
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Nisoldipine (ND) has low oral bioavailability (5%) due to first-pass metabolism. Previously, solid lipid nanoparticles (SLNs) of ND were reported. In this study, nanostructured lipid carriers (NLCs) of ND are developed to enhance the oral bioavailability. ND-NLCs were prepared using hot homogenization-ultrasonication method, using oleic acid and trimyristate as liquid lipid and solid lipid, respectively. Prepared NLCs are evaluated for an optimal system using measuring size, zeta potential, entrapment efficiency, in-vitro release and in-situ absorption studies. Further, in vivo pharmacokinetic (PK) studies of NLC were conducted in rats comparison with SLN and suspension as controls. Size, ZP and EE of optimized NLCs were found to be 110.4 ± 2.95 nm, –29.4 ± 2.05 mV and 97.07 ± 2.27%, respectively. Drug loaded into NLCs was converted to amorphous form revealed by differential scanning calorimeter (DSC) and X-ray diffractometry (XRD) technique and nearly spherical in shape by scanning electron microscopy (SEM) studies. Drug release and absorption of ND were prolonged from ND-NLCs and ND-SLNs. From the PK results, NLCs showed 2.46 and 1.09-folds improvement in oral bioavailability of ND compared with suspension and SLNs formulations, respectively. Taken together, the NLCs and SLNs are used as carriers for the enhancement of oral bioavailability of the ND.
Introduction
In the present scenario, oral drug delivery is continuously looking in to newer avenues due to realization of the factors such as poor drug solubility and/or absorption, rapid metabolism, high fluctuation in drug plasma level and variability due to food effect which are playing major role in disappointing in-vivo results leading to failure of the conventional delivery system. Since the last decade, the oral drug delivery has taken a new dimension with the increasing application of lipid as a carrier for the delivery of poorly water-soluble and lipophilic drugs to promote lymphatic uptake [Citation1–3].
Among different approaches, solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) have been developed as an important strategy to deliver conventional drugs especially biopharmaceutical classification systems II (BCS class II) [Citation4,Citation5]. SLNs are colloidal drug delivery systems that modify the kinetics [Citation6], pharmacodynamics [Citation7] and body distribution and drug release [Citation8]. Other effects are tissue or cell-specific targeting of drugs [Citation9] and the reduction of unwanted side effects by controlled release [Citation10].
NLCs are the second generation of lipid nanoparticles [Citation11,Citation12]. The main difference between SLNs and NLCs is the fact that the concept of these latter is performed by nanostructuring the lipid matrix, in order to increase the drug loading and to prevent its leakage, giving more flexibility for modulation of drug loading, further for modulation of drug release. This approach is achieved by mixing solid lipids with liquid lipids (oils) in NLCs, instead of highly purified lipids with relatively similar molecules in SLNs. This mixture has to be solid at least at 40 °C. The NLCs or oil-loaded SLNs contain lipid droplets that are partially crystallized and have a less-ordered crystalline structure or an amorphous solid structure, which were developed for overcoming the limitations of SLNs by Müller et al. at the end of the 1990s. The result is less ordered lipid matrix with many imperfections, which can accommodate a higher amount of drug [Citation13,Citation14]. Furthermore, the lipid blend in NLCs has undergone slower polymorphic transition and along with existing oil in the NLC-core, the spherical shape of the particle may account for these improving properties [Citation15,Citation16].
Nisoldipine (ND) is (1,4-dihydropyridine derivative) a calcium channel blocker. It has a very poor oral bioavailability (< 5%) as it undergoes extensive first-pass metabolism in gut wall as it is a known CYP3A substrate [Citation17,Citation18] and lipophilic drug with logP of 3. Hence, the bioavailability of ND can be improved by deliver in the form NLCs, is an alternative approach. Previously, we developed and reported the SLNs of ND using central composite design. From the results, approximately 2.17-folds enhancement in the oral bioavailability and prolonged pharmacodynamic effect for 36 h was observed when compared with suspension [Citation19]. Nekkanti et al. [Citation20] reported the proliposomes and self-micro-emulsifying drug delivery systems of ND for the enhancement of oral bioavailability. But, the NLCs of ND was not developed so far. Therefore, an attempt was to develop NLCs of ND to improve oral bioavailability.
The purpose of this study was to formulate and characterize the NLCs of ND for an optimal system. Accordingly, a comparative pharmacokinetic (PK) study of ND-NLC were conducted in Wistar rats against ND-SLN and ND-CS as controls. According to this, NLCs of ND were prepared using hot homogenization followed by ultrasonication technique. The prepared NLCs were characterized for size, size distribution (PDI), zeta potential (ZP), entrapment efficiency (EE), in-vitro release and stability studies. Solid-state characterization of optimized NLCs formulation using differential scanning calorimeter (DSC) and X-ray diffractometry (XRD) studies and surface morphology using SEM are performed. Further, in-vivo PK studies in comparison with SLNs and suspension formulation in Wistar rats were conducted.
Materials
ND was gifted sample from Orchid Labs (Chennai, India). Trimyristin (dynasan-114; glyceryl trimyristate) was purchased from Sigma-Aldrich Chemicals (Bangalore, India). Oleic acid and dialysis membrane (70 dm) were purchased from Himedia Chemicals (Hyderabad, India). Egg lecithin E-80 was a gift sample from Lipoid (Germany). Poloxamer-188 was a gift sample from Dr. Reddy’s Labs (Hyderabad, India). Methanol, acetonitrile and chloroform of high-performance liquid chromatography (HPLC) grade were purchased from Merck, Mumbai, India. Centrisart filters (molecular weight cut-off 20,000 Da) were purchased from Sartorius (Goettingen, Germany).
