
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Poor aqueous solubility of chemotherapeutics such as SN38 (7-ethyl-10-hydroxycamptothecin) and the associated systemic adverse effects are serious limitations of their clinical use. To improve the drug delivery efficiency of such compounds, they were covalently conjugated to hydrophilic macromolecular carriers that specifically deliver the drug moiety to the tumour cells. In the current study, we developed a PEGylated acetylated carboxymethylcellulose conjugate of SN38 which was covalently attached to an aptamer against a cancer stem cell marker, CD133. Then, the designed nanoplatform was used to specifically deliver SN38 to colorectal cancer cells. The results demonstrated that the synthesized conjugate was self-assembled to nanoparticles with 169 nm in size and poly dispersity index of 0.11. Besides, the targeted self-assembled nanoparticles could significantly enhance the cellular uptake by CD133-expressing HT29 cell line confirmed by fluorescent microscopy and flow cytometry. Moreover, our results revealed that the targeted self-assembled nanoconjugate exhibited significantly lower IC50 in HT29 cells overexpressing CD133 compared to non-targeted self-assembled nanoconjugate. The promising data suggest that the prepared targeted self-assembled drug conjugate nanoparticles possess the potential to offer the desirable physicochemical properties thereby enhancing the solubility and the therapeutic index of poorly soluble cytotoxic agents.
Introduction
Colorectal cancer is the third most deadly and commonly diagnosed malignancy in both men and women. It is estimated that approximately 135,430 new cases were diagnosed with colorectal cancer in 2017 in USA and it caused death of about 50,260 individuals in 2017 [Citation1]. To fight against colorectal cancer, there are local and systemic treatments which their usage is dependent on the stage and the type of the cancer. One of the chemotherapeutics used as the first-line therapy of metastatic colorectal cancer is irinotecan (CPT-11, a derivative of camptothecin) which its antineoplastic effect is through inhibiting DNA-topoisomerase I complex [Citation2]. Carboxylesterases available in liver and tumour convert irinotecan to its active metabolite named 7-ethyl-10-hydroxycamptothecin (SN38) which is approximately 1000 folds more cytotoxic than irinotecan [Citation3]. Thus, direct administration of SN38 could enhance the therapeutic index of the prodrug [Citation4]. However, therapeutic use of SN38 is limited by two factors: (1) its hydrophobicity which affects the aqueous solubility and (2) its low stability at physiologic pH (i.e. pH: 7.4) in which the lactone ring of SN38 is hydrolysed and inactivated [Citation5]. Considering the aforementioned limitations, administration of high doses of SN38 and irinotecan is associated with severe diarrhoea and myelosuppression [Citation6]. To overcome the previously mentioned problems, several approaches were developed such as encapsulation of the lipophilic drug moiety into nanoparticles such as micelles [Citation7], liposomes [Citation8], solid lipid nanoparticles (SLN) [Citation9], and dendrimers [Citation10].
Another approach is to enhance the solubility and stability of the hydrophobic drug moieties through conjugation of the drug to hydrophilic polymers. In fact, there are several polymer-drug conjugates entering clinical trials such as N-(2-hydroxypropyl)methacrylamide (HPMA)-conjugated doxorubicin PK-1 (FCE-28068) [Citation11], HPMA-conjugated paclitaxel (PNU-166945), polyglutamic acid (PGA)-conjugated paclitaxel (paclitaxel polyglumex, CT-2103) [Citation12], PGA-conjugated camptothecin (CT-2106), and polyethylene glycol (PEG)-conjugated camptothecin [Citation13]. It was shown that the development of polymer-drug conjugates led to enhanced aqueous solubility and stability of the drug, improved pharmacokinetics and biodistribution, probably due to longer blood circulation time, and enhanced permeation and retention (EPR) effect [Citation14]. In a recent study, a novel polymeric backbone made of PEG and acetylated carboxymethylcellulose (CMC) was conjugated to docetaxel (DTX) via ester linkage. The results of the study demonstrated its superior safety and effectiveness compared to currently approved Taxane chemotherapeutics such as DTX and Abraxane which is albumin bound paclitaxel [Citation15].
Although nanoparticle-based drug delivery systems improved the drug delivery efficiency and reduced the adverse effects of the free drug, there are still problems with nonspecific transportation of the chemotherapeutics and the associated severe systemic toxicity [Citation16].
Targeted delivery of therapeutics specifically to the tumour cell population would be a great approach to combat such problems. In fact, recent studies revealed that a small subpopulation of tumour cells named as cancer stem cells (CSCs) is responsible for tumour initiation and recurrence [Citation17,Citation18]. Transportation of the cytotoxic agents directly to such cells could help to eradicate cancers especially recurrent metastatic ones. CSCs possess stem-like properties which are multi-drug resistant as they overexpress drug efflux proteins [Citation19] and proteins related to DNA repair [Citation18]. Growing evidence demonstrated that CD133 (prominin-1) is an important cell surface receptor of CSCs which is claimed to be related with tumorigenesis [Citation20]. It was previously proved that CD133 activity is linked to Notch signalling pathway which is in relation with cell fate and differentiation of intestinal epithelium [Citation21]. Recently, an RNA aptamer against CD133 was developed which indicated promising targeted delivery efficiency [Citation22].
In the current study, we conjugated SN38 to PEG-acetylated CMC polymer backbone to improve the cellular uptake and therapeutic efficiency of SN38 as an anticancer agent. After evaluating the physicochemical properties of the designed nanoparticles, we covalently conjugated the self-assembled nanoparticle to RNA aptamer against CD133 which is an important cell surface receptor overexpressed in CSCs. In fact, the main goal of our study was to enhance the delivery efficiency of SN38 to CD133 overexpressed HT-29 cells. Thereafter, the therapeutic effectiveness of the final drug delivery platform was investigated in vitro.
