
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
New integrin-targeted nanoparticles made of chitosan-stabilized PLGA matrix was developed to specifically target colon adenocarcinoma. To this aim, SN38-encapsulated chitosan-coated PLGA NPs were conjugated with tetrac for integrin receptor-guided delivery. To provide a sustained release pattern for SN38, it was loaded into nanoparticles using single emulsion method. The size of NPs were 174.23 ± 6.12 nm with drug encapsulation efficiency and loading content of 73.16 ± 11.15 and 4.45 ± 0.31, respectively. The in vitro results confirmed that the designed nanoplatform showed specific cellular uptake and cytotoxicity in integrin overexpressing cancer cells and provided a sustained release profile for SN38. Additionally, an increased therapeutic potency of targeted formulation over both non-targeted and free drug was shown in vivo.
Introduction
Colorectal cancer is the third most diagnosed cancer in both men and women worldwide and it is estimated that 135,430 new cases be diagnosed in 2017 in USA. Also, it was reported that about 50,260 people would die from colorectal cancer in 2017 [Citation1]. Irinotecan, 5- fluorouracil, oxaliplatin and capecitabine are the chemotherapeutics used to fight against colorectal cancer. However, most anti-neoplastic agents are non-selective and possess serious adverse effects.
One of the attractive approaches to minimize the accumulation of chemotherapeutics in non-target organs is to benefit from targeted delivery systems using homing agents such as folic acid [Citation2,Citation3], transferrin [Citation4], small chemical molecules [Citation5], aptamers [Citation6] and peptides [Citation7]. Despite many promising findings reporting the efficient uptake and activity of simple targeted delivery systems, the extravasation of targeted agents from tumour vasculature to the parenchyma is still the rate-limiting stage for specific drug delivery [Citation8].
One of the emerging targets for drug delivery is the ɑvβ3-integrin expressed on the surface of endothelial cells and vascular smooth muscles which stimulation by thyroid hormones leading to induction of proliferation of tumour cells and angiogenesis [Citation9].
Tetrac (tetraiodothyroacetic acid) is a deaminated thyroid hormone analogue which specifically binds to ɑvβ3 integrin overexpressed on tumour endothelial cells and inhibits the binding of its agonists which are thyroid hormones [Citation10]. It was demonstrated that l-thyroxine (T4) induces the proliferation of cancer cells by activation of MAPK (ERK1,2) signalling pathway. Also, it was claimed that T4 is a proangiogenic factor by releasing FGF (fibroblast growth factor) through binding to ɑvβ3 integrin leading to angiogenesis [Citation11]. It was shown that tetrac downregulates the vascular supply to human tumour tissue by blocking the binding of T4 to ɑvβ3 integrin or angiogenesis induced by vascular endothelial growth factor (VEGF) [Citation12]. Such observations proved the fact that tetrac possesses particular anti-angiogenesis properties.
SN38 (7-ethyl-10-hydroxy camptothecin) is an antineoplastic agent belonging to camptothecin (CPT) family which is a highly potent topoisomerase 1 inhibitor [Citation13] which disrupts the structure and function of DNA [Citation14] . Previous studies demonstrated that SN38 is the active metabolite of irinotecan (CPT-11) and it is 100–1000 folds more potent than CPT-11 [Citation15–17]. So that liver activation is not required, thereby interpatient variability is eliminated [Citation18]. Despite the promising chemotherapeutic effects of SN38, its clinical application is relatively hindered by its very low solubility in pharmaceutically accepted media and little stability in physiological pH [Citation16,Citation19]. To overcome such limitations, studies are moved towards attachment of SN38 to hydrophilic polymeric moieties such as EZN-2208 which is PEGylated SN38 in phase 2 clinical trials [Citation20] or its loading into nanoparticles such as liposomes [Citation21], polymeric micelles (NK012 in phase 1 clinical studies) [Citation22] or carbon nanotubes [Citation23]. Such strategies could preserve the stability of the drug moiety while prolonging its blood circulation to enhance the tumour accumulation [Citation15]. In the present study, we loaded SN38 into a poly (lactide-co-glycolide) (PLGA) matrix which its surface was coated with chitosan. Previous studies demonstrated that such polymers preserve the drug moieties from enzymatic degradation and provide physicochemical stability [Citation24]. PLGA is a synthetic polymer which is FDA approved for use in biomedical devices [Citation25]. In various studies, it was also used as a carrier for cytotoxic drugs [Citation24,Citation26,Citation27]. Mehdizadeh et al. [Citation28] designed a biotin-decorated PLGA nanoparticles encapsulating SN38. However, the main challenges of PLGA-based formulations is the burst release of the cargo. To overcome the limitation, we coated the particle surface with chitosan as previously suggested by Chen et al. [Citation29]. Chitosan is a natural polysaccharide polymer [Citation30] extensively studied in drug delivery applications [Citation25–27]. In addition to provide a sustained release pattern while coated onto the surface of PLGA nanoparticles, its positive charge enhances the cellular uptake of the drug-loaded particles according to the negative charge of the cell membrane [Citation31].
Herein, with the aim of enhancing the therapeutic efficiency of SN38 and designing a tumour vasculature targeted delivery system, SN38 was loaded into PLGA nanoparticles coated with chitosan. Thereafter, the nanoparticle was conjugated to tetrac as the targeting moiety. After modifying the physicochemical properties of the designed platform, its capability in targeting ɑvβ3 integrin thereby facilitating cellular uptake and cytotoxicity in vitro and in vivo were investigated.
Materials and methods
Materials
SN38 was procured from AdooQ Bioscience Co., Ltd. (Irvine, CA). A low molecular weight chitosan, poly(lactic-co-glycolic acid) (PLGA) (MW ∼17,000 Da; lactic acid: glycolic acid =50:50), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysulphosuccinimide (NHS) and 3,3′,5,5′-tetraiodothyroacetic acid (tetrac), were purchased from Sigma-Aldrich (Schnelldorf, Germany). Dulbecco’s Modified Eagle’s Medium (DMEM), Roswell Park Memorial Institute (RPMI-1640) medium, fetal bovine serum (FBS), penicillin–streptomycin, and trypsin were purchased from Gibco (Darmstadt, Germany). Other solvent and chemical reagents were procured from Merck & Co. (Darmstadt, Germany).