Methods
Preparation of ND-loaded nanostructured lipid carriers (ND-NLCs)
The NLCs of ND were prepared by hot homogenization followed by ultrasonication method. ND (10 mg), solid lipid, liquid lipid and egg lecithin were dissolved in 10 mL of chloroform and methanol (1:1) mixture. The organic solvent was evaporated by rotary evaporator (Heidolph, Germany). Lipid phase was heated at above 5 °C of its melting point for drug embedment. The aqueous phase was prepared by dissolving the poloxamer-188 (150 mg) in double distilled water (Millipore, India) sufficient to produce 10 mL and heated at same temperature as that of molten lipid phase. Hot aqueous phase was added to molten lipid phase and homogenized (Diax 900, Heidolph, Germany) for 5 min at 12,000 rpm, whereas the system was maintained at 5 °C above melting point of the liquid throughout the homogenization process. The resulted coarse hot oil in water emulsion was ultrasonicated to get nanoemulsion using a 12-T probe sonicator (Vibra-Cell, Sonics, CT, USA) for 20 min. The nanoemulsion was cooled at room temperature to obtain ND-NLCs.
Preparation of ND-SLNs
The ND-SLNs were prepared as per the earlier reported composition of Narendar and Kishan [Citation19] using central composite design. SLNs composed of 10 mg of drug, 100 mg of dynasan-114, 75 mg of egg lecithin and 1.5% w/v of poloxamer-188 were used and prepared as per the above-mentioned method.
Preparation of nisoldipine suspension (NDS)
About 10 mg of ND was mixed with 50 mg of sodium carboxy methyl cellulose (Na CMC) in a mortar and mixed for 3 min by trituration. Then, 10 mL of double distilled water was added and further triturated for 3 min. The formed suspension was used for further studies.
Characterization of ND-NLCs and ND-SLNs
Determination of particle size and ZP
Particle size, PDI and ZP of NLCs and SLNs of ND were determined by photon correlation spectroscopy (PCS) using a Zetasizer Nano ZS90 (Malvern Instruments, UK). The prepared lipid nanoparticle formulations were diluted in 1:50 ratio with double distilled water and size was measured at 90° [Citation21]. All measurements were performed using triplicate. Simultaneously, the particle size of the suspension formulation was also measured.
Measurement of EE and drug content
ND content and EE of prepared ND-NLCs and SLNs were determined using HPLC. To determine EE, centrifugation was performed using Centrisart (Sartorius, Goettingen, Germany) tubes at 10,000 rpm for 30 min, which consisted of a filter membrane (molecular weight cut-off 20,000 Da) to separate the ultrafiltrate [Citation22]. The EE of the system was determined by measuring the concentration of free drug in the dispersion medium using HPLC, along with following formula:
where Wtotal was the weight of drug added in the system.
For total drug content, about 0.1 mL of formulation was dissolved in chloroform and methanol (1:1) mixture. The final dilution of solution was made with mobile phase. ND content was determined using HPLC.
The chromatographic conditions of the HPLC were system consisting of a SPD-20AD solvent delivery system containing double reciprocating plunger pump (Shimadzu, Japan), a SPD-20A UV-visible variable wavelength detector with deuterium lamp using a RP C18 analytical column, Merck (250 mm ×4.6 mm i.d., 5 µm) and the mobile phase consisting of acetonitrile: water (80:20) ratio at a flow rate of 1 mL/min was employed for the detection of drug at λmax of 237 nm [Citation19]. The method was linear in the range of 5–100 µg/mL with r2 value of 0.999. Limit of detection and limit of quantification of the method were found to be 2 and 4 µg/mL, respectively.
In-vitro drug release studies
Drug release studies of ND-NLCs, ND-SLNs and NDS formulations were performed using dialysis method in 0.1 N HCl (pH1.2) for 2 h followed by pH 6.8 phosphate buffer for 22 h [Citation22]. During in-vitro release studies, dialysis membrane (Himedia, Hyderabad, India) with a molecular weight cut-off ranging from 12,000 to 14,000 Da and 70 DM was used. Prior to the study, the dialysis membrane was soaked by kept in double distilled water for overnight. The apparatus had two compartments, a donor and a receptor compartment. In donor compartment, about 2 mL of formulation (ND-NLCs, ND-SLNs and NDS) was taken for release study, which consisted of a boiling tube with opening at the one end and tied with dialysis membrane at the other end. A beaker of 250 mL capacity was used as receptor compartment with 100 mL release medium and maintained at the temperature of 37 ± 0.5 °C. About 2 mL of samples was withdrawn from receiver compartment and was replaced with fresh medium periodically at predetermined time intervals and analyzed for drug content using UV-visible spectrophotometry (SL-210, ELICO, Hyderabad, India).
Stability studies
ND-loaded optimized NLCs (N4) were stored at room temperature and refrigerated temperature for 3 months. The changes in the formulation were observed by measuring the particle size, PDI, ZP, assay and EE periodically after 1st day, 15th day, first, second and third months after the storage [Citation21].
