
Abstract
Direct conversion is a powerful approach to safely generate mature neural lineages with potential for treatment of neurological disorders. Astrocytes play a crucial role in neuronal homeostasis and their dysfunctions contribute to several neurodegenerative diseases. Using a single-cell approach for precision, we describe here a robust method using optimized DNA amounts for the direct conversion of mouse fibroblasts to astrocytes. Controlled amount of the reprogramming factors Oct4, Sox2, Klf4 and cMyc was directly delivered into a single fibroblast cell. Consequently, 2500 DNA molecules, no more or less, were found to be the optimal amount that dramatically increased the expression levels of the astrocyte-specific markers GFAP and S100b and the demethylation gene TET1, the expression of which was sustained to maintain astrocyte functionality. The converted astrocytes showed glutamate uptake ability and electrophysiological activity. Furthermore, we demonstrated a potential mechanism whereby fibroblast was directly converted into astrocyte at a single-cell level; this was achieved by activating BMP2 pathway through direct binding of Sox2 protein to BMP2 gene. This study suggests that nanotechnology for directly injecting plasmid DNAs into cell nuclei may help understand such a conversion at single-cell level.
Introduction
Direct conversion technology has recently attracted attention for cell-based therapy in regenerative medicine because of its ability to convert somatic cells into tissue-specific cell types, including neurons, cardiomyocytes and beta cells [Citation1–4] as well as low risks of teratoma formation from using pluripotent stem cells due to undifferentiated cells [Citation5,Citation6]. In addition, compared to the use of embryonic stem cells and human fetal and/or adult neural stem cells, that of somatic cells is relatively free of ethical controversy and immune rejection [Citation7,Citation8] as they can be obtained from patients themselves [Citation9,Citation10]. However, a method that can achieve cell conversion in a single cell level without using a viral vector has not been developed yet, although many of reports on non-viral methods have been published in recent years [Citation11,Citation12]. Recently, it was reported that transplantation of astrocytes showed robust survival in the host brain and ameliorated the disease symptoms in animal models of Alzheimer’s disease and amyotrophic lateral sclerosis [Citation13,Citation14]. Astrocytes are a crucial neural lineage, which is responsible for the maintenance of brain homeostasis [Citation15–17].
Yamanaka’s factors have been used to generate induced pluripotent stem cells and other types of tissue-specific cells [Citation18–21]. For many years, viral vectors and liposomal transfection systems have been used to introduce Yamanaka’s factors into somatic cells [Citation21–23]. However, the conventional methods are not suitable for reprogramming of a desired single cell, because they could not control the introduction of viruses or plasmid DNAs amount into a single cell (Figure S1). Previously, we found that the expression of certain genes could be controlled by regulating the quantity of exogenous DNAs [Citation24]. Different levels of gene expression caused by different quantities of exogenous plasmid DNA reflect that a certain optimal quantity of plasmid DNA for gene expression may effectively reprogram the somatic cells delivered with plasmid DNAs and convert them into the desired cell type (Figure S2). The development of an optimal injection technology for injecting plasmid DNAs into a single cell will contribute to understanding the mechanisms of direct cell conversion, which will be useful for treating currently incurable diseases, such as diabetes and some neurological and cardiovascular diseases. However, the precise correlation between the quantity of the delivered gene and its expression levels has not been reported yet. Moreover, conventional transfection methods are not able to introduce consistent quantities of the plasmid DNAs into a single cell [Citation24,Citation25]. Therefore, it is necessary to establish a controllable method injecting precise amount of plasmid DNA(s) in a single cell to develop a cell conversion technology and an understanding of cell conversion mechanisms in coming years.
Here, we optimized a quantity, i.e. 2500 molecules of plasmid DNAs to be delivered into a single fibroblast cell by our precision injection system for directly converting it into an astrocyte (). Also, we demonstrated that the direct conversion for a functional astrocyte is regulated by the activation of BMP2 pathway via sustained expression of an endogenous Sox2 gene. These findings suggest that a precise control of the injection volume in a single cell may be feasible to convert somatic cells into a tissue-specific cell type for cell-based therapy of the incurable diseases.
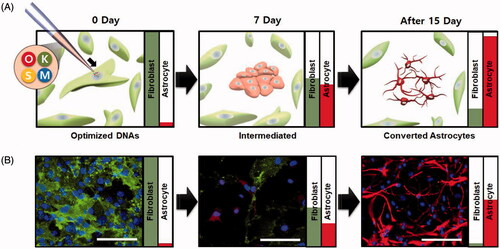
Figure 1. Overview of the direct conversion of a single fibroblast into astrocytes by quantitative injection system. (A) A scheme for astrocyte induction from a single fibroblast by quantitatively controlled injection of plasmid DNAs. (B) Representative fibroblast expressing a marker CD90 and astrocytes expressing a marker GFAP. Astrocytes converted from fibroblast cell which induced by single cell injection. Fibroblast marker CD90 and Astrocyte marker GFAP was labeled by immunofluorescence method. Scale bar = 100 μm.