Cell lines
Caco-2, C26, HUVEC and CHO cell lines were obtained from the National Cell Bank of Iran at the Pasteur Institute. Caco-2 cells were cultured in DMEM (high glucose) C26, HUVEC and CHO were cultured in RPMI-1640 supplemented with 10% (v/v) FBS and penicillin/streptomycin (100 U/mL, 100 U/mL). All cell lines were incubated at 37 °C in a humidified atmosphere (95%) containing 5% CO2.
Fabrication of tetrac-conjugated chitosan
To fabricate chitosan-conjugated tetrac, epoxy-activated tetrac (10 mg) was dissolved in 5 ml DMSO. Thereafter, chitosan (100 mg) was added to the mixture and stirred for 24 h. To purify the synthesized material, deionized water (dH2O, 10 ml) was added to the mixture and dialyzed (cutoff = 3500 kDa) against dH2O (3 L) for 24 h. The obtained product was freeze dried and stored at −20 °C.
The structure of tetrac chitosan was confirmed by 1H-NMR spectroscopy (VarianINOVA-400 MHz NMR spectrometer) in DMSO-d6.
Preparation and characterization of chitosan-coated PLGA nanoparticles loaded with SN38
To load SN38 into PLGA nanoparticles, emulsification solvent evaporation method (single emulsion method) was used as described previously [Citation1]. Briefly, 20 mg of PLGA/PLGA with SN38 (2 mg) was dissolved in chloroform (1 ml). Then, it was mixed with PVA solution (4 ml, 3% W/V) and emulsified by ultra-sonication (20 KHz, 50% amplitude) for 5 min on ice. Thereafter, an excess of PVA (10 ml, 0.2% w/v) was added to the primary mixture and sonicated for another 10 min (20 KHz, 20% amplitude) to produce a secondary emulsion. Thereafter, the obtained emulsion was stirred at room temperature (RT) to evaporate chloroform.
Then, it was centrifuged at 15,000 rpm for 20 min, washed with deionized water, suspended in NaCl (150 Mm) and stored at 4 °C in ultrapure water.
In the next stage, we coated the plain or drug-loaded PLGA nanoparticles with either chitosan or tet-chitosan. Chitosan/tetrac-chitosan solution (0.5%, 2 ml) was put into a glass tube and stirred using a magnetic stirrer at 300 rpm at RT for 2 h. Then, SN38–PLGA (2.5 mg/mL, 2 ml) was added dropwise to the solution. After 2 h, unreacted chitosan and tetrac-chitosan was removed using centrifugation at 15,000 rpm for at 4 °C 20 min. The obtained pellet was resuspended in NaCl (150 mM).
Calculation of encapsulation efficiency and loading content
To determine the amount of loaded SN-38, DMSO was used to solubilize the loaded SN-38 and its concentration in DMSO was calculated using the established standard curve by UV spectroscopy at 366 nm using a Varian CARY 100 UV/Vis spectrophotometer (Palo Alto, CA). The absorption of blank solution was considered in all measurements.
The drug encapsulation efficiency (EE) and loading content (LC) of the CS-PLGA-SN38 and tet-CS-PLGA-SN38 formulations were calculated implementing EquationEquations (1)(1)
(1) and Equation(2)
(2)
(2) , respectively.
(1)
(1)
(2)
(2)
Characterization of nanoparticles
Particle size and particle size distribution (polydispersity index, PDI) was determined through dynamic light scattering method using a Zetasizer (NANO-ZS, Malvern, UK) at RT.
Field-emission scanning electron microscopy (FE-SEM, Mira III FEG; TESCAN-UK, Ltd., Cambridge, UK) operated at 10 kV was used to determine the morphology of blank tetrac-CS-PLGA NPs and drug-loaded tetrac-CS-PLGA-SN38 NPs. A droplet of nanoparticle suspension (1 mg/ml) was dehydrated on a metal stub and coated through sputtering a thin gold film, then used for imaging.
In vitro release
SN38-loaded tetrac-CS-PLGA/SN38-loaded CS-PLGA (1 ml of each formulation) was put into a dialysis bag (cutoff =3500 Da), then immersed into the phosphate-buffered saline (pH 7.4) comprising 0.1% Tween-80 and placed into the shaker incubator (70 rpm, 37 °C).
Samples (1 ml) were withdrawn at specific time points and replaced with 1 ml of fresh buffer. The amount of released samples was measured spectrophotometrically at 366 nm.
Formulations stability on storage
The lyophilized form of each formulation (SN38-loaded tet-CS-PLGA and SN38-loaded CS-PLGA) was stored at −20, 4 and 25 °C for 6 months. Afterwards, their stability was determined through evaluating the change in particle size distribution and drug encapsulation efficiency of the samples as a function of the storage time.
In vitro cytotoxicity studies
To investigate the cytotoxicity of each formulation (SN38, blank or tetrac-CS-PLGA, SN38-loaded CS-PLGA and SN38-loaded tetrac-CS-PLGA) against Caco-2, C26, HUVEC and CHO cells, MTT assay was used. Firstly, 5000 cells/well were cultured in 96-well plates for 24 h. Then, the cells were treated with different concentrations of each of the above-mentioned formulations for 6 h (37 °C, 5% CO2). Afterwards, the medium was removed, replaced with fresh media and incubated for another 48 h. Thereafter, MTT solution (5 mg/mL in PBS, 20 µL) was added to each well and incubated for 4 h. Finally, MTT solution was aspirated and formazan crystals were dissolved by addition of DMSO (100 µL). The absorbance was measured at a wavelength of 570 nm with a reference wavelength of 630 nm on a microplate reader.
Cellular uptake
The quantitative measurement of cellular uptake of SN38 was performed using spectrofluorometer (Shimadzu Corporation, Kyoto, Japan) at λEx = 379 nm and λEm = 531 nm.
Caco-2, C26 and CHO cells (1 × 106) were cultured in 24-well plates and incubated overnight. Then, they were treated with either CS-PLGA-SN38/tetrac-CS-PLGA-SN38 formulations or free SN38 (2 µg/mL). The cells were incubated for 3 h at 37 °C under 5% CO2.
At the final stage, the cells were washed with cold PBS and lysed through addition of a cocktail of 0.7% NP40; Tris–HCl (pH 7.4); EDTA (70 mM); NaCl (200 nM) on ice for 10 min. After protein quantitation (BCA assay), sample was extracted with acetonitrile and the fluorescence of the supernatants (13,000 rpm; 15 min) was measured.