Lyophilization of ND-NLCs and SLNs
The optimized ND-NLCs and SLNs containing 10% w/v trehalose as cryoprotectant were prepared and kept at deep freezer at –40 °C (Sanyo, Japan) for overnight. Then, the frozen samples of the formulations were transferred into freeze-dryer (Lyodel, Delvac Pumps Pvt. Ltd, India). Vacuum was applied and sample was subjected to drying for about 48 h to get powdered lyophilized product [Citation23]. The powdered formulation is used for further studies after measuring the particle size, PDI and ZP.
Solid-state characterization
Drug-excipient compatibility studies by DSC
DSC analysis of ND, trimyristin (TM), physical mixtures (PM in 1:1 ratio) and lyophilized ND-NLCs and SLNs were performed using Universal V4 TA Instruments (New Castle, DE, USA), to determine the drug interaction as well as changes in the crystallinity of the drug loaded into NLCs. Initially, the instrument was calibrated with indium. All the samples of about ∼8 mg were heated in aluminium pans and heating at the range of 20–300 °C and at a rate of 20 °C/min. For this purpose, dry nitrogen was used as the effluent gas [Citation24].
Characterization of crystallinity by powder X-ray diffractometry (PXRD)
PXRD (Multiflex, M/s. Rigaku, Japan) was used for diffraction studies and to determine the crystallinity of ND. PXRD studies were monitored by the samples exposing to nickel filtered CuKα radiation (40 kV, 30 mA) and scanned from 2° to 70°, 2θ at a step size of 0.045° and step time of 0.5 s. Samples used for PXRD analysis were pure ND, pure dynasan-114, physical mixtures of drug with lipids at 1:1 ratio and lyophilized ND-loaded NLCs and SLNs formulation [Citation22].
Morphology by SEM
The morphology of ND-loaded NLCs was studied using SEM (Hitachi, Japan). For this, the freeze-dried NLCs of ND was suitably diluted to 100 times with double distilled water and from this a drop of nanoparticle formulation was placed on sample holder and air dried. This was sputter coated with platinum-coating machine (JEOL, JFC-1600 Auto fine coater). Afterwards, the sample was observed at accelerating voltage of 15,000 volts at various magnifications by mounted in SEM. Generally, the imaging of SEM was carried out using high vacuum [Citation25].
In-situ intestinal absorption study
The in-situ single pass intestinal perfusion test was carried out in rats using established methods [Citation26]. Male Wistar rats with weighing 180 ± 30 g were used for study. Prior to the study, the rats were fasting overnight, retained without food and given water for drinking. This drains the intestine, making it free from food particulates or faeces. The rats were anaesthetized by i.p. injection of thiopentone sodium (10 mg/kg body weight). They were given a slit on the abdominal side and lower part of small intestine was exposed a 10-cm length of small intestine distal to duodenum was taken out and semi-circular cuts were given at the two ends. The two ends were tied with a PE tubes using silk thread. Cotton dripped in physiological saline and was used to keep the tissues moist. Before starting the experiment, the intestine was cleaned with Krebs-Ringer buffer solution. Firstly, the intestinal area was stabilized by passing saline for approximately 15–20 min. Next, the perfusion solutions, i.e., NDS, optimized ND-NLCs and ND-SLNs formulations, were passed sequentially each one followed by washing period. The flow rate was fixed at 0.2 mL/min for 120 min. The drug permeated was analyzed using HPLC method. The samples (ND-NLCs and ND-SLNs) were compared to NDS as control.
The effective permeability coefficient was determined using the below equation:
(1)
(1)
where Qin is the flow rate of perfusate entering the intestine (0.2 mL/min), A is the intestinal surface area which was assumed to be the area of a cylinder (2πrl) where the length was measured at the end of the study, Cin and Cout were the inlet and outlet (corrected) solution concentrations, respectively.
Cout (corrected) was given using the below equation:
(2)
(2)
where Qout is the flow rate of the perfusate moving out of intestine for a given time interval.
The slope from the plot, remaining amount of drug vs. time, gives the absorption rate constant (Ka).
Another parameter that is the enhancement ratio (ER) was calculated using the below equation.
(3)
(3)
PK study
Study protocol
A single dose bioavailability study was designed in male Wistar rats under fasting conditions. The study was approved by the Institutional Animal Ethical Committee (IAEC), Synapse Life Sciences, Warangal, India. Male Wistar rats weighing 180–240 g were taken for study (six animals per each group). The oral bioavailability of ND-NLCs (N4) formulation was estimated in comparison with ND-SLNs (optimized) and NDS formulation, with oral dose of 10 mg/kg body weight. Approximately 0.5 mL of blood samples was collected from retro orbital plexus into Eppendorf tubes at regular predetermined time intervals and serum was separated by centrifugation (Remi Equipments, Hyderabad, India) for 15 min at 10,000 rpm. The serum was stored at –20 °C until analysis using HPLC.
HPLC analysis
The HPLC column, Merck C18 (250 × 4.6 mm), was equilibrated with an eluent mixture of acetonitrile:water (62:38 v/v) at a flow rate of 1 mL/min. The peaks were eluted at 237 nm wavelength without any interference from serum. The retention times of ND and internal standard were 4.8 and 7.4 min, respectively.