Materials and methods
Plasmid preparation for microinjection
Mammalian expression reprogramming gene plasmids (pCX-OKS-2 A, pCX-cMyc) were purchased from Addgene (Cambridge, MA). The E. coli cells containing plasmid DNA were cultured in 100 ml LB medium supplemented with Ampicillin overnight at 37 °C in a shaking incubator at 300 g for 12–16 h. The E. coli cells were harvested by centrifugation at 5000 g for 10 min, and the supernatant was removed. The NucleoBond® Xtra Midi plasmid DNA purification kit (Macherey-Nagel, Düren, Germany) was used for DNA preparation according to the manufacturer’s instructions. The plasmid yield was determined using a Nanodrop2000 (Thermo Scientific, Waltham, MA).
Mouse embryonic fibroblast culture
Mouse embryonic fibroblast cells were used for the micro-injection experiment. To isolate fibroblasts without nerve tissue from embryonic mice, we remove head and spins from a 13-day-old mouse embryo. Skin tissue fragments from 13-day-old embryonic mice were collected using surgical tools. Then, to separate the tissue into single cells, it was trypsinized for 15 min and dissociated by vigorous pipetting. The trypsinized cells were filtered with 100-µm and 40-µm cell strainers (BD Biosciences, San Jose, CA) and centrifuged to obtain a cell pellet. After pellet formation, the supernatants were removed, and the cell pellet was resuspended with fibroblast culture medium containing Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT) with 10% FBS (HyClone, Logan, UT), 2-mM l-glutamine (GIBCO, Carlsbad, CA), 0.1-mM MEM NEAA (GIBCO, Carlsbad, CA) and penicillin–streptomycin (GIBCO, Carlsbad, CA). Mouse embryonic fibroblast cells were cultured in a cell culture plate at 37 °C with 5% CO2 condition for expansion and single cell injection.
Single cell injection
To prepare fibroblasts for injection in a single cell, a 60-mm culture dish was coated with CELLstart™ CTS™ (Thermo Scientific, Waltham, MA) for 1 h at 37 °C, 5% CO2 condition. After coating, approximately 2000 cells were seeded on the center of the coated dish. On the next day, 10 μl of the prepared plasmid DNAs was loaded into the Femtotips®II (Eppendorf, Hamburg, Germany) for direct delivery of the plasmids into the nucleus of the fibroblast. The Femtotips®II was precisely placed on the nucleus of the fibroblast cell using micromanipulator (MP-225, Sutter instrument Co., Novato, CA). The plasmids were injected into a nucleus with the Femtojet® microinjection instrument (Eppendorf, Hamburg, Germany). The dosage of the plasmid DNAs was determined through an empirical equation and was controlled through the injection pressure (1400 hPa) and duration (0.6–1.5 s) [Citation24,Citation26]. To minimize the effects of the Femtotip damage, the plasmids were injected into approximately 50 fibroblast cells.
Conversion of astrocyte by microinjection
At the first day after injection, the cells were cultured with fibroblast culture media. On the next day, the culture medium for the injected cells was changed into N2B27 medium containing Dulbecco’s modified Eagle’s medium-F12 (GIBCO, Carlsbad, CA) with N2 supplement, B27 supplement (GIBCO, Carlsbad, CA), 200-μM ascorbic acid (Sigma, St. Louis, MO), 40-mg/mL BSA or 0.125% FBS [Citation19], 10-ng/mL FGF2/basic FGF and 10-ng/mL EGF. These inhibitors of cell signaling pathways were also added for more efficient conversion: 2-μM SB431542 (CAYMAN, Ann Arbor, MI), 0.5-μM thiazovivin (Stemgent, Cambridge, MA) and 10-µM CHIR99021 (CAYMAN) [Citation27,Citation28]. The N2B27 medium was replaced on alternate days until astrocytes were observed. The size of the injected cells was reduced approximately seven days after injection, and neural cell-like morphology was observed 14 days after injection. For maturation of the astrocytes, the injected cells were cultured in the N2B27 medium containing 10-ng/mL FGF2/basic FGF, 10-ng/mL EGF, 200-μM ascorbic acid, 2-μM SB431542, 0.5-μM thiazovivin and 10-µM CHIR99021 for 14 days, and then, they were cultured with the N2B27 medium containing 200-μM ascorbic acid, 2-μM SB431542, 0.5-μM thiazovivin and 10-µM CHIR99021 for over 14 days for differentiation into matured astrocytes.