Chick chorioallantoic membrane model of angiogenesis
Neovascularization was examined as described previously [Citation4]. Ten-day-old chick embryos were incubated at 37 °C with 55% relative humidity. First of all, the shell over the air sac, on the broad side of the meta-ellipsoid shaped egg, was disinfected. In order not to break the shell, a hypodermic needle was used to drill the shell outside the air sac under aseptic conditions. Then, a square window was made and the vascular zone was observed on the chick chorioallantoic membrane (CAM).
FGF2 (1 μg/mL) was dropped on the CAM and used as a proangiogenic factor to produce new blood vessel branches. After 30 min, tetrac or tetrac-CS-PLGA NPs (2 µg) was further added to the FGF2-treated CAMs. PBS-treated CAMs were used as a control group. CAMs were incubated for another 3 days and the progression of angiogenesis was investigated.
In vivo antitumour activity
All animal studies were performed according to the guidelines set by the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences.
BALB/c female mice (17–20 g) bearing C26 tumours were used for all chemotherapy experiments. Cells (3 × 105) per mouse suspended in PBS (pH 7.4) was subcutaneously injected to the right flank of the mice. After 14 days, 0.2 ml of irinotecan (25 mg/kg), CS-PLGA-SN38, tetrac-CS-PLGA-SN38 (equivalent SN38 concentration: 10 mg/kg) and blank tetrac-CS-PLGA (equal mass of carrier in drug-loaded formulation: 160 mg/kg) was injected to mice bearing 30 mm3 tumours (five mice in each group) through a single tail vein injection. NaCl (0.9% w/v) solution was served as a negative control.
The tumour volume was determined using a × b×w/2 formula, where “b” is the smallest diameter, “w” is the height of the tumour and “a” is the largest diameter.
Furthermore, the toxic effects of irinotecan, CS-PLGA-SN38, tetrac-CS-PLGA-SN38 and blank tetrac-CS-PLGA were evaluated through determination of mice body weight and survival rates which were measured for 35 days after the tumour initiation or it was interrupted if one of the following conditions was met: (i) the mouse lost more than 80% of the body weight; (ii) the tumour diameter became larger than 2 cm; (iii) they became ill and unable to feed; or (iv) they were found dead [Citation5,Citation6].
Statistical analysis
One-way analysis of variance (ANOVA) was used for statistical data analysis. p values <.05 was regarded as statistically significant. Results are expressed as mean ± SD.
Results and discussion
Synthesis of tetrac-conjugated chitosan
Epoxy-activated terac was conjugated with chitosan polymer. The tetrac molecules present in the chitosan backbone provided a versatile ligand on the surface of the NPs against integrin receptors.
In 1H-NMR spectrum of tetrac-CS, both chemical shifts corresponding to either chitosan backbone at 2–4 ppm or aromatic protons of tetrac mioeties at 7.2–7.9 ppm appeared (). Obtained results confirm the conjugation of tetrac to the chitosan polymer.
Figure 1. 1H-NMR spectrum of chitosan, tetrac-conjugated chitosan and tetrac in d6-DMSO.
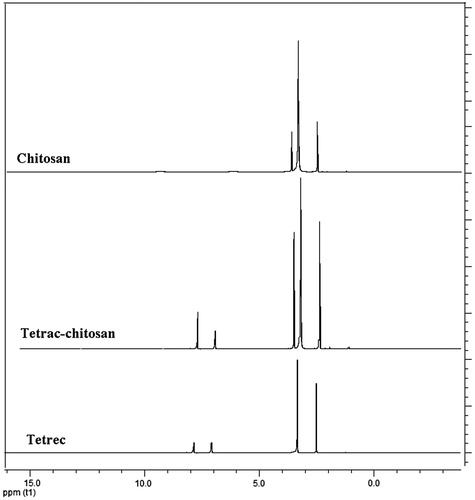
The conjugation yield of tetrac-CS was calculated through integration ratio of the 1H-NMR spectrum peaks of CS (δ 2.3 ppm) and the aromatic rings of tetrac (δ 7.2–7.9 ppm). The total conjugation yield was calculated 81.34 ± 2.14%.
Preparation and characterization of nanoparticles
Preparation of SN38-loaded tetrac-CS-PLGA nanoparticles includes two steps: (i) preparation of SN38-loaded PLGA nanoparticles implementing the emulsion-solvent evaporation method and (ii) coating tetrac-CS through the deposition technique in order to prepare SN38-loaded tet-CS-PLGA NPs.
Poly(lactic-co-glycolic acid) (PLGA) NPs, are biodegradable carriers for encapsulation of therapeutic agents comprising hydrophilic and hydrophobic drugs implementing various encapsulation methods such as solvent evaporation [Citation7].
In this research, SN38 was sufficiently encapsulated in PLGA NPs via single emulsion-solvent evaporation method with encapsulation efficiency and loading content of 75.34 and 9.1%, respectively. PLGA can provide the sustained release of encapsulated drug and sustain its release in vivo over a prolonged period [Citation8]. The negative surface charge of carboxyl-terminated PLGA was used to deposit strongly positive charged CS as a shell onto SN38-encapsulated PLGA core in order to prepare CS-PLGA core-shell NPs. In the aforementioned system, the negative zeta potential of SN38-loaded PLGA NPs becomes positive upon CS deposition on its surface. It should be noted that the negative zeta potential of SN38-loaded PLGA NPs becomes positive upon CS coating. However, the surface positive charge was lower when the NPS were coated with tetrac-CS, which decreased in comparison with CS coating due to the presence of tetrac moieties in chitosan back bone.
Chitosan as a coating polymer contains numerous amino groups for fictionalization with different ligands. In our study as tetrac served as ligand and therapeutic agent, then chitosan provide the ideal opportunity for abundant conjugation of tetrac molecules.
Previously, it was shown that the chitosan coating is a successful approach for preparation of sustained release system of various cargos, increase cellular uptake of PLGA-based NPs and finally enhance therapeutic index [Citation9,Citation10].
One of the important characteristics of nanomedicines is their dimensions. It was demonstrated that the size range of nanoscale cancer therapeutics must be 50–200 nm in order to provide tumour tissue accumulation through enhanced permeation and retention (EPR) effect [Citation11].
