
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of this study was to prepare two novel magnolol (MO)-loaded binary mixed micelles (MO-M) using biocompatible copolymers of Soluplus (SOL) and Solutol® HS15 (HS15), SOL and d-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS), to improve magnolol’s poor solubility and its oral bioavailability. The organic solvent evaporation method was used to obtain two MO-M by optimization; one was prepared by using SOL and HS15 (MO-H), and the other was prepared by using SOL and TPGS (MO-T). The entrapment efficiency (EE%) and drug loading (DL%) of MO-T were 94.61 ± 0.91% and 4.03 ± 0.19%, respectively, and the MO-H has higher EE% and DL% (98.37 ± 1.23%, 4.12 ± 0.16%). TEM results showed that the morphology of MO-M was homogeneous and was spherical in shape. The dilution stability of MO-M did not undergo significant changes. Permeability of MO-M across a Caco-2 cell monolayer was enhanced in Caco-2 cell transport models. The pharmacokinetics study showed that the relative oral bioavailability of MO-T and MO-H increased by 2.39- and 2.98-fold, respectively, compared to that of raw MO. This indicated that MO-H and MO-T could promote absorption of MO in the gastrointestinal tract. Collectively, the mixed micelles demonstrated greater efficacy as a drug delivery system. The development of these novel mixed micelles is valuable for resolving the poor solubility and bioavailability of drugs.
Introduction
Magnolol (MO), a lignin compound isolated from the root and stem bark of magnolia officinalis, has attracted much attention worldwide due to its extensive and significant biological activities [Citation1]. Owing to its anti-bacterial [Citation2], anti-inflammatory [Citation3], anti-anxiety [Citation4] and anti-oxidative effects [Citation5], it has wide clinical applications. In addition, compared to conventional anticancer drugs, the anti-cancer mechanism of MO is multi-targeted. For example, MO has been shown to inhibit DNA synthesis and activate apoptosis to suppress human colon and liver cancer cells [Citation6]. It also inhibits the growth of gallbladder cancer cells through the p53 pathway [Citation7]. Furthermore, MO induces H460 cell death via autophagy [Citation8]. Therefore, MO is a promising and potential anti-cancer drug; however, low solubility and bioavailability limit its applications. Therefore, it is essential to develop a novel delivery system (DDS) that can improve the solubility of MO.
A lot of formulation technologies such as solid dispersions [Citation9–11], nanoparticles [Citation12,Citation13] and liposomes [Citation14,Citation15] have been improved the oral bioavailability of hydrophobic drugs. The use of micelles as a carrier for oral drug administration to overcome the low solubility and bioavailability of hydrophobic drugs has recently received significant attention among many preparation techniques. Amphiphilic polymers can be self-assembled in aqueous medium to form a unique, stable and core–shell structure that can significantly improve the solubility of hydrophobic drugs [Citation16,Citation17]. The mixed micelles offer an excellent nano-drug carrier system due to its ability to enhance drug solubility, control drug release to avoid drug inactivation in the biological environment and allow specific targeting [Citation18], thereby enhancing drug bioavailability [Citation19].