Extraction procedure
About 100 µL of serum, 100 µL of mobile phase and 100 µL of 3 µg/mL of nimodipine solution as internal standard were added and vortexed for 3 min. Then, 25 µL of 0.1 M NaOH was added and vortexed for 2 min. To this, 1 mL of ether was added and vortexed for 3 min followed by centrifugation at 6000 rpm for 20 min. Organic phase was separated and evaporated. The residue was reconstituted with 100 µL of mobile phase and 20 µL of this solution was spiked on to the HPLC column [Citation27].
PK parameters
The PK parameters of ND-NLC, ND-SLNs and NDS formulation following oral administration, such as maximum serum concentration (Cmax), time for maximum serum concentration (tmax), AUCtotal, biological half-life (t½) and mean residence time (MRT), were calculated using Kinetica software (Version 5.0, Innaphase Corporation, Philadelphia, PA, USA). The values were expressed as mean ± SD.
Statistical treatment of data
The statistical comparison of data of three samples was done with one-way ANOVA followed by Dunnett’s test of significance using Graphpad prism software (Version 5.02.2013, Graphpad software, San Diego, CA, USA) and p < .01 was considered as statistically significant probability level.
Results and discussion
ND belongs to BCS class II drug. It has oral bioavailability of about less than 5% due to poor solubility and first-pass metabolism. Previously, ND-loaded SLNs were developed using central composite design to enhance the oral bioavailability. Now, the NLCs of ND were developed to improve the oral bioavailability and also compared with optimized SLNs formulation as well as suspension formulation as control. Accordingly, the NLCs of ND was developed using hot homogenization followed by ultrasonication method. Oleic acid and dynasan-114 were used as liquid lipid and solid lipid respectively for the preparation of ND-NLCs. Egg lecithin (75 mg) and poloxamer-188 (1.5% w/v) were used as surfactant and co-surfactant, respectively. From the literature, the homogenization and sonication time of 4 min (12,000 rpm) and 20 min, respectively, were selected for the preparation of ND-loaded NLCs and SLNs.
Measurement of size, PDI and ZP of ND-NLCs and ND-SLNs
ND-NLCs were prepared by two varying concentration of oleic acid with three different concentrations of dynasan-114 as per the composition in . Simultaneously, ND-SLNs (optimized) formulation was prepared. The prepared formulations were subjecting to size analysis. From the results, formulations N1–N5 (ND-NLCs) showed particle size of 110.4 ± 2.95 to 265.9 ± 4.69 nm, PDI of 0.202 ± 0.006 to 0.283 ± 0.007 and ZP of –22.6 ± 2.47 to –29.4 ± 2.05 mV. As the concentration of liquid lipid increased from 0.05 to 0.06 mL (N1 and N2) with constant solid lipid concentration (75 mg), size and PDI of formulations were decreased and ZP was increased. This might be due to the less availability of the solid lipid for the orientation of the drug. The amount of solid lipid was changed from 75 to 100 mg, with same amount of liquid lipids also resulted in reduced size with increased ZP (for N4 formulation) was observed. Further, increasing the amount of solid lipid, the size of the NLCs was increased ().
Table 1. Composition of nisoldipine-loaded NLCs, SLNs and suspension formulation.
Table 2. Physical characters – size, PDI, ZP, drug content and EE of ND-NLCs and SLNs formulations (mean ± SD, n = 3).
The ND-NLCs formulations exhibited negative surface charge which clearly suggested the orientation of ND in the lipid metric. The surface charge is a critical factor for the stability of colloidal preparations. From the results, the ZP values of NLCs formulations were found to be within range of –22.6 ± 2.47 to –29.4 ± 2.05 mV. It is an indirect measure of physical stability of NLCs and even influences the release kinetic and the biological fate of nanoparticles. To achieve a nanoformulation with good stability for an electrostatically stabilized, a minimum ZP of ±30 mV is required whereas in the case of a combined electrostatic and steric stabilization, a minimum ZP of ±20 mV is desirable [Citation28].
In this study, poloxamer-188 was used as surfactant. It is a non-ionic surfactant and decreased the electrostatic repulsion between the particles following sterical stabilization of the nanoparticles by forming a coat around their surface for maintaining the stability of nanoparticles [Citation29]. A surfactant mixture, i.e., phosphatidylcholine and poloxamer-188 was employed in the formulations, because NLCs stabilized by combination of surfactants were reported to have lower particle size and higher storage stability when compared to formulations stabilized with only one surfactant [Citation30]. The size, PDI and ZP of ND-SLNs were found to be 105.2 ± 3.81 nm, 0.233 ± 0.004 and –25.3 ± 2.84 mV, respectively, and were reproducible as of earlier study (104.4 ± 2.13 nm of size, 0.241 ± 0.02 of PDI and –25.33 mV of ZP).