As represented in , the average hydrodynamic size of the SN38-loaded PLGA NPs was 143 nm, and the CS or tet-CS modification of SN38-loaded PLGA NPs resulted in 171 and 178 nm particles, respectively.
Table 1. Physicochemical properties of CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs in terms of encapsulation efficiency (EE%), loading content (LC%), size, zeta potential and polydispersity index (PDI) of NPs.
The average increase of 32 nm of CS-PLGA-SN38 and tet-CS-PLGA-SN38 ascribed to chitosan and tetrac-conjugated chitosan coating.
The configuration of CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs and their morphology were also observed by SEM ().
Figure 2. SEM image of CS-PLGA-SN38 NPs (A) and tet-CS-PLGA-SN38 NPs (B).
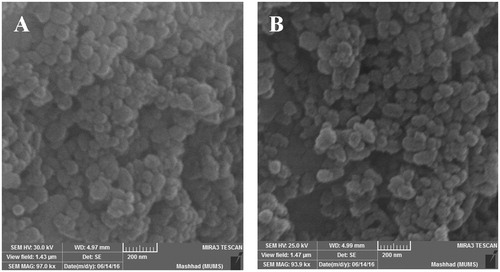
It was demonstrated that the CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs in the form of individual spherical NPs were dispersed with the size of near 100–180 nm. Tetrac attachment to the surface of NPs did not change the morphological properties.
In this system, the negative surface charge of the SN38-loaded PLGA NPs was used to electrostatically deposit CS or tet-CS on to the surface of PLGA NPs.
Therefore, the negative zeta potential of PLGA-SN38 NPs becomes positive upon CS or tet-CS coating, which slightly lower for tet-CS coating.
Numerous studies presented that chitosan coating of PLGA NPs through electrostatic deposition is an effective approach implemented for providing sustained release of drugs and improve cell internalization of PLGA-based nanoparticles while increase therapeutic index of cargo in vivo [Citation12,Citation13].
In vitro release
Biodegradable PLGA-based NPs can encapsulate hydrophobic drug molecules with high efficiency and provide sustained release manner of cargo.
Providing sustained release capability is pivotal for preparation of the versatile therapeutic nanomedicines as it provides constant, continuous transportation of anti-cancer agents to tumour cells.
Previously, it has been reported that continuous low-dose administration of anti-cancer drugs reduced side effects while significantly increase therapeutic index and survival rates.
As demonstrated in , the release profiles of SN38 from CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs were identical.
Figure 3. Release profiles of tet-CS-PLGA-SN38 NPs in PBS pH 7.4 and PBS pH 7.4 containing 30% FBS.
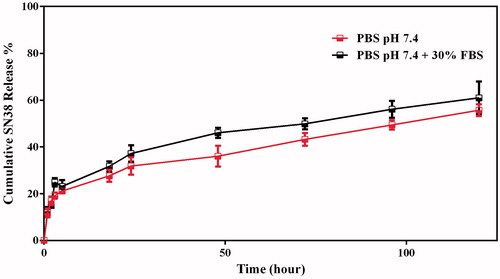
The obtained results also verified that the tetrac moieties on the CS back bone showed no impact on the release rate of the SN38 from CS-coated NPs.
In this regard, both formulations showed 9–13% release during first 6 h suggesting a slight burst release which ascribed to diffusion of the SN38 adsorbed on the NPs surface and to the release of the encapsulated SN38 close to the NPs surface.
The release rate of SN38 from the formulation was then slow down as only 45–58% was released during 96 h.
This kind of release pattern controlled through polymeric matrix erosion. In this regard, hydrophilic nature of chitosan caused water penetration to the NPs which promote degradation of polymeric matrix and loose the NPs integrity and consequently SN38 diffuse form the NPs.
On the other hand, no marked differences in the SN38 release were observed between PBS pH 7.4 and PBS pH 7.4 supplemented with 30% FBS for both CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs. The obtained results exhibited that the developed vehicles provides ideal characteristic for efficient SN38 delivery in vivo.
It should be noted that the release pattern of SN38 from tet-CS-PLGA-SN38 and CS-PLGA-SN38 NPs was identical and no significant difference was observed (data not shown).
Preparation and characterization of nanoparticles
The prepared formulations CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs freeze dried and formed a dried spongy powder without implementing cyoprotectant.
The freeze dried formulations rehydrated easily with deionized water under mild shaking in 2 min. The shelf lives of the formulations in the form of dried powder were evaluated at 4 and 25 °C up to 6 months.
illustrates the results of the stability studies in 4 and 25 °C for CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 freeze-dried NPs.
Figure 4. Stability of lyophilized CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs in terms of size (A) and encapsulation efficiency (B) after 6 months of storage at 25 and 4 °C (mean ± SD, n = 3).
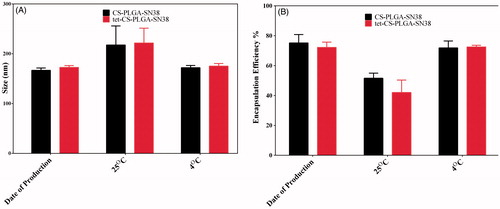
At day zero, the average size of NPs and encapsulation efficiency were 174.23 ± 6.12 and 73.16 ± 11.15 nm, respectively.
The obtained results demonstrated that the no considerable change in size and encapsulation efficiency was observed during 6 month storage at 4 °C while a significant increase in NPs size and decrease in encapsulation efficiency was detected for both formulations stored at room temperature.
Cellular uptake
The cellular uptake of free SN38, CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 is represented in in terms of intracellular concentration of SN38 which was assayed by fluorimetry in Caco-2, C26, HUVEC and CHO cells.
Figure 5. In vitro cellular uptake experiments of (A) Caco-2, (B) C26, (C) HUVEC and (D) CHO cell lines after 3 h exposure to free SN38, CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs (mean ± SD; n = 3). p > .5 is considered as non-significant (ns), *p < .05, **p < .01, ***p < .001.
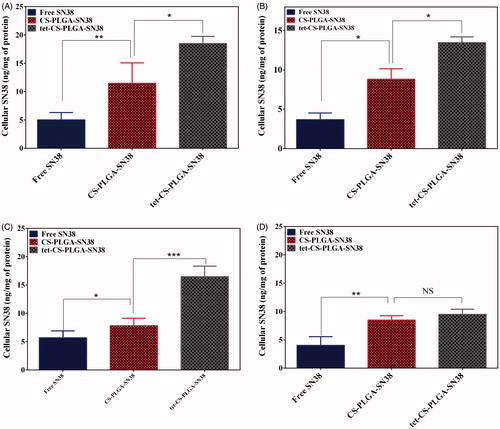
The obtained results demonstrated that cellular uptake of tet-CS-PLGA-SN38 was higher than that of CS-PLGA-SN38 NPs, as well as SN38, in Caco-2, C26 and HUVEC cell lines.