Materials and methods
Materials
7-Ethyl-10-hydroxyl camptothecin (SN38) was obtained from LC Laboratories (Woburn, MA). CMC sodium salt, Mw = 90,000 Da (Sigma-Aldrich, Oakville, ON), poly-(ethylene glycol) methyl ether (mPEG–OH, Mw = 2000 Da), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide HCl (EDC HCl), Rhodamine 6G, and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich (Oakville, ON).
Roswell Park Memorial Institute (RPMI) 1640 medium, fetal bovine serum (FBS), penicillin-streptomycin, and trypsin were purchased from GIBCO (Darmstadt, Germany). All solvent and chemical reagents were purchased from Merck (Darmstadt, Germany). A single-strand RNA aptamer for CD133 reported previously was employed as the targeting ligand [Citation23]. The 15-mer CD133 RNA aptamer (sequence: 5’-amine-C6 linker-(2’-F-C) (2’-F-C) (2’-F-C) (2’-F-U) (2’-F-C) (2’-F-C) (2’-F-U)A (2’-F-C) A (2’-F-U) AGG G 3’; where F =2’-fluoro, molecular weight: 4907.3 Da) was custom synthesized by MicroSynth (Balgach, Switzerland) [Citation20].
Cell lines
Cell lines human colon adenocarcinoma cell line HT29 and Chinese hamster ovary (CHO) cells were procured from the National Cell Bank of Iran, Pasteur Institute of Iran. The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) high glucose medium for HT29 and caco-2 and RPMI 1640 for CHO cells which was supplemented with 10% (v/v) heat-inactivated FBS and penicillin/streptomycin (100 U/mL) at 37 °C in a humidified atmosphere (95%) containing 5% of CO2.
Polymer synthesis
Calculation of stoichiometry for the CMC polymer
The CMC-Na polymer was modified with carboxymethyl groups, and was supplied by the manufacturer with an analyzed DS value of 0.7: each galactose monomer unit contained 0.7 mol carboxylic acid and 2.3 mol hydroxyl. The molecular weight of each monomer was approximated as 162 g/mol (galactose) + (59.01 g/mol (acid group) × 0.7) = 203.31 g/mol.
CMC acetylation
CMC was acetylated with a small modification of a previously reported protocol [Citation24]. Briefly, sodium carboxymethylcellulose (CMCNa, 90 kDa, 3 g) was carboxylated by addition of 200 mL sulfuric acid (20% v/v) and stirred vigorously for 2 h at room temperature (RT). To neutralize the acidic pH of the resultant, CMC-COOH, it was repeatedly washed with dH2O using centrifugation. To completely remove the remained water, the precipitated CMC-COOH was washed three times with glacial acetic acid and finally suspended in a round-bottom balloon in ice bath containing of glacial acetic acid (50 mL). Thereafter, acetic anhydride (30 mL) and sulfuric acid (100% v/v, 1.2 mL) were added, respectively. Then, the mixture was stirred at 50 °C overnight until a transparent solution was obtained. Finally, the solvent was evaporated using rotary evaporator, and the precipitate was repeatedly washed with dH2O until neutral pH was reached. The resultant powder of acetylated CMC (CMC-Ac) was lyophilized and stored at –20 °C for further use.
The 1H-NMR spectrum of the acetylated CMC in deuterated chloroform was recorded at RT using a Bruker Avance 400 MHz NMR spectrometer (Rheinstetten, Germany).
Synthesis of conjugated di-tert-butyl dicarbonate with SN38 (BOC-SN38)
To produce SN38-di-tert-butyl dicarbonate conjugate, SN38 (450 mg) was dissolved in dichloromethane (46 mL). Thereafter, di-tert-butyl dicarbonate (320 mg) and pyridine (2.8 mL) were added to the mixture and stirred at RT for 24 h, because it was protected from light. Then, the solution was filtered and washed with HCl solution (0.5 M). The organic phase was separated and dried using magnesium sulphate and then vacuum.
Synthesis of polymer-drug conjugate
CMC-Ac (80 mg) was dissolved in a mixture of MeCN (5 mL) and DMSO (3 mL). Then, EDC HCl (135 mg) and DMAP (174 mg) were added to the polymer solution. Thereafter, mPEG − OH (23 mg) and SN38-Boc (6 mg) were dissolved in reaction mixture and stirred overnight, because it was protected from light. The mixture was concentrated using rotary evaporator (55 °C, 5 mbar). Then, the product was dissolved in MeCN (5 mL) and precipitated using 40 mL diethyl ether. Afterwards, the solvent was removed by vacuum and the remaining solid material was washed twice with dH2O. The final product was lyophilized. To investigate the presence of SN38 and PEG in the final product and to analyse the amount of acetylation and pegylation of the synthesized product, we used 1HNMR. To this aim, the sample was dissolved in DMSO-d6 and the spectra were evaluated at 25 °C. The composition was estimated using integration of selected signals.
Preparation and characterization of nanoparticles
The polymer-drug conjugate (CMC-Ac-SN38-PEG) (2 mg) was dissolved in chloroform [0.6 mL/acetonitrile (0.4 mL) and emulsified with 3 mL PVA (5% v/v)]. Then, the mixture was added dropwise to a solution of PVA (5 mL, 1% v/v) with or without Rhodamine 6G (200 µg/mL), because ultrasonicated for 30 min. To remove the organic solvent, the mixture was stirred overnight at RT. The final solution was centrifuged at 14,000 rpm for 20 min and washed three times with dH2O, then dispersed in NaCl solution (150 mM, 1 mL).