With the goal of increasing solubility and oral bioavailability of MO, we aimed to establish two novel nano DDS by using SOL + HS15 and SOL + TPGS (). SOL, HS15 and TPGS have been reported to be drug solubilizers and absorption and permeation enhancers in solid dispersions, micelles and liposomes [Citation20–23]. They contain different proportions of PEG as hydrophilic moieties, and the structures of lipophilic groups vary. SOL can augment the solubility of MO, whereas HS15 and TPGS can enhance the stability of micelles. SOL, an amphiphilic nonionic surfactant composed of polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymers, has been used to enhance solubility of drugs [Citation24,Citation25]. Yang et al. have shown that SOL can be used as a stabilizer in the preparation of nanosuspensions to improve the bioavailability of fenofibrate [Citation26]. Another study used SOL to enhance the dissolution of Celecoxib [Citation27]. HS-15, a non-ionic surfactant consisting 70% polyglycol mono and diesters of 12-hydroxystearic acid and 30% free polyethylene glycol could enhance the solubility of poor water-soluble drugs [Citation28]. TPGS is a water-soluble derivative of natural vitamin E, and is composed of PEG and tocopherol succinate to form an amphiphilic structure. Additionally, it is very stable and does not hydrolyze under normal conditions [Citation29,Citation30]. As a result, it has been used as a component in mixed micelles to increase micelle stability, achieve controlled drug delivery and increase drug encapsulation efficiency [Citation31]. In addition, HS-15 and TPGS are effective p-gp inhibitors, which can enhance adsorption to induce significant effects on drug pharmacokinetics, and show no human toxicity [Citation32,Citation33].
Figure 1. Chemical structure and structural representation of MO-M.
In this study, two novel mixed micelle systems were prepared by the organic solvent evaporation method using various surfactants (HS15, SOL and TPGS) in order to enhance the aqueous solubility, permeability and oral bioavailability of MO. One micelle system (MO-H) was prepared by SOL and HS15, whereas the other (MO-T) was prepared by SOL and TPGS. To evaluate the drug delivery systems, we estimated drug content, storage and dilution stability, and drug release in vitro. The absorption and efflux characteristics of binary mixed micelles were evaluated in Caco-2 cell monolayers. Finally, oral bioavailability was assessed in vivo through pharmacokinetic studies.
Materials and methods
Materials and reagents
MO (purity >98%) and honokiol (purity >98%) were used as an internal standard, and were purchased from Meilun Biological Technology Co., Ltd (Dalian, China). HS15 was purchased from BASF Ltd. (Shanghai, China). SOL was purchased from BASF Ltd. (Shanghai, China), and TPGS was purchased from Aladdin Industrial Co. Ltd. (Shanghai, China). The Caco-2 cell lines were purchased from The American Type Culture Collection (Manassas, VA). Dulbecco’s modified Eagle medium (DMEM) was purchased from Thermo Fisher Scientific (Bridgewater, NJ). Milli-Q water (Millipore, Bedford, MA) was used throughout the study. Chromatographic grade methanol and acetonitrile (Tedia Company Inc., Fairfield, CT) were used for high performance liquid chromatography (HPLC) analysis. All other reagents were of analytical grade.
Animals
Male Sprague–Dawley rats (200 ± 20 g) were purchased from the SLAC Lab Animal Center of Shanghai (Shanghai, China). All rats were provided distilled water ad libitum. Animals were housed under a temperature of 25 ± 0.5 °C and relative humidity of 45 ± 5% for two weeks. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the Jiangsu Provincial Academy of Chinese Medicine. Prior to the experiment, animals were fasted for 12 h, and were only provided water.
Preparation of MO-H and MO-T
MO-loaded binary mixed micelles (MO-M) were prepared by an organic solvent evaporation method [Citation34]. MO and excipients were co-dispersed in anhydrous ethanol and stirred until completely dissolved. The transparent solution formed was transferred to a round bottom flask and concentrated under reduced pressure (50 rpm, 50 °C) to remove the ethanol using a rotary evaporator (IKA®RV10, Germany). After the film was formed completely in the flask, deionized water was added to the solution. The solution was then completely wetted by shaking, after which it was allowed to stand for about 12 h in the flask. MO-M was stored at 4 °C for the following experiments.
Characterization of MO-H and MO-T
Particle size, zeta potential and morphology analysis
The particle size and zeta potential of MO-M were measured by dynamic light scattering using a Malvern system (ZEN-3600, Malvern Instruments, Worcestershire, UK). Polydispersity index (PDI) was determined to assess particle size distribution. The morphology of MO-M was observed by transmission electron microscopy (TEM, JEM-200CX, JEOL, Japan) [Citation35].