Nevertheless, there was no significant difference in uptake of CS-PLGA-SN38 NPs compared to tet-CS-PLGA-SN38 in CHO cells which is integrin-negative cell line.
Obtained results clearly demonstrate the higher uptake of tet-CS-PLGA-SN38 by Caco-2, C26 and HUVEC cell lines is related to integrin receptor-mediated endocytosis.
Many studies have been evaluated the integrin receptor-mediated uptake of chemotherapeutics. In this regard, we previously confirmed the higher uptake of tetrac-conjugated campthotecin-loaded polymersomes in comparison with non-targeted polymersomes in HT29 and C26 cells. We verified that using tetrac ligand and targeting integrin increases camtothecin uptake by the target cells that overexpress integrin protein on their surfaces [Citation4]. Moreover, the enhanced cellular uptake level of tetrac-conjugated formulation may be attributed to the reduced exocytosis of tet-CS-PLGA-SN38 formulation in comparison with CS-PLGA-SN38 NPs [Citation15].
On the other hand, the lower cellular uptake of SN38 in comparison with that of CS-PLGA-SN38 NPs was mainly due to the strong absorption of positively charged NPs on the cell surface which increases the drug concentration next to the cell membrane, thus a concentration gradient was formed that provides the drug influx into the cell [Citation16]. Moreover, free SN38 molecules which passively diffused into the cytoplasm, were transported out by P-glycoprotein (P-gp) pumps. Then NPs which are taken up by cells via endocytosis pathway could escape from P-gp pumps and consequently resulted in a higher SN38 accumulation in the cell cytoplasm [Citation20].
In vitro toxicity assay
In order to evaluate the cellular toxicity of free SN38, CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 f formulations, MTT assay was performed for 48 h post-incubation in Caco-2, C26, HUVEC and CHO cell lines.
clearly demonstrated the efficacy of the targeted tet-CS-PLGA-SN38 formulation.
Figure 6. Cytotoxicity of free SN38, CS-PLGA-SN38 NPs and tet-CS-PLGA-SN38 NPs on the Caco-2, C26 and CHO cell lines (mean ± SD, n = 4). p > .5 is considered as non-significant (ns), *p < .05, **p < .01, ***p < .001.
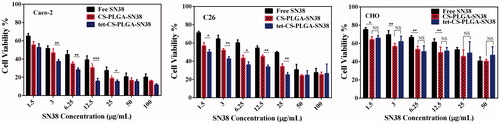
CS-PLGA-SN38 at SN38 concentrations of 4.46 and 3.29 µg/ml exhibited ∼50% cell survival in Caco-2 and C26, respectively, whereas at the same concentration, free SN38 showed 58% and 65% cell survival in Caco-2 and C26 cell lines, respectively. As shown in , the lower cellular uptake of free SN38 in comparison with CS-PLGA-SN38 is largely attributed to the enhanced endocytosis of the positively charged NPs.
Whereas free SN38 showed comparable toxicity in comparison with CS-PLGA-SN38 which was expected, as the SN38 release from nanoparticles is slow and controlled. tet-CS-PLGA-SN38 at SN38 concentrations of 0.95 µg/ml (for the Caco-2 cell line) and 1.61 µg/ml (for the C26 cell line) showed strong cytotoxicity, since only ∼50% cell survival was evident. In this system, tetrac moieties augmented the cytotoxicity of nanoparticles into integrin receptor-overexpressing cell lines (Caco-2 and C26) which is in agreement with cellular uptake experiment. Furthermore, the tet-CS-PLGA-SN38 significantly enhanced the cytotoxicity to the Caco-2 and C26 tumour cells, while cell toxicity against integrin-negative cell line (CHO cell line) resulted in unchanged cytotoxicity of the targeted formulation compared to non-targeted one.
The overall increase in cell toxicity of tet-CS-PLGA-SN38 for integrin-overexpressed cell lines demonstrates that the accumulation of SN38 is enhanced with integrin-targeted capability of tetrac-conjugated formulation compared to non-targeted delivery, resulting in corresponding toxicity levels.
We also performed the MTT experiments for blank CS-PLGA and tet-CS-PLGA NPs (equal amount of carrier which used for MTT experiments of SN38-loaded formulations).
Interestingly, we found that the conjugation of tetrac to CS-PLGA provide the antiproliferative effect even in the low-dose (0.5 µM) tetrac (∼13% inhibition) (Supplementary Figure S1).
Then, the terac moieties act as an antiproliferative agents and also targeting ligand which significantly enhanced toxicity of the tet-CS-PLGA-SN38 NPs in comparison with CS-PLGA-SN38 NPs.
Obtained results are in agreement with previous studies which reported the antiproliferative action of tetrac against breast adenocarcinoma [Citation21].
It was found that tetrac can disrupt numerous functions or events which are vital to cancer cells through the known thyroid hormone–tetrac receptor on the plasma membrane integrin αvβ3 [Citation22]. These functions include regulation of cell division, local stimulation of angiogenesis, chemo-resistance and resistance to radiation [Citation23]. So that it is favourable to produce nanocarriers decorated with tetrac as a ligand to target the cell surface integrin.
In vivo efficacy using the CAM model of angiogenesis
The in vivo efficacy of tet-CS-PLGA NPs was evaluated implementing the CAM model of angiogenesis. demonstrated CAMs treated with CS-PLGA NPs, free tetrac or tet-CS-PLGA NPs.
Figure 7. The anti-angiogenesis effects of tet-CS-PLGA NPs, CS-PLGA-NPs and free tetrac on FGF2-induced angiogenesis evaluated implementing the chick chorioallantoic membrane model.