Determination of EE and drug content
The total drug content in the NLCs formulations was determined and to be ranging from 9.89 ± 0.18 to 9.97 ± 0.20 mg. EE of the NLCs formulations was found to be 82.71 ± 0.31% to 97.07 ± 2.27%. High lipophilicity of ND resulted in high EE of drug in lipid nanoparticles (). This might be because of the long chain fatty acids attached to the glyceride resulting in increased accommodation of lipophilic drugs. The less ordered lipid matrix of liquid lipid created imperfections leading to void spaces in which drug molecules could be entrapped [Citation12]. In this method of preparation, drug was dissolved in molten lipid at temperature above the melting point of solid lipid and there was no drug leakage or precipitation of drug during the preparation. Further, the EE also influences the release characteristics of NLCs and depends on the NLCs ingredients, which influence factors such as the solubility of drug in the lipid phases, nanoparticle crystallinity index and thereby diffusion coefficient of drug, production method and conditions used. The solid state of lipidic nanoparticles in NLCs immobilizes the incorporated drug and leads to higher EE. High encapsulation efficiency of drug in lipid nanoparticles can cause high amount of drug to pass through the lymphatic transport, which in turn bypasses the first pass metabolism.
In-vitro release studies
In-vitro release studies of ND from ND-NLCs, ND-SLNs and NDS were performed in 0.1 N HCl (2 h) followed by pH 6.8 phosphate buffer (22 h) by dialysis method and profiles were showed in . From the results, in-vitro release was found to be prolonged both in pH 6.8 and in 0.1N HCl. Drug release of ND was found to be 63.64 ± 3.1, 69.83 ± 2.8, 75.88 ± 2.6, 83.91 ± 2.4 and 61.74 ± 2.8% from N1 to N5, 80.72 ± 2.7% from SLNs and 95.73 ± 2.4 from NDS during 20 h in 0.1 N hydrochloric acid followed by pH 6.8 phosphate buffer.
Figure 1. In-vitro drug release of ND from ND-NLCs, ND-SLNs and NDS in 0.1 N HCl followed by pH 6.8 phosphate buffer (mean ± SD, n = 3).
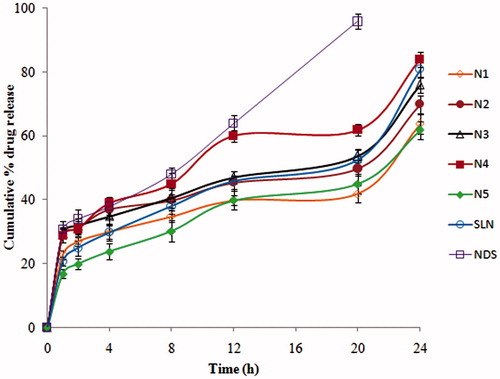
Drug release studies from ND-NLCs and ND-SLNs were conducted for a period of 24 h, since gastric transport is much longer than intestinal transit. ND belongs to BCS class-II, and is a lipophilic drug and this may be a reason for the slow release from the lipid matrix. ND showed more solubility in pH 6.8 phosphate buffer than in 0.1 N HCl. Hence, initially the release was found to be less. But, the unentrapped drug in aqueous portion of the NLCs dispersion got released very quickly. During the initial period, the release rate was found to be affected by the concentration of lipid and surfactant in aqueous phase. The release rate increased with the increasing of lipid concentrations. This could be due to the ND location within the outer shell of lipid rather than in the inner core. However, the increase of lipid concentration prolonged the ND release from NLCs, attributed to the good solubility of ND in all formulations and to the homogenous distribution of drug within the lipid matrix.
From the results, the N4 NLCs formulation had better size, low PDI, high ZP with more EE and high (83.91 ± 2.4%) drug release than other formulations. Hence, it was considered as optimized one and used for further studies.
Lyophilization of ND-NLCs
The optimized NLCs (N4) formulation was subjecting to freeze drying, using 10% w/v trehalose as cryoprotectant. The lyophilized NLCs powder was further diluted to 50 times with double distilled water and measured the physical characters. From the results, size, PDI and ZP of the N4 formulation were 113.8 ± 3.3 nm, 0.202 ± 0.006, –29.4 ± 2.05 mV and 354.8 ± 2.86 nm, 0.634 ± 0.013, –29.7 ± 3.2 mV for NLC; 107.4 ± 2.7 nm, 0.246 ± 0.005, –25.1 ± 3.2 mV and 313.7 ± 3.6 nm, 0.586 ± 0.008, –25.8 ± 2.7 mV for SLNs before and after lyophilization, respectively. This increased size and PDI of the formulations are might be due to the aggregation of the particles by lyophilization process.
Stability studies
Stability studies were conducted for optimized NLCx formulation (N4) at room temperature and refrigerated temperature for 3 months. Some changes were noticed in size and ZP values, but statistically insignificant (), which indicated the susceptibility for stability problems during storage at room temperature and 4 °C.
Table 3. Effect of storage at refrigerated and room temperature on size, PDI, ZP, assay and EE of optimized NLCs (N4) formulation for 3 months (mean ± SD, n = 3).
In-situ intestinal absorption study
In-situ intestinal absorption study was emphasized to know the absorption potential of optimized NLCs (N4) and SLNs formulation in comparison to suspension formulation (NDS) across the gastrointestinal tract (). The parameters such as permeability coefficient (Peff), absorption rate constant (Ka) and ER were calculated and are shown in . The Peff of N4, SLNs and NDS samples were found to be 2.95 ± 0.025 × 10−2, 2.36 ± 0.018 × 10−2 and 1.02 ± 0.015 × 10−2, respectively. The increased permeability coefficient of ND in NLCs and SLNs formulation compared to suspension formulation indicated that NP prevent barrier properties of GIT, thereby increasing the drug absorption. The rate of absorption indicated by the absorption rate constant was higher for lipid nanoparticles of ND when compared to NDS (). The ER was observed to be above 1, thus indicating an increased perfusion of SLNs and NLCs of ND. Thus, signifying the potential of lipid nanocarriers was to be given through oral route. From the results, it was concluded that drug permeation was increased by more than two-folds when compared to NDS. This might be due to the intestine is thrown into many folds. During the pass of lipid nanoparticle formulation, there could be the accumulation or holding of drug-loaded particles within or among the folds. This could be a reason for the lower drug quantity in the volume collected at the outlet in the in-situ intestinal permeation studies [Citation31].