Drug content and entrapment efficiency
The concentration of MO in mixed micelles was measured by HPLC [Citation36], which consisted of a Waters 2695 separation module, a Waters 2489 UV/Vis detector and a reversed-phase C18 column (250 mm × 4.6 mm × 5 μm) maintained at 30 °C; detection was conducted at 290 nm. The mobile phase was mixture of methanol–water (75:25, v/v), and the flow rate was 1.0 ml/min. The injection volume was 10 µL. The EE% and DL% were determined by the microporous membrane filtration method. When measuring the encapsulation efficiency (EE%) and drug loading (DL%), 1 ml freshly prepared micelle solution was either filtered through a 0.22 μm microporous membrane or unfiltered. Next, methanol was used to dilute the samples to the same concentration, after which the structure of micelles was completely destroyed by ultrasonication. The EE% and DL% were calculated by EquationEquations (1)(1)
(1) and Equation(2)
(2)
(2) respectively [Citation31]. All samples were analyzed three times.
(1)
(1)
(2)
(2)
Dilution stability analysis
MO-M was diluted with Milli-Q water by 1–250-fold at room temperature (15 °C) to evaluate its dilution stability. Changes in particle size and PDI were analyzed by the methods mentioned above; particle size and PDI after each dilution fold were also measured.
In vitro release study
The in vitro release behavior of MO-M was studied using the dialysis method [Citation37]. In short, 1 ml (at dose equivalents of 3.5 mg/ml MO) each of MO-M and equal amount of MO methanol solution was introduced into dialysis membrane bags (molecular weight cut-off of 3500 g/mol; Green Bird Inc., Shanghai, China). The dialysis bags were immersed in 200 ml fresh PBS (pH 1.2 and pH 6.8, 37 °C) with 0.5% (v/v) Tween 80 (Sinopharm Chemical Reagent Co., Ltd) [Citation38]. At predetermined time intervals (0, 1, 2, 4, 6, 8, 10, 12, 24 h), 1 ml of media was collected and replaced with an equal volume of fresh media to maintain constant volume. The sample was centrifuged at 13,620g for 10 min to obtain a supernatant before HPLC analysis. The amount of MO released in the supernatant was determined by HPLC. All assays were performed in triplicates.
Caco-2 cells culture and transport experiment
Caco-2 cell monolayer model
Caco-2 cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS), 1% nonessential amino acid (NEAA), 1% l-glutamine, penicillin (100 U mL−1)/streptomycin (100 μg mL−1); cells were cultured in a 5% CO2 incubator at 37 °C. The culture medium was replaced once every two days, and cells were passaged by trypsin digestion every 5–7 days. Caco-2 cell suspension was grown in the Transwell culture medium with a cell density of 1 × 106 cells/cm2. Monolayer cells were formed for 21 days, and transmembrane resistance was measured by Millicell-ERS (Millipore, Bedford, MA). When transmembrane resistance reached more than 600 Ω cm−2, membranes were used for transmembrane transportation of drugs.
Transport experiment
Following a 21-day culture, Caco-2 cells that met the above requirements were collected, and the old culture medium was slowly discarded before the experiment [Citation39]. Transwell culture plates were washed with 37 °C preheated Hank’s balanced salt solution (HBSS) three times. Cells were then incubated for 30 min, and HBSS was discarded. The transport test was conducted from the apical side (AP, also known as mucosal side) to the basolateral side (BL, also known as the basal surface side). The supply pool, which was composed of 0.5 ml MO or MO-M solution, was added to the AP surface. On the BL surface, 1.5 ml HBSS (pH 7.4) was added to the receiving cell; each concentration group was set up with three parallel holes. Aliquots (200 μL) from the receiver side were taken at 120 min intervals. The experiment was conducted in triplicate. The apparent permeability (Papp) value of the Caco-2 cell monolayer model was calculated using EquationEquation (3)(3)
(3) :
(3)
(3)
where dQ/dt (µmol·L−1·s−1) was the transport rate, A (cm2) was the surface area of the transport film, and C0 (µmol·L−1·cm−3) was the initial concentration of MAG in the donor.