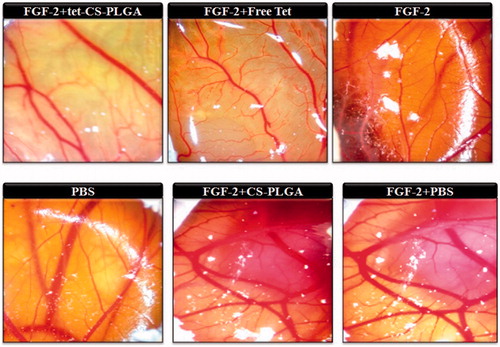
In this regard, CAMs were treated with FGF2 or PBS as a positive and a negative control. On the other hand, all treated CS-PLGA NPs, free tetrac or tet-CS-PLGA NPs CAMs model pretreated with FGF2 in order to evaluate their antiangiogenesis effects.
Obtained results illustrated that exposure of CAM to FGF2 resulted in a 2.4-fold increase in the number of vessel branch compared with PBS-treated CAM. The application of free tetrac resulted in ∼31% inhibition of FGF2-induced angiogenesis (). Interestingly, the equivalent concentration of tetrac in the form of tet-CS-PLGA NPs resulted in full inhibition of FGF2-induced angiogenesis while CS-PLGA NPs did not show any angiogenesis inhibition effect.
In the current study, it was verified that tet-CS-PLGA NPs even in low concentration of tetrac showed antiangiogenesis effect in CAM model and also antiproliferative effect in MTT assay in vitro.
The tetrac receptor is placed at the recognition site of the Arg-Gly-Asp (RGD) on integrin ɑvβ3 membrane protein, which is pivotal for the integrin interaction with growth factors and also extracellular matrix proteins [Citation31].
Previously it was reported that the activity of bFGF and VEGF as angiogenesis activator agents are blocked using tetrac [Citation25].
It was also demonstrated that tetrac disrupt a number of crucial functions in cancer cells comprising regulation of cell division, angiogenesis and chemo-resistance [Citation24].
In vivo anti-tumour effect
The chemotherapeutic efficacy of tet-CS-PLGA-SN38 (two times in week for 4 weeks, equivalent SN38 concentration of 10 mg/kg), CS-PLGA-SN38 (two times in week for 4 weeks, equivalent SN38 concentration of 10 mg/kg), blank tet-CS-PLGA and camptosar (two times in week for 4 weeks, equivalent irinotecan concentration of 25 mg/kg) was investigated implementing subcutaneous allograft models of C26 mice colon adenocarcinoma. Administration of CS-PLGA NPs and saline was served as control of the in vivo therapeutic experiments.
In our study, equivalent concentration of tetrac (1 mg/kg, two times in week for 4 weeks) in form of blank tet-CS-PLGA or tet-CS-PLGA-SN38 was administered in order to investigate the therapeutic potency of tetrac moieties in the prepared targeted nanoparticulate system.
After tumours reached to volume of 20 mm3, the therapeutic index of the tet-CS-PLGA-SN38, CS-PLGA-SN38 and camptosar (irinotecan) formulations and also blank tet-CS-PLGA/CS-PLGA NPs were investigated in the tumour-bearing mice.
illustrated the tumour growth rate in terms of tumour volume in all treatment groups. Our results showed that intravenous injection of either tet-CS-PLGA-SN38 or CS-PLGA-SN38 significantly exhibited more efficacy tumour growth inhibition compared with camptosar or control group. The aforementioned observation is probably because of the EPR effect.
Figure 8. Therapeutic efficacy of camptosar, CS-PLGA-SN38 NPs, tet-CS-PLGA-SN38 NPs, tet-CS-PLGA NPs and CS-PLGA-NPs in C26-tumour-bearing mice. BALB/c mice (five per group) bearing C26 tumours (20–30 mm3) received (IV) injections of CS-PLGA-SN38 NPs or tet-CS-PLGA-SN38 NPs (two times in week for 4 weeks, equivalent SN38 concentration of 10 mg/kg), Camptosar (two times in week for 4 weeks, equivalent irinotecan concentration of 25 mg/kg). Equivalent concentration of tetrac (1 mg/kg, two times in week for 4 weeks) in form of blank tet-CS-PLGA or tet-CS-PLGA-SN38 was administered in order to investigate the therapeutic potency of tetrac moieties in the prepared targeted nanoparticulate system. Control group received single IV injection of saline (NaCl 0.9%) (mean ± SD; n = 5).
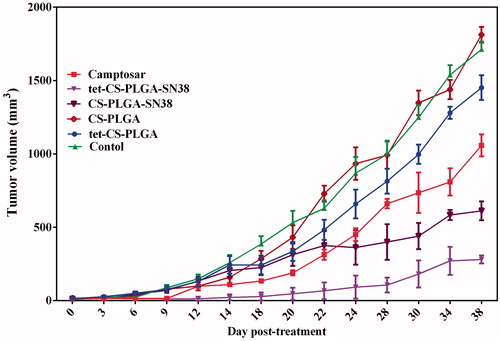
The higher tumour inhibitory effect of tet-CS-PLGA-SN38 compared with CS-PLGA-SN38 might ascribe to binding of tetrac moieties to the integrin receptor on the C26 cell surfaces which increase tumour residence time of the targeted formulation and hindering its extravasation from tumour tissue via fenestrated blood vessels.
It could be concluded that tet-CS-PLGA-SN38 were accumulated in tumour site and entered in to the tumour cells via receptor-mediated endocytosis upon binding to integrin receptor with high affinity.
Then, higher cellular transportation of SN38 implementing the tet-CS-PLGA-SN38 might enhance cellular toxicity and consequently decrease tumour growth rate while increasing therapeutic index in this treatment group.
While blank tet-CS-PLGA-SN38 was slightly effective in reducing tumour growth rate compared to CS-PLGA-SN38 or saline-treated groups, no significant therapeutic efficiency was observed for camptosar even in higher dose.
In another word, due to the MDR mechanism in tumour tissue after continuous exposure to anti-cancer drug then administration of nanoparticulate systems could act more efficiently compared to the free drug [Citation26].
The antitumour effect of blank tet-CS-PLGA NPs was marginal, whereas the therapeutic efficacy of tet-CS-PLGA-SN38 NPs was notably higher which demonstrated the crucial role of encapsulated SN38.
In this regard, Bharali et al. [Citation21] confirmed that the intravenous administration of the terac-conjugated PLGA daily at dose of 1 mg/kg for 16 days resulted in significant reduction in tumour volume of drug-resistant breast cancer orthotropic tumour model in nude mouse.