Critical micelle concentration (CMC) of the self-assembled nanoparticles was investigated according to the previously reported protocol by Zhang et al. [Citation25]. Briefly, PEG-AcCMC-SN38 (5 mg) was dissolved in a solution of 1,6-diphenyl-1,3,5-hextriene (DPH, 1.2 mg/mL) in acetonitrile as a solvent. Thereafter, a serial dilution of the prepared solution was made in acetonitrile and 100 µL of each dilution was added dropwise to 900 µL of NaCl (150 mM). After incubation for 6 h in a dark place, the absorption was evaluated by measuring UV absorbance at 313 nm.
Particle size analysis was done through dynamic light scattering with a particle size analyser at scattering angle 90°, 4-mW He-Ne laser, 25 °C (Zetasizer Nano-ZS, Malvern Instruments Ltd., Malvern, UK). The amount of SN38 was determined by measuring UV absorbance at 366 nm (Varian CARY 100 UV/Vis Spectrophotometer, CA) and using a SN38 calibration curve.
The amount of SN38 grafting efficiency (GE%) and drug content (DC%) of PEG-AcCMC-SN38 was calculated as follows:
Aptamer conjugation
PEG-AcCMC-SN38 or Rhodamine 6 G-loaded PEG-AcCMC-SN38 self-assembled nanoparticles were suspended in DNase/RNase free water (2 mg/mL) and incubated with EDC (25 mg) and NHS (10 mg) while gently stirred for 1 h at RT. To prepare aptamer-tagged nanoparticles, the final N-hydroxysuccinimide-activated particles were further incubated with 20 µL of the stock solution of aptamer (1 µM) and stirred for 18 h at 4 °C. Finally, to purify the aptamer-tagged self-assembled NPs, the reaction was centrifuged at 13,000 rpm for 15 min and washed three times with dH2O.
Physiochemical properties of self-assembled nanoparticles
Field emission scanning electron microscopy (FE-SEM, Mira III FEG, TESCAN-UK, Ltd.) was performed. Sample preparation was done as follows: the sample was dissolved in dH2O (1 mg/mL), dehydrated on a metal stub, coated with 40 nm gold, and used for imaging.
In vitro release of SN38
Four milligram of the synthesized self-assembled nanoparticles (PEG-AcCMC-SN38) were suspended in PBS (pH: 7.4, 10 mL) containing 30% fetal bovine serum (FBS) or citrate buffer (pH: 5.5, 10 mL). Then, 500 µL of samples was placed into several microtubes in a shaker incubator (37 °C, 70 rpm) and at specified time points, the sample in a specific microtube was withdrawn and mixed with 500 µL of ethyl acetate and centrifuged at 13,000 rpm for 15 min to separate the layers containing free SN38. Then, the amount of SN38 was determined spectrophotometrically at 366 nm.
Cellular uptake using flow cytometry
HT29 (ATCC, Manassas, VA) and CHO cells (ATCC® CCL-61™) were seeded in six-well plates at density of 1 × 105 cells/well. After 24 h, the cells were incubated for 2 h with either targeted or non-targeted Rhodamine 6 G-loaded PEG-CMC-Ac-SN38 NPs. Then, the culture medium was removed; the cells were detached and centrifuged at 1400 rpm for 10 min. The cell pellet was washed three times with cold PBS and suspended in an FCM buffer (0.1% of sodium azide and 2% of FBS in PBS). The intensity of the Rhodamine 6G fluorescence of cells was determined on a BD FACSCalibur equipped with 488 lasers in the FL2 channel. The data were analyzed using WinDMI 2.9 analysis software.
Cellular uptake using fluorescence microscopy
To evaluate the specific targeting efficiency of the aptamer-conjugated nanoparticles, HT-29 and CHO cells were seeded at a density of 1 × 105 per well in a six-well plate for 24 h. Then, targeted or non-targeted Rhodamine 6G-loaded PEG-CMC-Ac-SN38 NPs were incubated with the cells for 2 h. Afterwards, the cells were washed five times with cold PBS and analyzed using JULI inverted-fluorescence microscope (Oxford, UK).
NP cytotoxicity studies
MTT assay
Both CD133 positive (HT29 cells) and negative (CHO cells) cell lines were seeded into 96-well plates at 5 × 103 cells/well and incubated for 24 h. Then, different concentrations of SN38 were prepared in DMEM containing 10% of FBS (0.25, 0.5, 1, 2, 5, 10, 20, and 30 µg/mL). Afterwards, targeted and non-targeted CMC-Ac-SN38-PEG NPs containing equivalent amounts of SN38 were also dispersed in DMEM containing 10% of FBS and added to each well. After incubation for 6 h, the media was changed with fresh, complete medium, and was further incubated in a humidified incubator for 24 h at 37 °C. Thereafter, MTT (20 µL, 5 mg/mL in PBS) solution was added to each well and further incubated for another 4 h. The MTT solution was then aspirated from the wells and the formazan crystals were solubilized with DMSO (100 µL). The absorbance was measured at 570 nm with a reference wavelength of 630 nm using the aforementioned Infinite® 200 PRO multimode microplate reader from Tecan Group Ltd. (Männedorf, Switzerland).
Calcein assay
Moreover, we studied the cytotoxicity of the designed nanoplatforms using fluorescent microscopy according to a previously reported procedure [Citation26]. Briefly, cells were seeded at a density of 1 × 105 per well in a six-well plate. After 24 h, they were treated with both targeted and non-targeted CMC-Ac-SN38-PEG NPs and free SN38 at different concentrations of SN38, in DMEM containing 10% of FBS (0.25, 0.5, 1, 2, 5, 10, 20, and 30 µg/mL). After 6 h, the media was changed and the cells were incubated for another 24 h at 37 °C. Thereafter, the cells were incubated with calcein-AM (2 µL, 25 µM) for 30 min. Then, they were washed several times with PBS (pH: 7.4, 100 mM) and analyzed using JULI inverted-fluorescence microscope (Oxford, UK).