Immunofluorescence assay
For immunofluorescence assay, first, the culture media was removed and the cells were washed on a culture dish with PBS. Cells were fixed with 4% formaldehyde solution in PBS at RT for 1 h or at 4 °C overnight. After fixation, the cells were washed with PBS at least three times. Then, the cells were permeabilized with a detergent solution containing PBS and 0.1% triton X-100 (Sigma, St. Louis, MO) for 5 min at RT. Next, the cells were blocked with a blocking solution containing PBS and 10% normal donkey serum (Sigma, St. Louis, MO) for at least 1 h at RT. After blocking, the cells were treated with specific antibodies (primary antibody) diluted in 2% normal donkey serum with PBS. The reaction between cells and each primary antibody solution was performed overnight at 4 °C. We used the following markers as primary antibodies: the reprogramming factor antibody Oct4 (1:200; Abcam, Cambridge, MA), Sox2 (1:200; Millipore, Billerica, MA), Klf4 (1:200; Millipore, Billerica, MA) and cMyc (1:500; Sigma, St. Louis, MO), fibroblast marker CD90 (1:200; Abcam, Cambridge, MA), astrocyte marker GFAP (1:500; Millipore, Billerica, MA) and S100b (1:200; Dako, Santa Clara, CA) [Citation29]. After primary antibody attachment, cells were washed with PBS and incubated with fluorescence-conjugated secondary antibody solution for 1 h at RT. Alexa Fluor 488- and 594-conjugated secondary antibodies (1:1000; Invitrogen) were used as secondary antibodies. After washing the residual antibody solution, nuclei were counterstained with DAPI (1:10,000) for 5 min. All immunofluorescence samples were observed with the EVOS fluorescence microscope (AMG, Bothell, WA) and confocal microscope (Olympus, Tokyo, Japan).
Real time RT-PCR
Total RNA was prepared by cell lysis buffer (Signosis, Santa Clara, CA) following the manufacturer’s instructions [Citation24]. Approximately 500 ng of total RNA was transcribed to cDNA using RevertAid reverse transcriptase (Thermo Scientific, Waltham, MA) and oligo (dT) primers. The cDNAs were subjected to PCR in a 2720 thermal cycler (Applied Biosystems, Carlsbad, CA). The primer sequences used are listed in Supplementary Table 1. Pre-PCR conditions were 2 min at 50 °C, 10 min at 95 °C, 60 cycles of 15 s at 95 °C and 60 s at 60 °C. Real-time RT-PCR was performed with the StepOnePlus™ Real-Time PCR System (Applied Biosystems) with KAPA SYBR FAST ABI prism qPCR kit reagents (KAPA Biosystems, Woburn, MA).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were preformed according to the EZ-ChIP™ kit (Millipore, Billerica, MA) manufacturer’s protocol. MEF cells were plated in 100-mm plates and cultured for 1 day and then transduced with a retroviral vector expressing the Sox2 gene. Three days after transduction, approximately 3 × 106 cells were cross-linked with 1% formaldehyde for 10 min and harvested in the presence of protease inhibitor cocktail (Millipore, Billerica, MA). An EZ-ChIP™ kit (Milipore, Billerica, MA) and a provided protocol were used for cell lysis and immunoprecipitation process. Sox2 antibody (Millipore, Billerica, MA) was used to precipitate Sox2 protein–DNA complexes. After precipitation, the DNA was reversed from protein–DNA complexes by treatment with NaCl and then recovered by phenol extraction. PCR was performed to detect specifically bound DNA. Used primer sequence is listed in Supplementary Table 2.
Glutamate uptake assay
Conditioned medium was harvested from the converted astrocytes and control fibroblasts that were cultured with NC medium, and was analyzed with colorimetric glutamate assay kit (Abcam, Cambridge, MA). The assay was performed by provided procedure manual. The concentration of glutamate was detected by SpectraMax340 microplate leader (Molecular devices, Sunnyvale, CA) at OD450 nm.
Electrophysiology assay
The injected cells were seeded onto coverslips for electrophysiology and converted into astrocytes in N2B27 media containing 10-ng/mL FGF2/basic FGF, 10-ng/mL EGF, 200-μM ascorbic acid, 2-μM SB431542, 0.5-μM thiazovivin and 10-µM CHIR99021 for 14 days. The standard solution for pipette contained 150-mM KCl, 1-mM CaCl2, 1-mM MgCl2, 5-mM EGTA and 10-mM HEPES (pH 7.2 was adjusted with KOH). The bath contained in 150-mM NaCl, 3-mM KCl, 2-mM CaCl2, 1-mM MgCl2, 10-mM HEPES, 5.5-mM D-glucose and 20-mM sucrose (pH 7.4 was adjusted with NaOH). Patch pipettes were made from borosilicate glass capillaries (Warner Instruments, Inc.). The pipettes had 5–6-MΩ resistance. Whole-cell currents were recorded using a patch clamp amplifier (Axopatch 700B, Axon Instrument, Inc.). The current–voltage relations were measured by applying ramp pulses (from −150 mV to +50 mV over a 1000-ms period) from a holding potential of −60 mV. A Digidata 1550A interface was used to convert digital–analog signals between amplifier and computer. Data were sampled at 5 kHz and filtered at 1 kHz. Currents were analyzed with Clampfit software (Axon instruments, Inc.). All experiments were conducted at room temperature.