(4)
(4)
where Papp (AP–BL) was absorption permeability, and Papp (BL–AP) was secretory permeability.
Oral bioavailability study
Collection of plasma samples
For the pharmacokinetic study, SD rats were randomly divided into three groups (n = 6). In short, MO-H, MO-T and free MO were orally administered at a dose of 80 mg kg−1. Blood samples were collected into heparinized tubes from the orbital vein at predetermined time points (0.17, 0.33, 0.50, 0.75, 1, 2, 4, 6, 8, 10 and 12 h); blood samples were immediately centrifuged to obtain the supernatant plasma.
Treatment of plasma samples
Plasma (100 μL) and 10 μL of the internal standard solution were vortexed for 15 s. Methanol (500 μL) was then added, and the solution was vortexed for 3 min, followed by centrifugation at 13,620 g and 4 °C for 5 min. The supernatant was then transferred to a clean vial, and was evaporated to dryness under a nitrogen atmosphere. The dried residue was rinsed with 100 μL of methanol and water (1:1), and was centrifuged at 13,620 g for 5 min. After centrifugation, 20 μL of the samples was injected into the HPLC system with the abovementioned chromatographic conditions. The mobile phase was replaced with acetonitrile and 0.2% formic acid (50:50) (v/v). Other chromatographic conditions were unchanged from the previous drug content analyses.
Statistical analysis
All experiments were conducted triplicates, and all values are expressed as mean ± standard deviation (SD). Statistical analysis of significance was performed using the SPSS 19.0 statistical software (SPSS Inc., Chicago, IL). Differences between various groups were evaluated using Student’s t-test, and p < .05 indicated statistical significance.
Results and discussion
Characterization of MO-H and MO-T
Particle size, zeta potential, morphology, DL, and EE of MO-H and MO-T
A range of single factor experiments was performed to investigate the effect of different proportions of SOL, HS15 and TPGS on the preparation of MO-M. After pre-prescription optimization screening, the final ratio of SOL to HS 15 was 40:10 (mg/ml), and the final ratio of SOL to TPGS was 50:6 (mg/ml). The solution had a light blue appearance; the sizes and zeta potentials of MO-H and MO-T are shown in . PDI indicated that particle size distribution fell within a narrow range. The spherical and homogeneous morphology of MO-H and MO-T were confirmed by TEM analysis (). Based on these findings, we predict that the micelle systems composed of SOL and HS15, as well as SOL and TPGS, can effectively encapsulate MO into the core of micelles. The structure formed by amphiphilic block copolymers can self-aggregate, exposing their hydrophilic heads outside and hiding their hydrophobic segments in the interior core region to increase the solubility of MO. The amount of MO entrapped in the mixed micelles was 3.5 mg/ml, which demonstrated that the micelle system could effectively deliver the MO.
Figure 2. TEM micrographs and image of MO-H (A); TEM micrographs and image of MO-T (B); Scale bar = 100 nm. Size distribution of MO-H (C); Size distribution of MO-T (D).
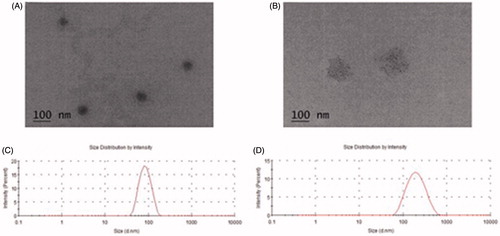
Table 1. Characteristics of MO loaded mixed micelles system.