Statistical analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) test, and p value ≤.05 was considered significant.
Results and discussion
Synthesis and characterization of polymer-drug conjugate
To fabricate PEG-AcCMC-SN38 conjugate as shown in , acetylated CMC was produced. CMC acetylation helps to decrease the gelling property, enhance solvent solubility, and produce high yield of conjugation to SN38 [Citation27]. To this aim, CMC was oxidized to CMC-COOH and incubated with acetic anhydride. illustrates 1HNMR spectra of CMCNa and acetylated CMC (AcCMC). The resonance peak at 3–5 ppm was assigned to protons of CMC structure (). The resonance peak at 2 ppm was assigned to the protons of the acetyl group of AcCMC structure (). By calculating the relative molecular weights and considering the integration of the peaks, the acetylation efficiency was estimated to be 91.3%.
Figure 1. Chemical structure of PEG-AcCMC-SN38.
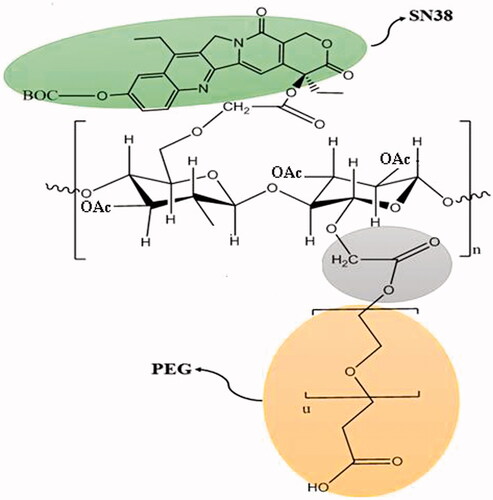
Figure 2. (A) 1HNMR spectra of Na-CMC in deionized water, (B) 1HNMR spectrum of Ac-CMC in DMSO-d6, and (C) 1HNMR spectrum of PEG-AcCMC-SN38 in chloroform.
SN38 has two hydroxyl groups: one of them is phenolic and more reactive for functionalization. Moreover, due to the stabilized lactone ring of SN38, 20-OH group was more reactive. Therefore, SN38 phenolic group was protected via reaction with di-tert-butyl dicarbonate and a single peak approximately 1.6 ppm in 1HNMR spectrum confirmed it ().
Thereafter, PEG/BOC-SN38 was conjugated to AcCMC backbone via esoteric bonds using EDC/NHS chemistry to obtain PEG-AcCMC-SN38. 1HNMR spectrum of PEG-AcCMC-SN38 is shown in . The high-intensity resonance peak at 3.6 ppm was related to methylene protons of PEG. Using the relative molecular weights and peaks integrals, the amount of CMC PEGylation was estimated to be approximately 9.8%. Also, the presence of CMC in the construct was confirmed by the resonance peak at 3–5 ppm. The resonance related to methylene groups of SN38 was observed at 1.2 ppm. On the other hand, the resonance peak at 8.3 ppm which related to aromatic protons of SN38 confirmed the drug conjugation to the AcCMC backbone.
Drug content and polymer grafting
Grafting efficiency of BOC-SN38 to PEG-AcCMC was 24.43%, and the amount of conjugated SN38 was found to be 1.33% which was determined by UV spectrophotometry at 366 nm. The drug content of the prepared drug-conjugate could be considered acceptable in comparison with previously reported SN38 prodrugs comprising SN38-glucuronide, IMMU-130, and EZN-2208 which have 3.7% of drug loading. It should be noted that in the aforementioned prodrugs, polymer-drug conjugation [Citation28].
Critical micelle concentration
CMC of PEG-AcCMC-SN38 was investigated using the fluorescent dye 1,6-diphenyl-1,3,5-hextriene (DPH). The absorption intensity of DPH enhanced significantly above the CMC of the synthesized PEG-AcCMC-SN38 as it was incorporated into hydrophobic core of the micelles. Thus, the abrupt change in absorption intensity was regarded as the CMC value.
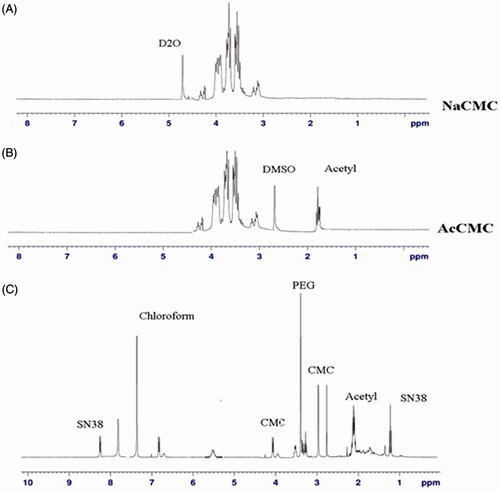
As shown in , CMC of our self-assembled structures was determined to be 0.006 mg/mL. In comparison to the CMC of the previously reported prodrugs such as hyaluronic − paclitaxel conjugates (7.8 μg/mL) and poly(glutamyl glutamine PTX) (25 μg/mL), the synthesized prodrug demonstrated low CMC in the same range of µg/mL.