Statistical analysis
Results were presented as the mean ± SD. Statistically significant differences were calculated using independent and paired Student’s t-tests for unpaired and paired samples. p < .05 was considered as statistically significant.
Results and discussion
Quantitation of plasmid DNAs for delivery into a single fibroblast
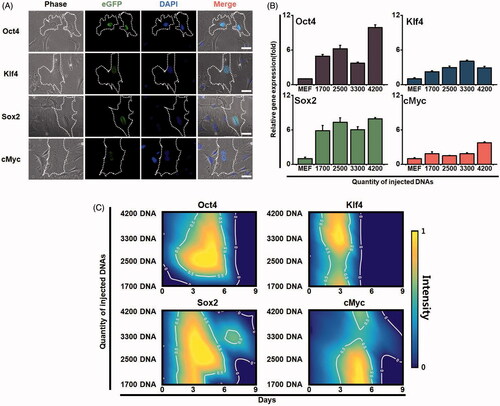
To introduce plasmid DNAs into a single mouse fibroblast cell, we used precision injection system as shown in our previous study [Citation24,Citation26]. A pCX-OKS-2A plasmid was used for Oct4, Klf4 and Sox2 genes, and a pCX-cMyc plasmid was used for the cMyc gene [Citation22]. The OKSM plasmid DNAs were directly injected into the nucleus of a single cell with 1700, 2500, 3300 and 4200 DNA molecules per injection (850–2100 molecules per plasmid). Three days after injection, using immunofluorescence microscopy, we observed the expression of each gene in the nuclei with the injected single cells (). The plasmids successfully produced Oct4, Klf4, Sox2 and cMyc proteins in the injected fibroblast. The expression level was much higher for the injected Oct4 and Sox2 genes than for the Klf4 and cMyc genes (). In addition, when we examined the intensity of gene expression (), at 3 to 5 days after injection, the Oct4 and Sox2 genes showed the highest expression levels under a condition wherein 2500 DNA molecules were injected. However, all of the injected plasmid DNAs were not observed approximately seven days after injection in all conditions (). Considering these results, we developed an optimal injection condition to enable efficient expression of the reprogramming factors in a single cell, which heightens the possibility for cell conversion.
Figure 2. Expression patterns of each reprogramming factor in a single cell. (A) Expression levels of the exogenous reprogramming factors in the injected fibroblasts. Oct4, klf4, Sox2 and cMyc genes were injected into a single cell. The expression of each gene was observed by immunofluorescence microscopy (green fluorescence). Cells were observed with optical fluorescence microscope. (B) Gene expression of reprogramming factors in the injected fibroblasts. Different amount of the exogenous reprogramming factors Oct4, klf4, Sox2 and cMyc were injected into a single fibroblast. The expression levels of each gene were analyzed by real-time RT-PCR. (C) Sustainability and intensity of the expression levels in the injected cells in time-dependent manner. Contour gradient indicates increase in gene expression from blue to yellow.