Dilution stability analysis of MO-H and MO-T
Since colloidals orally administered to the body undergo strong dilution in physiological fluids, a dilution stability test was carried out. The dilution stability analysis of MO-H and MO-T revealed no turbidity. In addition, layer separations were observed, and no significant change in particle size or PDI () was observed.
Table 2. Average size and PDI of MO-M diluted from 1 to 250 folds with Milli-Q water.
In vitro release
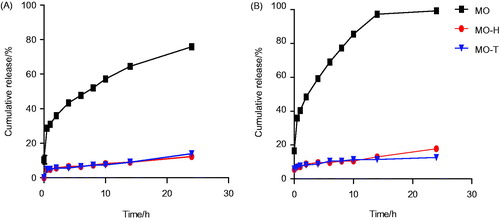
The in vitro release of MO-H and MO-T was investigated. The dialysis method was applied with PBS (pH 1.2 and pH 6.8) containing 0.5% (v/v) Tween 80 at 37 °C to simulate the stomach and the intestinal environments. The amount of cumulative release at set time points was calculated (). The cumulative release of free MO was always greater than that of MO-H and MO-T, both in pH 1.2 and pH 6.8 release media. For instance, in the first 14 h, MO-H, MO-T and free MO showed a release of 12.96%, 11.46% and 97.27% at pH 6.8, respectively. At pH 1.2, similar observation was found. These results suggested that MO-H and MO-T possess significant sustained-release. Based on the experimental data, the cumulative release of MO-T and MO-H in the dialysis medium at pH 6.8 was greater than that at pH 1.2. Therefore, we suspected that the harsh environment within the stomach could break the structure of hydrophilic segments of MO-M, but would not fully destroy the core-shell structure of the MO-M. Ultimately, the drug would be safely delivered to the small intestine, and be absorbed by the human body [Citation40]. From the above experiments, we hypothesized that MO-M is stable in the gastrointestinal tract, and increases the efficiency of gastrointestinal absorption.
Figure 3. Cumulative release of MO-M in vitro release study with phosphate buffered saline pH 1.2 (A) and pH 6.8 (B). Date are presented as mean ± SD (n = 3).
Transport experiment across caco-2 cell monolayers
In this study, Caco-2 cell monolayers were used to investigate the transport of MO-H, MO-T and MO through the intestinal epithelial barrier. As shown in , for AP-BL transport, the Papp value of MO was 2.25 ± 0.21 × 10−7 cm s−1. MO-T and MO-H significantly (MO-T, p < .01; MO-H, p < .05) increased the absorption of MO (4.16 ± 0.36 × 10−7 cm s−1 and 3.52 ± 0.44 × 10−7 cm s−1, respectively). This may primarily be due to the characteristics of SOL and TPGS, which affect cellular functions. BL-AP transport was analyzed to investigate secretory permeability. BL-AP permeability of MO-T and MO-H was 2.95 ± 0.27 × 10−7 cm/s and 1.71 ± 0.23 × 10−7 cm/s, respectively. The Papp value of MO-M was also greater than that of free MO. MO-T induced greater increase in drug transport across the Caco-2 cell monolayers than that induced by MO-H. This result may be attributed to the high percentage of SOL and TPGS in MO-T, as these solubilizers not only increase aqueous solubility but also permeation of MO [Citation41,Citation42]. These results indicate that MO-T and MO-H can be used as a novel delivery system to deliver MO through the gastrointestinal tract into the blood.
Table 3. Permeability and efflux ratio of MO-H, MO-T and MO in Caco-2 cell model.