Figure 3. (A) Critical micelle concentration of PEG-AcCMC-SN38. (B) Field emission-scanning electron microscopy (FE-SEM) image of PEG-AcCMC-SN38 self-assembled nanoparticles. (C) Release profile of SN38 from PEG-AcCMC-SN38 self-assembled nanoparticles in PBS (pH: 7.4) containing 30% of FBS and citrate buffer (pH: 5.5) for 96 h.
The low CMC values of the synthesized prodrug suggested that the self-assembled nanoparticles prepared from these prodrugs are thermodynamically stable and can better maintain micellar integrity upon dilution [Citation29,Citation30].
Preparation and characterization of nanoparticles
To prepare self-assembled nanoparticles, PEG-AcCMC-SN38 solution in MeCN and DMSO emulsified with PVA (3 mL, 5% v/v). Then, the mixture was added drop-wise to a 5 mL solution of 1% of PVA while ultrasonicated for 30 min. Then, the self-assembled nanoparticles were purified through centrifugation.
After self-assembly of the particles in aqueous media, particles size and zeta potential were determined. We assume that the hydrophobic and hydrophilic moieties rearrange in aqueous media, so that hydrophilic groups would stay in the surface while hydrophobic moieties would aggregate in the inner core to produce a water-soluble formulation with the least free energy level. The morphology of self-assembled nanoparticles is shown in . Scanning electron microscopy (SEM) image revealed that the designed nanoparticles were spherical and homogenously dispersed with size of 120–200 nm confirming the results obtained from dynamic light scattering.
indicates size distribution, particle size, and zeta potential of nanoparticles (PEG-AcCMC-SN38) and aptamer-conjugated nanoparticles. Mean particle size of nanoparticles was 169.9 ± 3.5 nm with polydispersity index (PDI) of 0.11. Zeta potential of the nanoparticles was determined in hydrophilic media. The results showed that the surface charge of nanoparticles was –21.6 mv.
Table 1. Characteristics of targeted and nontargeted self-assembled structures in terms of size, polydispersity index (PDI), and zeta potential.
Aptamer-conjugated nanoparticles
Afterwards, the aptamer against CD133 was conjugated covalently to the self-assembled PEG-AcCMC-SN38 nanoparticles through EDC/NHS chemistry. The Apt-PEG-AcCMC-SN38 NPs was 171.5 ± 2.6 in size with PDI of 0.13 (). The obtained results demonstrated that the aptamer conjugation do not have any impact on the size and polydispersity index of the NPs.
Compared to self-assembled PEG-AcCMC-SN38 NPs, the zeta potential of the self-assembled Apt-PEG-AcCMC-SN38 NPs was –12.1 mv which significantly increased after aptamer conjugation.
In vitro release of SN38
SN38 release profile of PEG-AcCMC-SN38 was investigated at physiological and acidic pH conditions. The results demonstrated that less than 10% of the conjugated drug was released after 96 h in both acidic and neutral environments () indicating the stability of the designed formulation. The obtained results verified that the ester linkage between AcCMC and SN38 would be stable during the transport in the blood circulation thereby increasing its stability.
It should be noted that the aforementioned ester linkage between drug and AcCMC backbone could easily be cleaved upon its exposure to intracellular esterase which may further enhance the therapeutic index of the conjugated drug while reducing its systematic toxicity [Citation31].
Cellular uptake using flow cytometry
One of the important features of the targeted delivery systems is to bind specifically to the target cells. To evaluate the ability of our designed system to bind to CD133 positive cells (HT-29 cell line) [Citation32,Citation33], we incubated the cells with either Rhodamine-loaded PEG-AcCMC-SN38 or Apt-PEG-AcCMC-SN38 for 2 h.
Moreover, as for negative control, excess amount of free CD133 aptamer (2 μg/well) was added to the wells 30 min before the cellular uptake experiment.
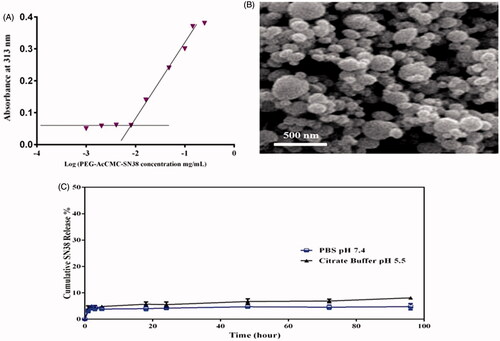
The obtained results demonstrated that the fluorescent intensity of HT29 incubated with Rhodamine-loaded PEG-AcCMC-SN38 was similar to that of treated cells with free aptamer prior to the addition of Rhodamine-loaded Apt-PEG-AcCMC-SN38 in amplitude ().
Figure 4. Flow cytometry histogram showing the cellular uptake of Rhodamine 6G encapsulated PEG-AcCMC-SN38, Apt-PEG-AcCMC-SN38 and Apt-PEG-AcCMC-SN38 + free aptamer and untreated cells as control in CD133 positive cells, HT-29 cells (A) CD133 negative cell, (B) CHO cells.
Addition of an excess amount of CD133 aptamer prior to cellular uptake experiment, as a free competing ligand for Rhodamine-loaded Apt-PEG-AcCMC-SN38 significantly decreased the internalization efficiency of aptamer-targeted self-assembled NPs. However, for HT29 cells treated with Rhodamine-loaded Apt-PEG-AcCMC-SN38, the fluorescent intensity was increased by 71%, whereas the fluorescence intensity of the cells treated with Rhodamine-loaded PEG-AcCMC-SN38 was increased by 48%. The difference between fluorescent intensity of the experiment groups was probably due to the conjugated CD133 aptamers on the surface of NPs, which bound to the CD133 receptor on the surface of HT29 cells, resulting in enhanced cell attachment and internalization.