Figure 2. Drug permeation profiles of ND from ND-NLCs, ND-SLNs and NDS by in-situ absorption studies in rat intestine (mean ± SD, n = 3).
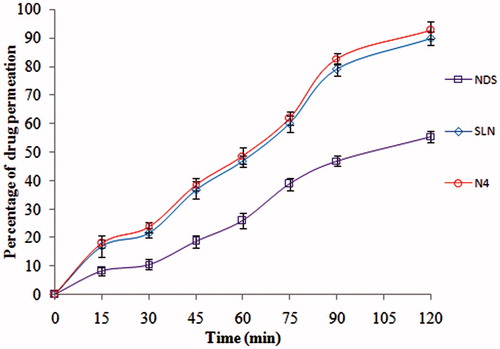
Table 4. Release coefficients and kinetic parameters of drug permeation studies of nisoldipine from ND-NLCs, SLNs and NDS formulation across rat intestine (mean ± SD, n = 3).
Solid-state characterization
Drug-excipient compatibility studies by DSC
The compatibility status of the lipids in the NLCs formulation was investigated using DSC. DSC thermograms of pure drug, pure lipid, physical mixture of drug and lipid (1:1) and lyophilized NLCs formulation are shown in . The degrees of crystallinity of the samples were calculated and are shown in . The DSC thermogram of pure ND showed a sharp endothermic peak at 158.85 °C with 545.84 J/g enthalpy, which corresponding to the 100% of crystallinity of the drug. The pure lipid dynasan-114 exhibited an endothermic peak at 61.64 °C. Physical mixture of drug with dynasan-114 showed drug peak at 161.74 °C and lipid peak at 65.83 °C, respectively, and enthalpy values were found to be 487.67 J/g. Crystallinity was reduced to 89.34%. In this mixture, melting endotherm of drug was well preserved with slight changes in terms of broadening or shifting in the temperature of the melt. It is known that the quantity of material used, especially in drug-excipient mixtures, could influence the peak shape and enthalpy. Thus, these minor changes in the melting endotherm of drug could be due to the mixing of drug and excipient, which lowered the purity of each component in the mixture and this, might not necessarily indicate potential incompatibility. The absence of endotherm peak of drug in lyophilized NLCs and SLNs formulations unravels the conversion of native crystalline state of the drug to amorphous state. The crystallinity of NLCs and SLNs were found to be 25.04% and 28.74%, respectively. Similar observations were also reported in earlier study [Citation32].
Figure 3. DSC thermograms of (A) pure nisoldipine, (B) pure lipid, (C) physical mixture of ND and lipid (1:1 ratio), (D) lyophilized ND-NLCs and (E) lyophilized ND-SLNs.
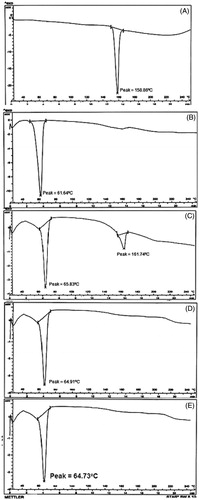
Table 5. Degree of crystallinity of pure drug, physical mixture and lyophilized NLCs and SLNs formulation.
Powder X-ray diffractometry
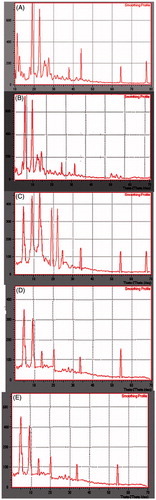
PXRD studies were carried out for pure drug, pure lipid, physical mixture of drug and lipid at 1:1 ration and lyophilized NLCs formulation and are shown in . PXRD patterns of pure ND showed sharp peaks at 2θ scattered angles of 11.31°, 12.54°, 18.86°, 22.78°, 26.10°, 27.86° and 45.17°, these were indicative of the crystalline nature of drug. In case of physical mixture, characteristic peaks of ND were preserved with reduced intensity, and drug peaks were almost absent in the lyophilized sample. This indicated that the drug was not in crystalline form after lyophilization of NLCs and SLNs. Further, the intensity of the lipid peaks also reduced in lyophilized sample. This reduced intensity indicated the decreased crystallinity of lipid. The change in crystallinity of lipid and drug would influence the release of ND from nanoparticles. This decrease in crystallinity was also noticed in DSC investigation of samples.
Figure 4. XRD spectra of (A) pure nisoldipine, (B) pure lipid, (C) physical mixture of ND and lipid (1:1 ratio), (D) lyophilized ND-NLCs and (E) lyophilized ND-SLNs.
Morphology of NLCs using scanning electron microscopy
The surface morphology of ND-NLCs was examined by scanning electron microscopy and is shown in . ND-NLCs formulation was studied for surface morphology at ×2k, ×8k, ×15k and ×30k magnification times. From the microscopic images, the particles possessed smooth surface with nearly spherical in shape with increasing in particle size and polydispersity due to lyophilization method, the agglomeration phenomenon was increased.