Pharmacokinetic studies
The pharmacokinetic parameters of MO and its formulation are shown in ; shows the plasma drug concentration–time curve in rats following oral administration of 80 mg/kg of MO-H, MO-T and MO. Significant differences in pharmacokinetic parameters were observed between the MO and MO-H or MO-T (p < .01 or p < .05). The average maximum concentration (Cmax) of MO-H, MO-T and free MO was 0.837 ± 0.050, 0.918 ± 0.040 and 0.397 ± 0.045 µg/L, respectively. This result indicates that formation of micelles markedly increased Cmax of MO in the blood. The average maximum time (Tmax) after oral administration of MO, MO-H and MO-T were 0.917 ± 0.129, 0.708 ± 0.188 and 0.750 ± 0.158 h, respectively. Tmax values showed no obvious differences between the MO and MO-M groups, whereas their half-life (t1/2) values were significantly different (p < .01). Although both MO-T and MO-H improved the systemic performance of MO, they were markedly different. For example, the relative oral bioavailability of MO-T and MO-H increased by 2.98-fold and 2.39-fold, respectively, compared to that of raw MO. Furthermore, MO-M can prolong drug retention in the gastrointestinal tract and increase passive drug absorption due to their high adhesion to the intestinal wall. We concluded that one reason for the improved bioavailability of free MO in mixed micelle system is the increased solubility of MO. In addition, in vivo bioavailability experiments further confirmed that the sustained-release behavior of micelles increases retention time of the drug, and further enhances intracellular uptake by enterocytes. Our findings indicate that both MO-T and MO-H could significantly increase the bioavailability of MO.
Figure 4. The plasma drug concentration–time curve in rats after oral administration of 80 mg/kg of MO-H, MO-T and MO. Date are presented as mean ± SD (n = 6).
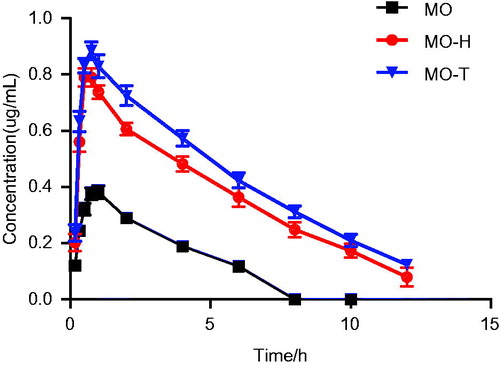
Table 4. Pharmacokinetic parameters of MO-H, MO-T and MO.
Conclusion
An oral drug delivery system based on a binary mixed micelle system was developed to enhance the solubility and oral absorption of MO. In this study, an organic solvent evaporation method was used to prepare MO-M, which exhibited high drug loading and entrapment efficiency. MO was encapsulated in spherical micelles, and demonstrated sustained release behavior. MO-H and MO-T significantly enhanced permeability of MO across the Caco-2 cell monolayer, resulting in increased bioavailability of MO. Therefore, we conclude that using MO-H and MO-T as a drug delivery system is a good strategy for overcoming the poor solubility and bioavailability of MO.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Yang C, Zhi X, Xu H. Advances on semisynthesis, total synthesis, and structure-activity relationships of honokiol and magnolol derivatives. Mini Rev Med Chem. 2016;16:404–426.
- Ho KY, Tsai CC, Chen CP, et al. Antimicrobial activity of honokiol and magnolol isolated from Magnolia officinalis. Phytother Res. 2001;15:139–141.
- Kou DQ, Jiang YL, Qin JH, et al. Magnolol attenuates the inflammation and apoptosis through the activation of SIRT1 in experimental stroke rats. Pharmacol Rep. 2017;69:642–647.
- Alexeev M, Grosenbaugh DK, Mott DD, et al. The natural products magnolol and honokiol are positive allosteric modulators of both synaptic and extra-synaptic GABA(A) receptors. Neuropharmacology. 2012;62:2507–2514.
- Dong L, Zhou S, Yang X, et al. Magnolol protects against oxidative stress-mediated neural cell damage by modulating mitochondrial dysfunction and PI3K/Akt signaling. J Mol Neurosci. 2013;50:469–481.
- Lin SY, Liu JD, Chang HC, et al. Magnolol suppresses proliferation of cultured human colon and liver cancer cells by inhibiting DNA synthesis and activating apoptosis. J Cell Biochem. 2002;84:532–544.