Conversion of the OSKM-injected cells into a neural lineage
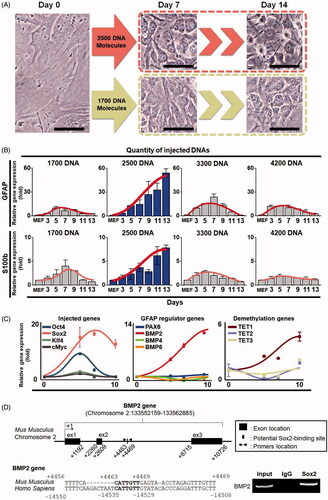
To examine the functional activity of the injected plasmid DNAs, we analyzed the expression levels of the marker genes Tuj1, GATA4 and BMP4 for three germ layers including neurons, oligodendrocytes and astrocytes, which are not expressed in fibroblast cells (Figure S3). Total RNAs were isolated from the fibroblasts that were cultured for four days after injection with 2500 DNA molecules in fibroblast culture condition. In our real-time RT-PCR analysis, the marker genes for three germ layers were expressed more in the injected cells than in the fibroblasts. Interestingly, the neural ectodermal marker Tuj1 gene was highly expressed in all analyzed cell populations; however, endodermal marker gene was expressed in only three out of the analyzed cell populations and mesodermal marker gene was relatively low, which prompted us to convert the OSKM-injected fibroblast cells into neural lineages. To do this, we established a protocol for neural cell culture using a neural converting (NC) medium supplemented with N2 and B27 supplements, 10-ng/mL fibroblast growth factor 2 (FGF2/basic FGF), 10-ng/mL epidermal growth factor (EGF) and a combination of small molecules, including 2-μM SB431542 (TGF-β inhibitor), 0.5-μM thiazovivin (ROCK inhibitor), 200-μM ascorbic acid (antioxidant) and 10-μM CHIR99021 (GSK3 inhibitor). The NC medium stimulates a mesenchymal-to-epithelial transition (MET) and contributes to more efficient and safe conversion, as shown in our previous report [Citation19,Citation27,Citation28]. The OSKM-injected cells were cultured in the medium, which was changed once every two days until the neural cell morphology appeared (Figure S4). Approximately nine days after injection, the morphology of the injected cells was distinctively different from that of a fibroblast. Furthermore, neural cell-like cells were observed in the cells injected with 2500 DNA molecules at about 14 days after injection; however, they were not observed under other conditions of plasmid DNA injection ( and Figure S5). These results suggest that the injected cells undergo direct conversion and only an optimal amount of DNA molecules could convert somatic cells to a tissue-specific type of cells. In our previous study [Citation24], however, the efficiency of gene expression was not likely dependent on the amount of the DNA molecules transfected by a single cell injection system; this is probably because of the changes in the self-defense system of the cells that have been known to affect the expression of exogenous materials, such as plasmids [Citation30,Citation31]. We could not identify the reasons for the damages within nucleus that may have occurred by DNA injection in this experiment.
Figure 3. Changes of cell morphology and gene expression of the injected cells during a direct conversion process. (A) Morphological changes of plasmid DNA-injected cells. We injected 1700 DNA and 2500 DNA molecules into different single cells, and morphology was observed at seven and 13 days after injection, respectively. Cells were observed with a phase contrast microscope. The scale bar size is 50 μm. (B) The relative RNA expression levels of astrocyte marker genes GFAP and S100b in converted cells. Gene expression was analyzed by real-time RT-PCR. Total RNAs were collected from the cells injected with 1700, 2500, 3300 and 4200 DNA molecules. Statistically significant differences were calculated using independent and paired Student’s t-tests for unpaired and paired samples. p < .05 was considered statistically significant. (C) The relative RNA expression levels of the exogenously injected genes, GFAP regulator genes, and the demethylation-related TET family genes TET1, TET2 and TET3 under the condition wherein 2500 DNA molecules were injected. Each gene expression level was analyzed by real-time RT-PCR. Total RNAs were collected at days 5, 7 and 10. (D) Sox2 directly binds to BMP2 gene from MEF cells. One Sox2 binding site was identified in mouse BMP2 gene. Bar indicates the motif that is conserved between mouse and human genes. ChIP analysis shows that Sox2 bound to the site. Input DNA was used for positive control.
Characterization and mechanism study of neural lineage converted cells
To characterize the effectiveness of conversion to neural lineage cells, we first isolated total RNAs from these cells and analyzed the mRNA expression levels of the neural stem cell marker genes Sox1, Sox2, Musashi and Nestin and the astrocyte marker genes GFAP and S100b using real-time RT-PCR. The expression levels of both astrocyte marker genes were dramatically increased in the converted neural cells in a time-dependent manner, while they were decreased at seven days after injection in conditions wherein 1700, 3300 and 4200 DNA molecules were injected (). Interestingly, however, the expression levels of the neural stem cell marker genes were not significantly increased under any condition, but the Sox2 gene was transiently expressed only for 3 to 7 days in the condition wherein 4200 plasmid DNA molecules were injected, which may have been due to exogenous plasmid DNAs (Figure S6). Although unlike the expression levels of Sox1 and Musashi, those of Tuj1, Nestin and Sox2 were high at 7–9 days, 9–10 days and 7–9 days after injection, respectively, in the cells injected with 4200 DNAs, they were not sustained till later stages of the direct conversion process, suggesting that high amounts of DNA molecules may not be necessary to convert the somatic cells into a desired type of cells, such as neural stem cells. However, we cannot exclude the possibility that somatic cells converted to neural stem cells only if the expression levels of Sox2 and other neural marker genes are sustained till later stages of the direct conversion process. These results show that the converted neural lineage cells are an astroglial cell type rather than neural stem cells or neuronal and oligoglial cell types in our well-established cell culture conditions supplemented with growth factors and small molecules, such as bFGF, EGF, ascorbic acid, SB431542, thiazovivin and CHIR99021.