Besides, we studied the targeting efficiency of Rhodamine-loaded PEG-AcCMC-SN38 and Rhodamine-loaded Apt-PEG-AcCMC-SN38 towards the CD133 negative cells (CHO cell line) (). The obtained results illustrated that there was no difference between the two treated groups in cellular uptake efficiency suggesting that the receptor-mediated endocytosis provided higher uptake of the aptamer-conjugated system in CD133 positive cell line (HT29).
Cellular uptake using fluorescent microscopy
To further study, the specific targeting efficiency of aptamer-conjugated nanoparticles to both CD133 positive and negative cell lines, fluorescence microscopy was performed to determine if the targeted nanoparticles remain bound to the surface of the cells or they were internalized into cytoplasm. clearly indicates that the Rhodamine-loaded Apt-PEG-AcCMC-SN38 was mainly accumulated in the HT29 cells. Accordingly, Apt-PEG-AcCMC-SN38 could be internalized into the target cells and carried the anticancer drugs into the cell. Compared to Rhodamine-loaded Apt-PEG-AcCMC-SN38, the amount of Rhodamine-loaded PEG-AcCMC-SN38 that entered HT29 cells were much less suggesting again that the CD133 aptamer enhanced the uptake of nanoparticles into the CD133 + cells (HT29).
Figure 5. Fluorescent microscopy images showing the cellular uptake of Rhodamine 6G encapsulated Apt-PEG-AcCMC-SN38, PEG-AcCMC-SN38 and Apt-PEG-AcCMC-SN38 + free aptamer as a competitive ligand in (A) CD133 positive cells, HT-29 cells (B) CD133 negative cells, CHO cells.
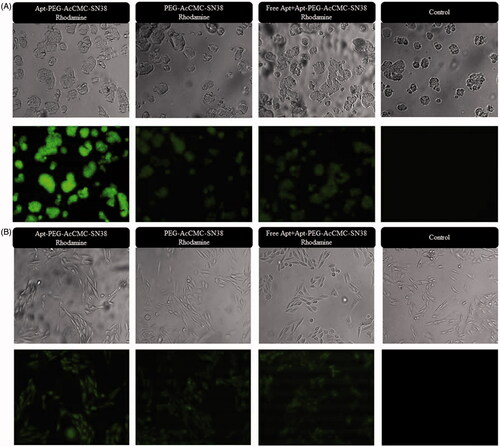
On the other hand, the fluorescence intensity of free aptamer-treated HT29 cells prior to the Rhodamine-loaded Apt-PEG-AcCMC-SN38 incubation was identical to that of Rhodamine-loaded PEG-AcCMC-SN38 incubated with HT29 cells.
It should be noted that there was no difference between all groups (Rhodamine-loaded Apt-PEG-AcCMC-SN38 vs. Rhodamine-loaded PEG-AcCMC-SN38 treatments) in CD133-CHO cell line which further proved the selectivity and specificity of the CD133 RNA aptamer ().
Cytotoxicity studies
MTT assay
The cellular uptake experiment showed that the CD133 aptamer-conjugation enhanced the cellular uptake of self-assembled NPs by CD133-overexpressing HT29 tumour cells. However, it is not known that how this would influence the transportation of conjugated SN38 to the cells. To investigate the issue, in vitro cytotoxicity of free SN38, PEG-AcCMC-SN38 or Apt-PEG-AcCMC-SN38 was evaluated in HT-29 and CHO cells.
The cytotoxicity results of either Apt-PEG-AcCMC or PEG-AcCMC did not exhibit pronounced cytotoxicity (data not shown), suggesting that the synthesized polymeric backbone was relatively nontoxic to the cells and that the detected cytotoxicity was caused by the conjugated SN38.
As shown in , Apt-PEG-AcCMC-SN38 was significantly more cytotoxic to HT29 cells compared to the PEG-AcCMC-SN38 indicating that the targeted formulation could specifically bind to the CD133 receptors present on the surface of HT29 cells and successfully be internalized via receptor-mediated endocytosis (p ≤ .05).
Figure 6. Cytotoxicity of Apt-PEG-AcCMC-SN38, PEG-AcCMC-SN38 and the free drug in (A) HT-29 cells as CD133 positive cell and (B) CHO cells as CD133 negative cell.
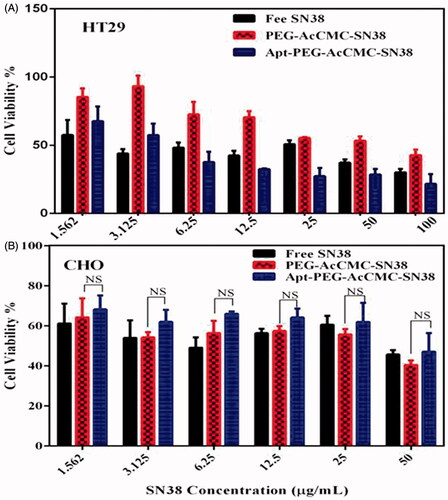
In contrast to HT29 cell line, there was no significant difference between the Apt-PEG-AcCMC-SN38 and PEG-AcCMC-SN38 in CHO cell line as CD133 negative cells (). Obtained results demonstrated that the RNA aptamer against CD133 increased the cytotoxicity of Apt-PEG-AcCMC-SN38 into CD133 overexpressing HT29 tumour cells. Moreover, the Apt-PEG-AcCMC-SN38 enhanced the SN38 cytotoxicity to the HT29 tumour cells while showing no efficacy towards the CHO cells.