- Li M, Zhang F, Wang X, et al. Magnolol inhibits growth of gallbladder cancer cells through the p53 pathway. Cancer Sci. 2015;106:1341–1350.
- Li HB, Yi X, Gao JM, et al. Magnolol-induced H460 cells death via autophagy but not apoptosis. Arch Pharm Res. 2007;30:1566–1574.
- Wang W, Cui C, Li M, et al. Study of a novel disintegrable oleanolic acid-polyvinylpolypyrrolidone solid dispersion. Drug Dev Ind Pharm. 2017;43:1178–1185.
- Zhang Q, Polyakov NE, Chistyachenko YS, et al. Preparation of curcumin self-micelle solid dispersion with enhanced bioavailability and cytotoxic activity by mechanochemistry. Drug Deliv. 2018;25:198–209.
- Swain RP, Subudhi BB. Effect of semicrystalline polymers on self-emulsifying solid dispersions of nateglinide: in vitro and in vivo evaluation. Drug Dev Ind Pharm. 2018;44:56–65.
- Zhang ZH, Wang XP, Ayman WY, et al. Studies on lactoferrin nanoparticles of gambogic acid for oral delivery. Drug Deliv. 2013;20:86–93.
- Hallan SS, Kaur P, Kaur V, et al. Lipid polymer hybrid as emerging tool in nanocarriers for oral drug delivery. Artif Cells Nanomed Biotechnol. 2016;44:334–349.
- Panahi Y, Farshbaf M, Mohammadhosseini M, et al. Recent advances on liposomal nanoparticles: synthesis, characterization and biomedical applications. Artif Cells Nanomed Biotechnol. 2017;45:788–799.
- Deng J, Zhang Z, Liu C, et al. The studies of N-octyl-N-arginine-chitosan coated liposome as an oral delivery system of cyclosporine A. J Pharm Pharmacol. 2015;67:1363–1370.
- Chen G, Jaskula-Sztul R, Harrison A, et al. KE108-conjugated unimolecular micelles loaded with a novel HDAC inhibitor thailandepsin-A for targeted neuroendocrine cancer therapy. Biomaterials. 2016;97:22–33.
- Tang L, Fu L, Zhu Z, et al. Modified mixed nanomicelles with collagen peptides enhanced oral absorption of cucurbitacin B: preparation and evaluation. Drug Deliv. 2018;25:862–871.
- Jin X, Zhang Y, Zhang Z, et al. Juglone loaded poloxamer 188/phospholipid mixed micelles evaluated in vitro and in vivo in breast cancer. Int J Pharm. 2016;515:359–366.
- Mazzaferro S, Bouchemal K, Ponchel G. Oral delivery of anticancer drugs III: Formulation using drug delivery systems. Drug Discov Today. 2013;18:99–104.
- Shubber S, Vllasaliu D, Rauch C, et al. Mechanism of mucosal permeability enhancement of CriticalSorb(R) (Solutol(R) HS15) investigated in vitro in cell cultures. Pharm Res. 2015;32:516–527.
- Xia D, Yu H, Tao J, et al. Supersaturated polymeric micelles for oral cyclosporine a delivery: the role of Soluplus-sodium dodecyl sulfate complex. Colloids Surf B Biointerfaces. 2016;141:301–310.
- Fan J, Dai Y, Shen H, et al. Application of soluplus to improve the flowability and dissolution of baicalein phospholipid complex. Molecules. 2017;22:pii: E776. doi: 10.3390/molecules22050776.
- Choudhury H, Gorain B, Pandey M, et al. Recent advances in TPGS-based nanoparticles of docetaxel for improved chemotherapy. Int J Pharm. 2017;529:506–522.
- Jin X, Zhou B, Xue L, et al. Soluplus(®) micelles as a potential drug delivery system for reversal of resistant tumor. Biomed Pharmacother. 2015;69:388–395.