Since we observed that 2500 DNA molecules can stimulate the expression of the astrocyte marker GFAP gene and protein during the conversion process, we next examined a pathway that may activate the GFAP gene in the OSKM plasmid-injected cells by analyzing the expression levels of GFAP regulator genes, such as PAX6, BMP2, BMP4 and BMP6. We found that only BMP2 gene was activated during the cell conversion process ( and Figure S7). Next, we examined whether Sox2 protein directly binds to BMP2 gene by ChIP-PCR assay. Toward this goal, we analyzed the mouse and human BMP2 gene from 0.6 kb upstream (−0.6 kb) to 11.3 kb downstream (+11.3 kb) of transcriptional start site using the Sox2 binding motif (5′-CATTGTT-3′) [Citation32] with the ECR browser program [Citation33]. Interestingly, we identified one conserved site residing at +4463 to +4469. Sox2 protein directly bound to BMP2 gene (), which indicates that the direct conversion of a single fibroblast into an astrocyte is regulated through a Sox2–BMP2 pathway [Citation34–36]. Next, we examined the epigenetic states of the converted astrocytes by analyzing the expression levels of the demethylation genes TET1, TET2 and TET3. Consequently, we found that TET1 expression continuously increased during cell conversion process ( and Figure S7). Therefore, these findings suggest that our precision injection technology without using viral vectors may be feasible for the direct conversion of a single cell to a tissue-specific cell type; this would be useful in cell-based therapy for various currently incurable diseases.
In particular, we demonstrated a mechanism in which a single fibroblast is converted to an astrocyte; this was achieved by the activation of a Sox2-triggered GFAP regulator BMP2 but not of BMP4 or BMP6 and by the increase of an active CpG demethylase TET1 but not of TET2 and TET3. Since Yamanaka’s factors OSKM have been used to generate induced pluripotent stem cells as well as other types of tissue-specific cells [Citation19,Citation37], we also used OSKM to convert a fibroblast into an astrocyte although the transcription factors (NFIA, NFIB and SOX9) to define astroglial fate have been identified in a previous study [Citation29], which suggests an alternative pathway for astroglial fate decision.
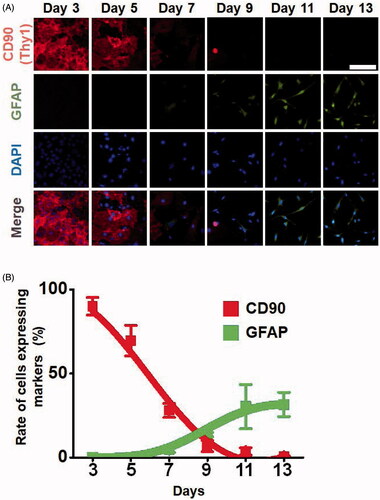
Next, we examined the dynamic changes in gene expression that occur during the direct conversion process of fibroblast to astrocyte. First, we analyzed the expression patterns of the fibroblast marker CD90 and astrocyte marker GFAP under the condition wherein 2500 DNA molecules were injected, as shown in . Interestingly, CD90 gradually disappeared from day 3 to day 5, whereas GFAP gradually increased after day 7 of the direct conversion process (). Next, we analyzed the percentage of CD90 + and GFAP + cells during direct conversion. The CD90 + cells completely disappeared at day 14 in most cells, while approximately 30% of cells were GFAP-positive ().
Figure 4. Expression levels of fibroblast and astrocyte markers in a time-dependent manner during the direct conversion process. Cells that were injected with 2500 DNA molecules were converted into astrocytes in a culture condition for neural cells. (A) Expression levels of the marker proteins, the fibroblast marker CD90 and astrocyte marker GFAP were analyzed by immunofluorescence assays. (B) Rate of cells expressing markers. Cells expressing the fibroblast marker CD90 and astrocyte marker GFAP were counted.
We also examined whether the precision injection system can convert fibroblasts into other types of neural cells, such as neurons and oligodendrocytes. According to our immunofluorescence assays, most neural cell-like cells were astrocyte marker GFAP+. However, only few cells were Tuj1 + neurons (Figure S8), and O4-positive oligodendrocytes were not found (Figure S8) in our NC medium.
Functionality of converted astrocytes
To examine the functionality of the converted astrocytes with exon filament structure [Citation38,Citation39], we cultured the injected fibroblasts in the NC medium for 14 days, and then, the medium was changed, and the culture resumed for 7–14 days using NC medium without bFGF or EGF to allow for full differentiation into the mature astrocytes. Fully differentiated astrocytes expressed the astrocyte marker proteins GFAP and S100b, which were co-localized () in our immunofluorescence assays.