Figure 5. SEM images of optimized ND-NLCs at different magnifications.

PK study
ND has poor oral absorption (<5%) due to hepatic first-pass metabolism and poor aqueous solubility (BCS class-II). Previously, we attempted to develop and report enhanced oral bioavailability of ND using SLNs delivery system. But, this study emphasized to investigate the feasibility of NLCs for improved oral delivery of ND and also comparison of two lipid carrier systems. Previously, the oral bioavailability of vinpocetine, etoposide, carvedilol and atorvastatin [Citation21,Citation22,Citation33,Citation34] was improved by NLCs delivery system. The PK parameters of ND in individual rats for optimized NLCs (N4), optimized SLNs formulation and NDS were calculated by non-compartmental estimations using Kinetica 2000 software. The PK parameters AUCtotal, Tmax, Cmax, MRT and t1/2 were calculated. The statistical comparison of data was done using one-way ANOVA followed by Dunnett’s test at a significance level of p < .01. The serum concentration vs. time profiles following single dose administration of ND-NLC, SLNs and NDS formulation are shown in and . The relevant PK parameters were calculated. From the results, the Cmax for NLCs (12.27 ± 1.65 µg/mL) and SLNs formulation (11.94 ± 1.16 µg/mL) with respect to suspension (7.01 ± 1.03 µg/mL) were found to be higher and was statistically significant at p < .001. However, the time to reach the peak concentration was analogous to that of NDS. The MRT and half-life of the both NLCs and SLNs were almost double comparison with suspension formulation. This indicates the prolonged release as well as avoidance of the first-pass metabolism of the drug from the lipid nanoparticles. The AUCtotal was found to be 97.73 ± 3.81, 89.02 ± 3.16 and 39.72 ± 1.99 µg·h/mL for NLC, SLNs and NDS formulation. The obtained results were shown a higher plasma drug concentration (Cmax) and longer time taken to peak plasma concentration (Tmax) for NLCs and SLNs formulation compared to NDS formulation. The bioavailability of ND from optimized NLCs (N4) and SLNs formulation increased by 2.46 and 2.24-folds when compared to control, i.e., drug suspension. Further, about 1.09-folds improvement in the enhancement of oral bioavailability of NLCs was observed when compared with SLNs formulation. Due to the nanosize of the NLCs and SLNs, the effective surface would increase influencing the adhesion to GI tract. Subsequently, there is increased contact time of the lipid nanoparticles. In addition, the oleic acid, phosphatidylcholine and poloxamer-188 could alter the permeability characters of the GI membrane and also high drug loading in to the NLCs formulation. The fatty acid chain present in the solid lipid of NLCs improves the uptake by lymphatic transport. This lymphatic transport minimizes the first-pass effect of the drug [Citation35]. Overall, we propose the improved oral bioavailability of NLCs and SLNs could be due to the contribution of individual and/or combined mechanisms known till now.
Figure 6. Pharmacokinetic (PK) profiles of nisoldipine in rat serum following oral administration of ND-NLCs (N4), optimized SLNs and NDS formulation (mean ± SD, n = 6).
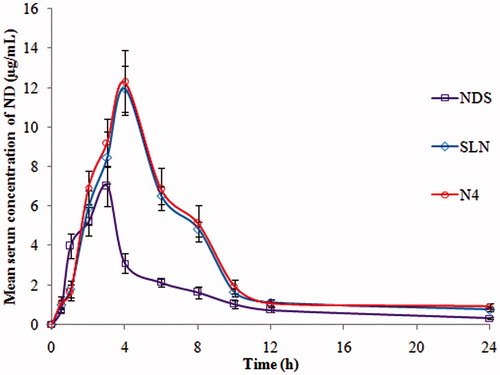
Table 6. Pharmacokinetic parameters of nisoldipine after oral administration of ND-NLCs, SLN and NDS in rats (mean ± SD, n = 6).
Conclusion
In this investigation, ND-loaded NLCs was developed and characterized to improve the oral bioavailability and a comparison was made by already reported SLNs preparation of ND. NLCs were prepared by well-known established methods, using oleic acid and dynasan-114 as liquid and solid lipid matrices. The solid-state characterization revealed the transformation of crystalline state of the drug to amorphous form in NLCs. Controlled release and enhanced drug release were accomplished by incorporating ND into the NLCs and SLN. The NLCs (N4) was stable at RT and 4 °C for 3 months. The in-situ absorption of ND was enhanced with NLCs as well as SLNs delivery system. The PK studies of optimized NLCs and SLNs showed 2.46 and 2.24-folds improvement in oral bioavailability when compared with drug suspension respectively in Wistar rats. Further, about 1.09-folds enhancement in the oral bioavailability of NLCs compare to SLN. Therefore, these results confirmed the prospective of NLCs and SLNs as suitable delivery system for oral delivery of poorly soluble drug in improving the oral bioavailability.
Acknowledgements
The authors would like to thank management of Synapse Life sciences and Kakatiya University, Warangal, India for providing facilities to carry out the work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Charman WN, Porter CJH. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv Drug Deliv Rev. 1996;19:149–169.
