the six treatment groups in terms of body weight and the living state during a span of 28 days (Figure S1). Furthermore, H&E staining of those major organs obtained at the end of study () revealed that almost no necrosis occurred in the six treatment groups. To sum up, these results indicated combination of tumour microenvironment modulation by CMNP and chemotherapy by PMNP might be rather safe in the present study.
Figure 8. H&E staining of slices of major organs including hearts, livers, spleens, lungs and kidneys from mouse models in different treatment groups (n = 5).
Discussion
Nanomedicine for tumour treatment has aroused great and extensive interest nowadays. However, PDA still represents as a highly intractable tumour due to the dense ECM which not only compresses tumour vessels to reduce tumour perfusion but also resists the effective penetration of nanomedicine. Besides, the dense ECM also compromises the tumour delivery of ECM regulators which is used to conquer the ECM barrier of PDA. Therefore, the crucial basic issue lies in the design of a new strategy to effectively deliver ECM regulators to PDA.
A lot of ECM regulators such as hyaluronidase [Citation16,Citation17], recombinant tissue plasminogen activator (rtPA) [Citation18], cyclopamine [Citation14], and IPI926 [Citation19] have been explored to disrupt ECM by degrading ECM directly or inhibiting ECM synthesis. However, these agents could not avoid the potential of driving tumour progression or causing serious adverse effects to normal organs because of their nonspecific biodistribution or unsatisfactory pharmacokinetics profile [Citation20]. Nanoparticles, especially long-circulation PEGylated nanoparticles were then exploited to shield these agents, improve their delivery to tumours, and protect normal organs from their potential adverse effect [Citation21]. In the present study, biomimetic nanoparticles with longer circulation time than traditional PEGylated nanoparticles were developed to deliver cyclopamine for the modulation of PDA microenvironment. To the best of our knowledge, it was the first time that biomimetic nanoparticles were integrated with ECM modulation to endow cycloparmine advantages including favourable biocompatibility, much longer circulation time, and therefore remarkable tumour ECM modulation effect and less safety concern.
To design the biomimetic nanoparticles, self-assembled PLGA nanoparticles were used to encapsulate lipophilic drug cyclopamine (or paclitaxel) with a relatively high-drug loading in a simple way with any surfactant. Moreover, instead of traditional synthetic material for nanoparticle modification, RBC membrane was collected to coat nanoparticles for imitation of biological characteristics of RBC. According to previous studies, the surface charge of polymeric nanoparticles could significantly influence the membrane cloaking process [Citation22,Citation23]. Surfactant might change the unstable and negative charge of the PLGA nanoparticle cores, leading to their strong affinity to negatively charged membranes, which could collapse the fluidic lipid bilayer and break the local arrangement necessary for cell membrane coverage on nanoparticles [Citation22]. Thus, Tris-HCl buffer (pH 8.0) was used as a base to ironize PLGA-COOH molecule and make it amphiphilic and self-assembled into nanopartilces without the presence of regular surfactant [Citation10]. This kind of biomimetic nanoparticle with superior long-circulation property, less clearance by immune system, and better biocompatibility is definitely a more hopeful choice for tumour delivery of ECM regulators and anti-tumour drugs.
As expected, CMNP injection could deliver cyclopamine to tumour tissues more effectively than oral administration of free cyclopamine because of the long circulation nature of RBC membrane-coated nanoparticles, which consisted well to previous study [Citation11]. Compared with free cyclopamine, CMNP could also disrupt ECM significantly and improve tumour perfusion remarkably at the cyclopamine dose of 5 mg/g, which was only 10% of that of free cyclopamine for oral administration. Moreover, data revealed that the TM modulation effect by CMNP was better than or at least comparable to that by free cyclopamine. The prolonged circulation time of CMNP might not completely explain the powerful effect of CMNP on PDA microenvironment. As PDA microenvironment modulation by CMNP could make the complex microenvironment barrier easier to overcome for the following CMNP, which could then deliver more CMNP to the tumour site to further modulate PDA microenvironment, similar to a powerful positive-feedback mechanism. However, more experiments should be well designed to further demonstrate the potential mechanism. Based on the powerful effect to modify the tumour microenvironment of PDA by CMNP, the combination of CMNP and PMNP treatment reasonably resulted in the most significant shrinkage of tumour xenografts in pharmacodynamics experiments. Finally, the changes of living state and body weight during the whole experiment and the histological evaluation of animal organs by H&E staining demonstrated the favourable biocompatibility and good safety of CMNP in our study. Thus, biomimetic nanoparticles may be valuable to enhance the clinical efficacy of a broad range of ECM modulators and drugs.