Figure 5. Functionality of the astrocytes converted from a single fibroblast by quantitative injection. (A) Astrocytes expressing marker proteins GFAP and S100b by immunofluorescence assays. (B) Glutamate uptake ability assay of the converted astrocytes. Glutamate concentration was analyzed in the conditioned medium of fibroblast (black bar) and in converted astrocytes (white bar). p < .01 was considered statistically significant. (C) Electrophysiology results of mouse embryonic fibroblasts as a negative control, astrocytes as a positive control, and converted astrocytes.
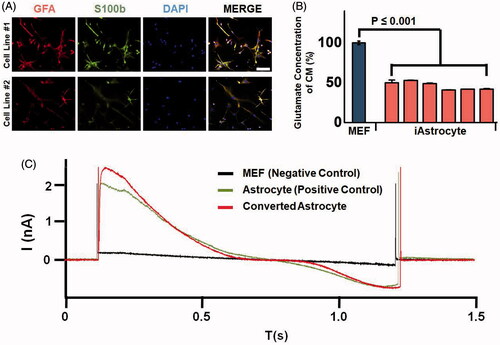
Next, to examine whether the converted astrocytes were functional, we analyzed the glutamate uptake ability by measuring the amount of glutamate metabolized by an enzymatic determination method in astrocyte-derived conditioned medium (). The amount of glutamate in this medium was much lower in converted astrocytes than in fibroblasts (control cells). These results indicated that the converted astrocytes obtained by the quantitative injection method were functional. In addition, we analyzed the expression levels of astrocyte ion channel marker genes, namely Aqp4 and Kir4.1, in the four different converted GFAP + astrocyte cell populations (Figure S9), and found that the expression of the potassium channel marker gene Kir4.1 was significantly high in all four cell populations. Moreover, to examine the electrophysiological activity of the converted astrocytes, we performed a patch test (). The patch results showed that the converted astrocytes were functional, suggesting that our quantitative and controllable injection method using a single cell can be a promising technology for the direct conversion of somatic cells into a different type of functional cell.
Cell conversion technology for cell transplantation therapy has attracted attention because astrocytes play a pivotal role in brain homeostasis owing to their participation in neuronal metabolism, synaptic plasticity and neuroprotection [Citation40–42]. Ultimately, producing astrocytes through a quantitative injection in a single cell without the use of viral vector systems could be a promising cell source for neural disorders, such as Alexander disorder, Alzheimer’s disease, Huntington’s disease and Parkinson’s disorder [Citation43–45]. However, there still remain a limitation for direct conversion of a single fibroblast to an astrocyte, including a direct conversion potential in human cells and in vivo animal models.
Conclusion
In this study, we revealed the potential of non-viral transfection system through direct conversion of fibroblast into astrocyte. Our experiments indicate that single cell transfection system could lead remarkable cellular changes as like as typical transfection methods when inducing reprogramming factors (Oct4, Klf4, Sox2 and cMyc) into single cell. Moreover, we successfully generate functional astrocyte from fibroblast with our non-viral single cell transfection system. Therefore, we demonstrated that the single cell transfection system is a potent tool in the generation matured astrocyte from fibroblast without any viral vector and tool in quantitative analyze of direct conversion mechanism study. With some improvement according to further studies, this single cell injection based transfection could replace the viral transfection. Those experimental results were giving a clear evidence that transfection with single cell injection, which can generate functional cells by direct conversion, could be a possible replacer of viral transfection, and could be a possible tool for quantitatively controlled analysis of transfection.
Hang-Soo_Park_et_al._Supplementary_files.pdf
Download PDF (800.7 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Wei R, Hong T. Lineage reprogramming: a promising road for pancreatic beta cell regeneration. Trends Endocrinol Metab. 2016;27:163–176.
- Ieda M, Fukuda K. Cardiomyocyte generation using stem cells and directly reprogrammed cells. Front Biosci. 2012;4:1413–1423.
- Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390–393.
- Yang N, Chanda S, Marro S, et al. Generation of pure GABAergic neurons by transcription factor programming. Nat Methods. 2017;14:621–628.
- Choi KA, Hong S. Induced neural stem cells as a means of treatment in Huntington’s disease. Expert Opin Biol Ther. 2017;17:1333–1343.
- Mitchell RR, Szabo E, Benoit YD, et al. Activation of neural cell fate programs toward direct conversion of adult human fibroblasts into tri-potent neural progenitors using OCT-4. Stem Cells Dev. 2014;23:1937–1946.
- Sordi V, Pellegrini S, Piemonti L. Immunological issues after stem cell-based β Cell Replacement. Curr Diab Rep. 2017;17:68.
- Rodriguez RM, Ross PJ, Cibelli JB. Therapeutic cloning and cellular reprogramming. Adv Exp Med Biol. 2012;741:276–289.