This is consistent with the results obtained by flow cytometry in which CD133 aptamer increased the uptake of Apt-PEG-AcCMC-SN38 into HT29 cells but not in CHO cells. The results suggest that the CD133 aptamer may selectively increase the transportation of conjugated SN38 (prodrug) to HT29 cells. The RNA aptamer against CD133 showed high selectivity against HT29 cells. In this regard, the versatility of MUC1, AS1411, and EpCAM conjugated NPs against AGS, HT29, and MCF-7 cell lines was previously reported and the obtained results verified the enhanced cytotoxicity and uptake of aptamer-targeted systems in comparison with non-targeted ones [Citation34–38].
Calcein assay
After treatment of cells with PEG-AcCMC-SN38, Apt-PEG-AcCMC-SN38 or free SN38, the cells viability was monitored using calcein assay (). The obtained results were in agreement with those obtained from MTT assay illustrating that the Apt-PEG-AcCMC-SN38 significantly decreased the number of viable HT29 cells in comparison with PEG-AcCMC-SN38. However, no difference was observed between CD133- negative CHO cells treated with either Apt-PEG-AcCMC-SN38 or PEG-AcCMC-SN38.
Figure 7. Fluorescent microscopy images showing the cytotoxic effect of different groups compared to control in HT-29 cells as CD133 positive cells and in CHO cells as CD133 as negative cells.
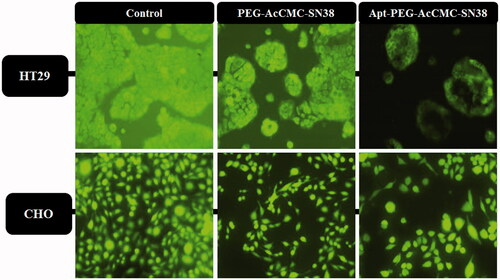
In this regard, untreated control cells were mostly fluorescently green. In order to compare the cytotoxic effects of Apt-PEG-AcCMC-SN38 and PEG-AcCMC-SN38, HT29 cells treated with Apt-PEG-AcCMC-SN38 showed lower green fluorescent intensity verifying the reduced cellular viability and, therefore, higher toxicity of Apt-PEG-AcCMC-SN38 compared to PEG-AcCMC-SN38 particles.
Higher levels of toxicity of Apt-PEG-AcCMC-SN38 for HT29 cells compared to those treated with PEG-AcCMC-SN38 were likely due to an increased uptake and higher accumulation of aptamer-conjugated prodrug in HT29 cells via receptor-mediated endocytosis while Apt-PEG-AcCMC-SN38 showed no similar effect in treated CHO cells.
Conclusion
In this article, we developed a targeted drug delivery system based on the acetylated carboxymethylcellulose (AcCMC). To improve the solubility and reduce the adverse effects of free SN38, we conjugated it to the PEGylated AcCMC backbone. The prepared prodrug could be self-assembled to the spherical NPs with size of 169 nm. Thereafter, the prepared self-assembled NPs were covalently conjugated to a RNA aptamer against CD133 which is a CSC marker overexpressed on HT-29 cell surfaces. The aptamer-conjugated PEG-AcCMC-SN38 nanoparticles were less than 200 nm in size and demonstrated enhanced cellular uptake in CD133 positive HT29 cell line. The in vitro cytotoxicity experiment revealed that aptamer-conjugated PEG-AcCMC-SN38 significantly increased the cytotoxicity of SN38 on HT-29 cells compared to the non-targeted PEG-AcCMC-SN38, whereas the cellular toxicity in CD133 negative cells (CHO cells) was not enhanced. In conclusion, based on the current findings, the prepared Apt-PEG-AcCMC-SN38 could be considered as a promising targeted delivery system for prodrug of SN38 as a poorly aqueous soluble drug with considerable unfavourable adverse effects.
Disclosure statement
The authors report no conflicts of interest.
Related Research Data
References
- Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–193.
- Fuchs C, Mitchell EP, Hoff PM. Irinotecan in the treatment of colorectal cancer. Cancer Treat Rev. 2006;32:491–503.
- Peng C-L, Lai P-S, Lin F-H, et al. Dual chemotherapy and photodynamic therapy in an HT-29 human colon cancer xenograft model using SN-38-loaded chlorin-core star block copolymer micelles. Biomaterials. 2009;30:3614–3625.
- Fang T, Dong Y, Zhang X, et al. Integrating a novel SN38 prodrug into the PEGylated liposomal system as a robust platform for efficient cancer therapy in solid tumors. Int J Pharm. 2016;512:39–48.
- Lee S-Y, Yang C-Y, Peng C-L, et al. A theranostic micelleplex co-delivering SN-38 and VEGF siRNA for colorectal cancer therapy. Biomaterials. 2016;86:92–105.
- Bala V, Rao S, Boyd BJ, et al. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172:48–61.
- Gu Q, Xing JZ, Huang M, et al. SN-38 loaded polymeric micelles to enhance cancer therapy. Nanotechnology. 2012;23:205101.
- Ebrahimnejad P, Dinarvand R, Sajadi A, et al. Preparation and in vitro evaluation of actively targetable nanoparticles for SN-38 delivery against HT-29 cell lines. Nanomedicine. 2010;6:478–485.
- Mosallaei N, Mahmoudi A, Ghandehari H, et al. Solid lipid nanoparticles containing 7-ethyl-10-hydroxycamptothecin (SN38): preparation, characterization, in vitro, and in vivo evaluations. Eur J Pharm Biopharm. 2016;104:42–50.
- Goldberg DS, Vijayalakshmi N, Swaan PW, et al. G3. 5 PAMAM dendrimers enhance transepithelial transport of SN38 while minimizing gastrointestinal toxicity. J Control Release. 2011;150:318–325.