Conclusions
In the present study, it was the first time that biomimetic nanoparticles encapsulating TM modulators, which not only possessed prolonged circulation than ordinary nanoparticles but also endowed cyclopamine powerful tumour microenvironment modulation effect, were developed. It was shown CMNP treatment successfully disrupted ECM and improved tumour perfusion of PDA even at a very low dose. Furthermore, CMNP treatment could significantly enhance the delivery of MNP to PDA tumours and thus the anti-tumour efficacy of PMNP treatment. Therefore, this study provides a general strategy to deliver TM modulator and drugs by biomimetic nanoparticles and improve enhance the clinical efficacy of these ECM modulators and drugs.
T._JIANG_ET_AL_supplementary_material.zip
Download Zip (115.3 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Feig C, Gopinathan A, Neesse A, et al. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276.
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumour microenvironment. Cancer Cell. 2012;21:309–322.
- Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–410.
- Jiang T, Zhang B, Shen S, et al. Tumour microenvironment modulation by cyclopamine improved photothermal therapy of biomimetic gold nanorods for pancreatic ductal adenocarcinomas. ACS Appl Mater Interfaces. 2017;9:31497–31508.
- Tremblay MR, Nevalainen M, Nair SJ, et al. Semisynthetic cyclopamine analogues as potent and orally bioavailable hedgehog pathway antagonists. J Med Chem. 2008;51:6646–6649.
- Park K. Facing the truth about nanotechnology in drug delivery. Acs Nano. 2013;7:7442–7447.
- Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. Acs Nano. 2009;3:16–20.
- Wu Z, de Avila BEF, Martin A, et al. RBC micromotors carrying multiple cargos towards potential theranostic applications. Nanoscale. 2015;7:13680–13686.
- Hu CMJ, Fang RH, Zhang LF. Erythrocyte-inspired delivery systems. Adv Healthcare Mat. 2012;1:537–547.
- Luk BT, Fang RH, Hu CM, et al. Safe and immunocompatible nanocarriers cloaked in RBC membranes for drug delivery to treat solid tumors. Theranostics. 2016;6:1004–1011.
- Hu CMJ, Zhang L, Aryal S, et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci USA. 2011;108:10980–10985.
- Copp JA, Fang RH, Luk BT, et al. Clearance of pathological antibodies using biomimetic nanoparticles. Proc Natl Acad Sci USA. 2014;111:13481–13486. [pii]
- Yang RN, Mondal G, Wen D, et al. Combination therapy of paclitaxel and cyclopamine polymer-drug conjugates to treat advanced prostate cancer. Nanomed-Nanotechnol. 2017;13:391–401.
- Zhang B, Jiang T, Shen S, et al. Cyclopamine disrupts tumour extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials. 2016;103:12–21.
- Bailey JM, Swanson BJ, Hamada T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429.
- Zhou H, Fan Z, Deng J, et al. Hyaluronidase embedded in nanocarrier PEG shell for enhanced tumour penetration and highly efficient antitumour efficacy. Nano Lett. 2016;16:3268–3277.
- Kirtane AR, Sadhukha T, Kim H, et al. Fibrinolytic enzyme cotherapy improves tumour perfusion and therapeutic efficacy of anticancer nanomedicine. Cancer Res. 2017;77:1465–1475.
- Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461.
- Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958–962.
- Yang S, Gao H. Nanoparticles for modulating tumour microenvironment to improve drug delivery and tumour therapy. Pharmacol Res. 2017;6618:30163–30169.
- Fischlechner M, Zaulig M, Meyer S, et al. Lipid layers on polyelectrolyte multilayer supports. Soft Matter. 2008;4:2245–2258.
- Luk BT, Hu CMJ, Fang RNH, et al. Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale. 2014;6:2730–2737.