- Porter CJH, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 2001;50:61–80.
- Driscoll MC. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15:405–415.
- Müller RH, Mehnert W, Lucks JS. Solid lipid nanoparticles (SLN): an alternative colloidal carrier system for controlled drug delivery. Eur J Pharm Biopharm. 1995;41:62–69.
- Mehnert W, Mäder K. Solid lipid nanoparticles production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196.
- Kopparam M, Vobalaboina V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Rel. 2005;107:215–228.
- Narendar D, Kishan V. Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Deliv. 2016;23:395–404.
- Schwarz C. Solid lipid nanoparticles (SLN) for controlled drug delivery II: drug incorporation and physicochemical characterization. J Microencapsul. 1999;16:205–213.
- Sahu PK, Mishra DK, Jain N, et al. Mannosylated solid lipid nanoparticles for lung-targeted delivery of Paclitaxel. Drug Dev Ind Pharm. 2015;41:640–649.
- Xu Z, Chen L, Gu W, et al. The performance of docetaxel-loaded solid lipid nanoparticles targeted to hepatocellular carcinoma. Biomaterials. 2009;30:226–232.
- Müller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54:S131–S155.
- Müller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242:121–128.
- Müller RH, Keck CM. Challenges and solutions for the delivery of biotech drugs – a review of drug nanocrystal technology and lipid nanoparticles. J Biotechnol. 2004;113:151.
- Varshosaz J, Eskandari S, Tabakhian M. Production and optimization of valproic acid nanostructured lipid carriers by the Taguchi design. Pharm Dev Technol. 2010;15:89–96.
- Jores K, Mehnert W, Drechsler M, et al. Investigations on the structure of solid lipid nanoparticles (SLN) an oil-loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95:217–227.
- Piyush J, Bina G, Amber V. Nanostructured lipid carriers and their current application in targeted drug delivery. Artif Cells Nanomed Biotechnol. 2016;44:27–40.
- Hamilton SF, Houle LM, Thadani U. Rapid-release and coat-core formulations of nisoldipine in treatment of hypertension, angina, and heart failure. Heart Dis. 1999;5:279–288.
- Hitomi T, Ayako O, Hideyasu M, et al. Relationship between time after intake of grapefruit juice and the effect on pharmacokinetics and pharmacodynamics of nisoldipine in healthy subjects. Clin Pharmacol Ther. 2000;67:201–214.
- Narendar D, Kishan V. Pharmacokinetic and pharmacodynamic studies of nisoldipine loaded solid lipid nanoparticles by central composite design. Drug Dev Ind Pharm. 2015;41:1968–1977.
- Nekkanti V, Rueda J, Wang Z, et al. Comparative evaluation of proliposomes and self micro-emulsifying drug delivery system for improved oral bioavailability of nisoldipine. Int J Pharm. 2016;505:79–88.
- Ganesh BP, Nandkishor DP, Prashant KD, et al. Nanostructured lipid carriers as a potential vehicle for Carvedilol delivery: application of factorial design approach. Artif Cells Nanomed Biotechnol. 2016;44:12–19.
- Zhuang CY, Li N, Wang M, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharma. 2010;394:179–185.
- Shete H, Chatterjee S, De A, et al. Long chain lipid base tamoxifen NLC. Part II: pharmacokinetic, biodistribution and in vitro anticancer efficacy studies. Int J Pharm. 2013;454:584–592.
- Shete H, Patravale V. Long chain lipid based tamoxifen NLC – part I: preformulation studies, formulation development and physicochemical characterization. Int J Pharm. 2013;454:573–583.
- Hu FQ, Jiang SP, Du YZ, et al. Preparation and characterization of stearic acid nanostructured lipid carriers by solvent diffusion method in an aqueous system. Colloids Surf B Biointerfaces. 2005;45:167–173.
- Beloqui A, Solinis MA, Gascon AR, et al. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release. 2012;166:115–123.
- Li M, Wang Q, Wang C, et al. Tissues distribution of R-(-)- and S-(+)-m-nisoldipine after single enantiomer administration in rats. Drug Dev Ind Pharm. 2009;35:65–72.
- Mitri K, Shegokar R, Gohla S, et al. Lipid nanocarriers for dermal delivery of lutein: preparation, characterization, stability and performance. Int J Pharm. 2011;414:267–275.
- Müller BG, Leuenberger H, Kissel T. Albumin nanospheres as carriers for passive drug targeting: an optimized manufacturing technique. Pharm Res. 1996;13:32–37.
- Zheng K, Zou A, Yang X, et al. The effect of polymer surfactant emulsifying agent on the formation and stability of a-lipoic acid loaded nanostructured lipid carriers (NLC). Food Hydrocoll. 2013;32:72–78.
- Karthik YJ, Raju J, Ashok V, et al. Bioavailability enhancement of zaleplon via proliposomes: role of surface charge. Eur J Pharm Biopharm. 2012;80:347–357.
- Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95:627–638.
- Zhang T, Chen J, Zhang Y, et al. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur J Pharm Sci. 2011;43:174–179.
- Kritika S, Supandeep SH, Bharat L, et al. Development and characterization of floating spheroids of atorvastatin calcium loaded NLC for enhancement of oral bioavailability. Artif Cells Nanomed Biotechnol. 2016;44:1448–1456.
- Arik D, Amnon H. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm. 2007;67:96–105.