- Iacovides D, Rizki G, Lapathitis G, et al. Direct conversion of mouse embryonic fibroblasts into functional keratinocytes through transient expression of pluripotency-related genes. Stem Cell Res Ther. 2016;7:98.
- An N, Xu H, Gao WQ, et al. Direct conversion of somatic cells into induced neurons. Mol Neurobiol. 2016;55:642–651.
- Hu W, Qiu B, Guan W, et al. Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell. 2015;17:204–212.
- Adler AF, Grigsby CL, Kulangara K, et al. Nonviral direct conversion of primary mouse embryonic fibroblasts to neuronal cells. Mol Ther Nucleic Acids. 2012;1:e32.
- Pihlaja R, Koistinaho J, Malm T, et al. Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia. 2008;56:154–163.
- Lepore AC, Rauck B, Dejea C, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11:1294–1301.
- Toy D, Namgung U. Role of glial cells in axonal regeneration. Exp Neurobiol. 2013;22:68–76.
- Tsai HH, Li H, Fuentealba LC, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358–362.
- Sandhu JK, Gardaneh M, Iwasiow R, et al. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol Dis. 2009;33:405–414.
- Matsui T, Takano M, Yoshida K, et al. Neural stem cells directly differentiated from partially reprogrammed fibroblasts rapidly acquire gliogenic competency. Stem Cells. 2012;30:1109–1119.
- Han DW, Tapia N, Hermann A, et al. Direct reprogramming of fibroblasts into neural stem cells by defined factors. Cell Stem Cell. 2012;10:465–472.
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676.
- Okita K, Nakagawa M, Hyenjong H, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953.
- Sommer CA, Stadtfeld M, Murphy GJ, et al. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549.
- Kwon H, Park HS, Yu J, et al. Spatio-temporally controlled transfection by quantitative injection into a single cell. Biomaterials. 2015;67:225–231.
- Yan R, Park JH, Choi Y, et al. Nanowire-based single-cell endoscopy. Nat Nanotechnol. 2011;7:191–196.
- Jewon Y, Hyosung K, Park H-s, et al. Femtoliter scale quantitative injection control by experimental and theoretical modeling. Biomed Eng Lett. 2016;6:250–255.
- Zheng J, Choi KA, Kang PJ, et al. A combination of small molecules directly reprograms mouse fibroblasts into neural stem cells. Biochem Biophys Res Commun. 2016;476:42–48.
- Park HS, Hwang I, Choi KA, et al. Generation of induced pluripotent stem cells without genetic defects by small molecules. Biomaterials. 2015;39:47–58.
- Caiazzo M, Giannelli S, Valente P, et al. Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Reports. 2015;4:25–36.
- Baumann JG. Intracellular restriction factors in mammalian cells– an ancient defense system finds a modern foe. Curr HIV Res. 2006;4:141–168.
- Lin SL, Sukasweang S, Chuong CM, et al. D-RNAi (messenger RNA-antisense DNA interference) as a novel defense system against cancer and viral infections. Curr Cancer Drug Targets. 2001;1:241–247.
- Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117.
- Ovcharenko I, Nobrega MA, Loots GG, et al. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32(Web Server issue):W280–W286.
- Favaro R, Valotta M, Ferri AL, et al. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat Neurosci. 2009;12:1248–1256.
- Zhang Z, Song Y, Zhao X, et al. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146.
- Dore JJ, DeWitt JC, Setty N, et al. Multiple signaling pathways converge to regulate bone-morphogenetic-protein-dependent glial gene expression. Dev Neurosci. 2009;31:473–486.
- Thier M, Worsdorfer P, Lakes YB, et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell. 2012;10:473–479.
- Bernardinelli Y, Muller D, Nikonenko I. Astrocyte-synapse structural plasticity. Neural Plast. 2014;2014:232105.
- Kacem K, Lacombe P, Seylaz J, et al. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia. 1998;23:1–10.
- Hertz L, Chen Y, Song D. Astrocyte cultures mimicking brain astrocytes in gene expression, signaling, metabolism and K + uptake and showing astrocytic gene expression overlooked by immunohistochemistry and in situ hybridization. Neurochem Res. 2017;42:254–271.
- Raivich G, Bohatschek M, Kloss CU, et al. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105.
- Sturrock RR. The beta astrocyte: its possible role as a central nervous system phagocyte. Anat Anz. 1988;166:331–340.
- Kumar A, Narayanan K, Chaudhary RK, et al. Current perspective of stem cell therapy in neurodegenerative and metabolic diseases. Mol Neurobiol. 2017;54:7276–7296.
- Colangelo AM, Alberghina L, Papa M. Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci Lett. 2014;565:59–64.
- Lindvall O, Barker RA, Brustle O, et al. Clinical translation of stem cells in neurodegenerative disorders. Cell Stem Cell. 2012;10:151–155.