In another study, Sudha et al. [Citation27] demonstrated that the daily subcutaneous administration of the tetrac-conjugated NPs (1 mg/kg equivalent tetrac) up to 10 days decreased 50% of tumour xenograft weight in comparison with untreated control group (p < .01).
Then, it should be considered that numerous factors including route of administration and regimen schedule of tetrac-conjugated NPs have crucial effect on therapeutic outcome.
Survival rates of C26-tumour-bearing mice treated with tet-CS-PLGA-SN38, CS-PLGA-SN38, blank tet-CS-PLGA/CS-PLGA NPs and camptosar during 50-day was represented in .
Figure 9. Survival rates of the BALB/c mice (five per group) bearing C26 tumours (20–30 mm3) received (IV) injections of CS-PLGA-SN38 NPs or tet-CS-PLGA-SN38 NPs (two times in week for 4 weeks, equivalent SN38 concentration of 10 mg/kg), Camptosar (two times in week for 4 weeks, equivalent irinotecan concentration of 25 mg/kg). Equivalent concentration of tetrac (1 mg/kg, two times in week for 4 weeks) in form of blank tet-CS-PLGA or tet-CS-PLGA-SN38 was also administered.
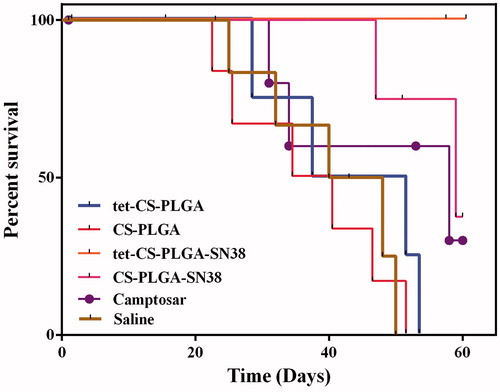
All mice which administrated with tet-CS-PLGA NPs were died after 53 days while four mice out of five mice that treated with tet-CS-PLGA died after 60 days.
Three mice out of five that received CS-PLGA-SN38 remained alive until Day 60. Meanwhile, all animals in the tet-CS-PLGA-SN38 NPs-treated group remained alive until Day 60.
On the other hand, all mice in saline-administered groups died after 50 days.
The difference in survival rate for the camptosar-treated group compared with either tet-CS-PLGA-SN38 or CS-PLGA-SN38-treated groups was statistically significant (p < .05) due to the lower systematic toxicity of nanoparticulate form of SN38 and also superior pharmacokinetic of nanoscale formulation.
In contrast, there was no statistically difference in survival rates between the camptosar, saline and blank tet-CS-PLGA/CS-PLGA treated groups.
Administration of tet-CS-PLGA-SN38 NPs (two times in week for 4 weeks, 10 mg/kg) demonstrated highest therapeutic efficacy in terms of tumour inhibition potency and survival rate compared to other treatments groups (p < .05).
In our experiment, CS-PLGA-SN38 NPs exhibited superior antineoplastic efficacy in comparison with camptosar while its therapeutic index was significantly less efficacious than tet-CS-PLGA-SN38 NPs. The obtained results revealed that after two times in week for 4 weeks intravenous injection of 10 mg/kg tet-CS-PLGA-SN38 NPs was the most potent treatment for suppressing C26 tumour growth in mice.
The cytotoxicity of all treated groups was evaluated based on body weight loss (BWL). Obtained results demonstrated, 30 days after treatment no remarkable BWL detected in any of the treatment groups (). However, slight BWL was observed in camptosar-treated group which is was owing to systematic toxicity of irinotecan as a routine chemotherapeutic agent [Citation29].
Figure 10. Body weight (g) of the BALB/c mice (five per group) bearing C26 tumours (20–30 mm3) received (IV) injections of CS-PLGA-SN38 NPs or tet-CS-PLGA-SN38 NPs (two times in week for 4 weeks, equivalent SN38 concentration of 10 mg/kg), Camptosar (two times in week for 4 weeks, equivalent irinotecan concentration of 25 mg/kg). Equivalent concentration of tetrac (1 mg/kg, two times in week for 4 weeks) in form of blank tet-CS-PLGA or tet-CS-PLGA-SN38 was also administered.
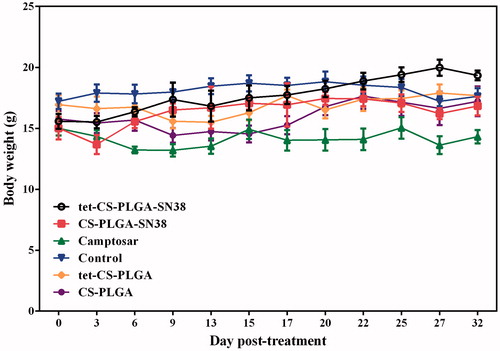
All evidence confirmed the ideal antineoplastic and reduced toxicity of the developed targeted SN38-encapsulated tet-CS-PLGA NPs.
Conclusion
In this study, we prepared a targeted nanoscale delivery system based on PLGA and chitosan, decorated with tetrac molecules for integrin-targeted transportation of SN38 to colon adenocarcinoma. Compared to CS-PLGA-SN38 NPs, tet-CS-PLGA-SN38 NPs increased cellular uptake and cytotoxicity against integrin overexpressed cell lines comprising Caco-2 and C26 while in CHO cell line with negligible integrin expression no apparent enhancement in toxicity was observed. The present study also demonstrated that tetrac-CS-PLGA is a potential delivery system, which had an anti-angiogenesis effect in CAMs model. In C26 tumour-bearing mice, tetrac-CS-PLGA-SN38 NPs demonstrated ideal tumour targeting and it notably enhanced therapeutic index of SN38 and CS-PLGA-SN38 NPs on inhibition of tumour growth while significantly improved survival rate of mice. Taken together, tetrac-CS-PLGA platform could provide the versatile vehicle for the targeted delivery of chemotherapeutics to integrin-overexpressed cancer cells.
Supplemental Material
Download JPEG Image (1.2 MB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Alibolandi M, Rezvani R, Farzad SA, et al. Tetrac-conjugated polymersomes for integrin-targeted delivery of camptothecin to colon adenocarcinoma in vitro and in vivo. Int J Pharm. 2017;532:581–594.
- Li Y, Song L, Lin J, et al. Programmed nanococktail based on pH-responsive function switch for self-synergistic tumor-targeting therapy. ACS Appl Mater Interfaces. 2017;9:39127–39142.