- Ju RJ, Mu LM, Li XT, et al. Development of functional docetaxel nanomicelles for treatment of brain glioma. Artif Cells Nanomed Biotechnol. 2018;46:1–11.
- Yang H, Teng F, Wang P, et al. Investigation of a nanosuspension stabilized by Soluplus(R) to improve bioavailability. Int J Pharm. 2014;477:88–95.
- Homayouni A, Sadeghi F, Varshosaz J, et al. Promising dissolution enhancement effect of soluplus on crystallized celecoxib obtained through antisolvent precipitation and high pressure homogenization techniques. Colloids Surf B Biointerfaces. 2014;122:591–600.
- Murgia S, Fadda P, Colafemmina G, et al. Characterization of the Solutol(R) HS15/water phase diagram and the impact of the Delta9-tetrahydrocannabinol solubilization. J Colloid Interface Sci. 2013;390:129–136.
- Guo Y, Luo J, Tan S, et al. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49:175–186.
- Muddineti OS, Vanaparthi A, Rompicharla S, et al. Cholesterol and vitamin E-conjugated PEGylated polymeric micelles for efficient delivery and enhanced anticancer activity of curcumin: evaluation in 2D monolayers and 3D spheroids. Artif Cells Nanomed Biotechnol. 2018;46:1–14.
- Huang S, Yu X, Yang L, et al. The efficacy of nimodipine drug delivery using mPEG-PLA micelles and mPEG-PLA/TPGS mixed micelles. Eur J Pharm Sci. 2014;63:187–198.
- Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm Res. 1999;16:1550–1556.
- Singh MS, Lamprecht A. P-glycoprotein inhibition of drug resistant cell lines by nanoparticles. Drug Dev Ind Pharm. 2016;42:325–331.
- Pawar A, Singh S, Rajalakshmi S, et al. Development of fisetin-loaded folate functionalized pluronic micelles for breast cancer targeting. Artif Cells Nanomed Biotechnol. 2018;46:1–15.
- Manaia EB, Abucafy MP, Chiari-Andreo BG, et al. Physicochemical characterization of drug nanocarriers. Int J Nanomedicine. 2017;12:4991–5011.
- Zhang Q, Hong B, Liu J, et al. Multiwalled-carbon-nanotubes-based matrix solid-phase dispersion extraction coupled with high-performance liquid chromatography for the determination of honokiol and magnolol in Magnoliae Cortex. J Sep Sci. 2014;37:1330–1336.
- Dai Y, Wang S, Shi W, et al. PH-responsive carboxymethyl chitosan-derived micelles as apatinib carriers for effective anti-angiogenesis activity: preparation and in vitro evaluation. Carbohydr Polym. 2017;176:107–116.
- Zhang Z, Cui C, Wei F, et al. Improved solubility and oral bioavailability of apigenin via Soluplus/Pluronic F127 binary mixed micelles system. Drug Dev Ind Pharm. 2017;43:1276–1282.
- Zhang Z, Lv H, Jia X, et al. Influence of vitamin E tocopherol polyethylene glycol succinate 1000 on intestinal absorption of icariside II. Pharmazie. 2012;67:59–62.
- Jiang L, Li X, Liu L, et al. Thiolated chitosan-modified PLA-PCL-TPGS nanoparticles for oral chemotherapy of lung cancer. Nanoscale Res Lett. 2013;8:66.
- Zhang Z, Chen Y, Deng J, et al. Solid dispersion of berberine-phospholipid complex/TPGS 1000/SiO(2): preparation, characterization and in vivo studies. Int J Pharm. 2014;465:306–316.
- Sharma S, Sahni JK, Ali J, et al. Effect of high-pressure homogenization on formulation of TPGS loaded nanoemulsion of rutin – pharmacodynamic and antioxidant studies. Drug Deliv. 2015;22:541–551.