- Duncan R. Development of HPMA copolymer-anticancer conjugates: clinical experience and lessons learnt. Adv Drug Deliv Rev. 2009;61:1131–1148.
- Singer JW, Shaffer S, Baker B, et al. Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anticancer Drugs. 2005;16:243–254.
- Avendaño C, Menéndez JC. Medicinal chemistry of anticancer drugs. Amsterdam: Elsevier; 2008. Drug targeting in anticancer chemotherapy; p. 351–385.
- Feng Q, Tong R. Anticancer nanoparticulate polymer‐drug conjugate. Bioeng Transl Med. 2016;1:277–296.
- Hoang B, Ernsting MJ, Roy A, et al. Docetaxel-carboxymethylcellulose nanoparticles target cells via a SPARC and albumin dependent mechanism. Biomaterials. 2015;59:66–76.
- Ajorlou E, Khosroushahi AY, Yeganeh H. Novel water-borne polyurethane nanomicelles for cancer chemotherapy: higher efficiency of folate receptors than TRAIL receptors in a Cancerous Balb/C Mouse Model. Pharm Res. 2016;33(6):1426–1439.
- Jiang J, Chen H, Yu C, et al. The promotion of salinomycin delivery to hepatocellular carcinoma cells through EGFR and CD133 aptamers conjugation by PLGA nanoparticles. Nanomedicine. 2015;10(12):1863–1879.
- Swaminathan SK, Roger E, Toti U, et al. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release. 2013;171(3):280–287.
- Chen Y, Chen C, Xiao Y, et al. Liposomes Encapsulating 10-Hydroxycamptothecin-Cyclodextrin Complexes and Their In Vitro Anti-Tumor Activities. J. Nanosci Nanotechnol. 2015;15(5):3786–3795.
- Shigdar S, Qiao L, Zhou S-F, et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330:84–95.
- Ulasov IV, Nandi S, Dey M, et al. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol Med. 2011;17(1-2):103.
- Ni M, Xiong M, Zhang X, et al. Poly (lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int J Nanomedicine. 2015;10:2537.
- Mongelard F, Bouvet P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Curr Opin Mol. 2010;12(1):107–114.
- Roy A, Murakami M, Ernsting MJ, et al. Carboxymethylcellulose-based and docetaxel-loaded nanoparticles circumvent P-glycoprotein-mediated multidrug resistance. Mol Pharm. 2014;11:2592–2599.
- Zhang X, Jackson JK, Burt HM. Determination of surfactant critical micelle concentration by a novel fluorescence depolarization technique. J Biochem Biophys Methods. 1996;31:145–150.
- Mueller H, Kassack MU, Wiese M. Comparison of the usefulness of the MTT, ATP, and calcein assays to predict the potency of cytotoxic agents in various human cancer cell lines. J Biomol Screen. 2004;9:506–515.
- Ernsting MJ, Tang W-L, MacCallum N, et al. Synthetic modification of carboxymethylcellulose and use thereof to prepare a nanoparticle forming conjugate of docetaxel for enhanced cytotoxicity against cancer cells. Bioconjugate Chem. 2011;22:2474–2486.
- Patnaik A, Papadopoulos KP, Tolcher AW, et al. Phase I dose-escalation study of EZN-2208 (PEG-SN38), a novel conjugate of poly(ethylene) glycol and SN38, administered weekly in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;71:1499–1506.
- Lee H, Lee K, Park TG. Hyaluronic acid-paclitaxel conjugate micelles: synthesis, characterization, and antitumor activity. Bioconjug Chem. 2008;19:1319–1325.
- Van S, Das SK, Wang X, et al. Synthesis, characterization, and biological evaluation of poly(L-gamma-glutamyl-glutamine)-paclitaxel nanoconjugate. Int J Nanomed. 2010;5:825–837.
- Zhang H, Wang J, Mao W, et al. Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: In vitro and in vivo studies. J Control Release. 2013;166(2):147–158.
- Sahlberg SH, Spiegelberg D, Glimelius B, et al. Evaluation of cancer stem cell markers CD133, CD44, CD24: association with AKT isoforms and radiation resistance in colon cancer cells. PLos One. 2014;9:e94621.
- Elsaba TMA, Martinez-Pomares L, Robins AR, et al. The stem cell marker CD133 associates with enhanced colony formation and cell motility in colorectal cancer. PLos One. 2010;5:e10714.
- Alibolandi M, Taghdisi SM, Ramezani P, et al. Smart AS1411-aptamer conjugated pegylated PAMAM dendrimer for the superior delivery of camptothecin to colon adenocarcinoma in vitro and in vivo. Int J Pharm. 2017;519:352–364.
- Taghavi S, Ramezani M, Alibolandi M, et al. Chitosan-modified PLGA nanoparticles tagged with 5TR1 aptamer for in vivo tumor-targeted drug delivery. Cancer Lett. 2017;400:1–8.
- Alibolandi M, Mohammadi M, Taghdisi SM, et al. Fabrication of aptamer decorated dextran coated nano-graphene oxide for targeted drug delivery. Carbohydr Polym. 2017;155(Suppl C): 218–229.
- Alibolandi M, Ramezani M, Abnous K, et al. AS1411 aptamer-decorated biodegradable polyethylene glycol-poly(lactic-co-glycolic acid) nanopolymersomes for the targeted delivery of gemcitabine to non-small cell lung cancer in vitro. J Pharm Sci. 2016;105:1741–1750.
- Alibolandi M, Ramezani M, Abnous K, et al. In vitro and in vivo evaluation of therapy targeting epithelial-cell adhesion-molecule aptamers for non-small cell lung cancer. J Control Release. 2015;209:88–100.