- Li Y, Wu H, Yang X, et al. Mitomycin C-soybean phosphatidylcholine complex-loaded self-assembled PEG-lipid-PLA hybrid nanoparticles for targeted drug delivery and dual-controlled drug release. Mol Pharm. 2014;11:2915–2927.
- Davis FB, Mousa SA, O’Connor L, et al. Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circulat Res. 2004;94:1500–1506.
- Alibolandi M, Abnous K, Mohammadi M, et al. Extensive preclinical investigation of polymersomal formulation of doxorubicin versus Doxil-mimic formulation. J Control Release. 2017;264:228–236.
- Alibolandi M, Taghdisi SM, Ramezani P, et al. Smart AS1411-aptamer conjugated pegylated PAMAM dendrimer for the superior delivery of camptothecin to colon adenocarcinoma in vitro and in vivo. Int J Pharm. 2017;19:352–364.
- Brandhonneur N, Hatahet T, Amela-Cortes M, et al. Molybdenum cluster loaded PLGA nanoparticles: an innovative theranostic approach for the treatment of ovarian cancer. Eur J Pharm Biopharm. 2018;125:95–105.
- Li X, Jiang X. Microfluidics for producing poly (lactic-co-glycolic acid)-based pharmaceutical nanoparticles. Adv Drug Deliv Rev. 2017;pii:S0169-409X(17)30318-6.
- Taghavi S, Ramezani M, Alibolandi M, et al. Chitosan-modified PLGA nanoparticles tagged with 5TR1 aptamer for in vivo tumor-targeted drug delivery. Cancer Lett. 2017;400:1–8.
- Babu A, Amreddy N, Muralidharan R, et al. Chemodrug delivery using integrin-targeted PLGA-chitosan nanoparticle for lung cancer therapy. Sci Rep. 2017;7:14674.
- Alibolandi M, Abnous K, Hadizadeh F, et al. Dextran-poly lactide-co-glycolide polymersomes decorated with folate-antennae for targeted delivery of docetaxel to breast adenocarcinima in vitro and in vivo. J Control Release. 2016;241:45–56.
- Park J, Pei Y, Hyun H, et al. Small molecule delivery to solid tumors with chitosan-coated PLGA particles: a lesson learned from comparative imaging. J Control Release. 2017;268:407–415.
- Kang BS, Choi JS, Lee SE, et al. Enhancing the in vitro anticancer activity of albendazole incorporated into chitosan-coated PLGA nanoparticles. Carbohydr Polym. 2017;159:39–47.
- Li Y, Lin J, Ma J, et al. Methotrexate–camptothecin prodrug nanoassemblies as a versatile nanoplatform for biomodal imaging-guided self-active targeted and synergistic chemotherapy. ACS Appl Mater Interfaces. 2017;9:34650–34665.
- Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm. 2005;2:373–383.
- Liang XJ, Chen C, Zhao Y, et al. Circumventing tumor resistance to chemotherapy by nanotechnology. Methods Mol Biol. 2010;596:467–488.
- Alibolandi M, Abnous K, Anvari S, et al. CD133-targeted delivery of self-assembled PEGylated carboxymethylcellulose-SN38 nanoparticles to colorectal cancer. Artif Cells Nanomed Biotechnol. 2018[Mar 8]; [1–11]. DOI:10.1080/21691401.2018.1446969
- Prasad S, Dangi JS. Development and characterization of pH responsive polymeric nanoparticles of SN-38 for colon cancer. Artif Cells Nanomed Biotechnol. 2016;44:1824–1834.
- Einafshar E, Asl AH, Nia AH, et al. New cyclodextrin-based nanocarriers for drug delivery and phototherapy using an irinotecan metabolite. Carbohydr Polym. 2018;194:103–110.
- Kibria G, Hatakeyama H, Harashima H. Cancer multidrug resistance: mechanisms involved and strategies for circumvention using a drug delivery system. Arch Pharm Res. 2014;37:4–15.
- Bharali DJ, Yalcin M, Davis PJ, et al. Tetraiodothyroacetic acid-conjugated PLGA nanoparticles: a nanomedicine approach to treat drug-resistant breast cancer. Nanomedicine. 2013;8:1943–1954.
- Davis PJ, Glinsky GV, Lin HY, et al. Cancer cell gene expression modulated from plasma membrane integrin alphavbeta3 by thyroid hormone and nanoparticulate tetrac. Front Endocrinol. 2014;5:240.
- Sudha T, Bharali DJ, Sell S, et al. Nanoparticulate tetrac inhibits growth and vascularity of glioblastoma xenografts. Horm Canc. 2017;8:157–165.
- Davis PJ, Hercbergs A, Luidens MK, et al. Recurrence of differentiated thyroid carcinoma during full TSH suppression: is the tumor now thyroid hormone dependent? Horm Canc. 2015;6:7–12.
- Yoshida T, Gong J, Xu Z, et al. Inhibition of pathological retinal angiogenesis by the integrin alphavbeta3 antagonist tetraiodothyroacetic acid (tetrac). Exp Eye Res. 2012;94:41–48.
- Alibolandi M, Ramezani M, Abnous K, et al. In vitro and in vivo evaluation of therapy targeting epithelial-cell adhesion-molecule aptamers for non-small cell lung cancer. J Control Release. 2015;209:88–100.
- Sudha T, Bharali DJ, Yalcin M, et al. Targeted delivery of paclitaxel and doxorubicin to cancer xenografts via the nanoparticle of nano-diamino-tetrac. Int J Nanomed. 2017;12:1305–1315.
- Mehdizadeh M, Rouhani H, Sepehri N, et al. Biotin decorated PLGA nanoparticles containing SN-38 designed for cancer therapy. Artif Cells Nanomed Biotechnol. 2017;45:495–504.
- Li F, Jiang T, Li Q, et al. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am J Cancer Res 2017;7:2350–2394.
- Dhanapal J, Balaraman Ravindrran M. Chitosan/poly (lactic acid)-coated piceatannol nanoparticles exert an in vitro apoptosis activity on liver, lung and breast cancer cell lines. Artif Cells Nanomed Biotechnol. 2018;3:1–9.
- Davis PJ, Incerpi S, Lin H-Y, et al. Thyroid hormone and P-glycoprotein in tumor cells. BioMed Res Int. 2015;2015:168427.