
Abstract
Exosomes are endogenous nanocarriers that can deliver biological information between cells. They are secreted by all cell types, including immune cells such as natural killer (NK) cells. However, mammalian cells release low quantities of exosomes, and the purification of exosomes is difficult. Here, nanovesicles were developed by extrusion of NK cells through filters with progressively smaller pore sizes to obtain exosome mimetics (NK-EM). The anti-tumour effect of the NK-EM was confirmed in vitro and in vivo. The morphological features of the NK-EM were revealed by transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA) and Western blot. In vitro, the cytotoxicity of the NK-EM to cancer cells (glioblastoma, breast carcinoma, anaplastic thyroid cancer and hepatic carcinoma) was assessed using bioluminescence imaging (BLI) and CCK-8 assay. For in vivo study, a xenograft glioblastoma mouse model was established. The anti-tumour activity of NK-EM was confirmed in vivo by the significant decreases of BLI, size and weight (all p < .001) of the tumour compared with the control group. Moreover, NK-EM cytotoxicity for glioblastoma cells that related with decreased levels of the cell survival markers p-ERK and p-AKT, and increased levels of apoptosis protein markers cleaved-caspase 3, cytochrome-c and cleaved-PARP was confirmed. All those results suggest that NK-EM exert stronger killing effects to cancer cells compared with the traditional NK-Exo, at the same time, the tumour targeting ability of the NK-EM was obtained in vivo. Therefore, NK-EM might be a promising immunotherapeutic agent for treatment of cancer.
Introduction
Natural killer (NK) cells are innate immune lymphocytes that destroy infecting or transformed cells without the need for activation, in contrast to T and B cells [Citation1–3]. They have been the focus of more than 150 cellular-based immunotherapy clinical trials as of March 2017 (clinicaltrials.gov). The anti-tumour behaviour of NK cells has been harnessed to treat hematologic malignancies that include acute myeloid leukaemia, acute lymphoblastic leukaemia multiple myelomas [Citation4], solid tumours such as melanoma [Citation5,Citation6], breast cancer [Citation7,Citation8], thyroid cancer [Citation2] and glioblastoma [Citation1,Citation9]. Because of the low number of NK cells in peripheral or cord blood, immune cell therapy with NK cells requires an ex vivo expansion process to achieve relevant numbers that express activation markers, natural cytotoxicity receptors (natural-killer group 2, member D [NKG2D], NKp44, Fas ligand [FasL], etc.) and cytokines (tumour necrosis factor-alpha [TNF-α], granzyme A and B, perforin) [Citation2,Citation4,Citation10].
Exosomes are nanovesicles released by cells. The vesicles can contain molecules and so have the potential of targeted cell delivery of the therapeutic payload [Citation11–13]. This potential has generated intense research interest in the onco-immunology field. Exosomes contain specific proteins depending on the cell of origin. As examples, exosomes from tumour cells contain tumour antigens, platelet-derived exosomes contain coagulation factors, and exosomes from dendritic cells express toll-like receptor ligands [Citation14,Citation15]. Exosome-mediated functions vary depending on the condition or the origin of the cells [Citation16]. However, they share a characteristic composition of the protein and lipids, which include the exosome markers, heat shock 70-Kda protein, CD63 and CD81 [Citation11,Citation14,Citation17,Citation18]. Exosomes function as signalling messengers between the donor cytosol and internal compartments of target cells [Citation19,Citation20]. Tumour exosome crosstalk with immune cells leads to attenuation of the cells by downregulation of their anti-tumour activity [Citation21,Citation22]. Immune cells such as NK cells can release exosomes (NK exosomes, NK-Exo) that express cytotoxic proteins including FasL and perforin [Citation11,Citation13,Citation23] and cytokines including granzyme A, granzyme B and TNF-α [Citation4,Citation11]. NK-Exo display anti-tumour activity.
This benefit of exosomes is tempered by their relatively low levels in mammalian cells, and their difficult and expensive purification [Citation24,Citation25]. In addition, since there is no “gold standard protocol” for preparing pure extracellular vesicles, the characteristics and functions of exosomes could differ according to the preparation methods [Citation16]. Therefore, the generation of exosome-mimetic vesicles (EM) with a substantially greater yield is attractive for the development of future immunotherapies. This is the first study to explore the EM produced with the NK cells (NK-EM) and confirm their anti-tumour ability in vitro and in vivo.
Materials and methods
Cell culture
Cells of the human NK cell line NK92-MI obtained from American Type Culture Collection (ATCC, Rockville, MD) were incubated in stem cell growth medium (Cellgro, Freiburg, Germany) supplemented with 2% exosome-depleted human serum obtained by ultracentrifugation at 120,000 × g for 18 h and 1% penicillin-streptomycin at 37 °C in a 5% CO2 atmosphere. Human glioblastoma (D54), breast carcinoma (MDA-MB-231), anaplastic thyroid cancer (CAL-62) and hepatic carcinoma (HepG2) cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, Logan, UT) supplemented with 10% foetal bovine serum (FBS; Gibco, Carlsbad, CA) and 1% penicillin-streptomycin (Hyclone). Cancer cells were transfected with recombinant retrovirus harbouring a plasmid containing both enhanced firefly luciferase (effluc) and thy1.1 driven by a long terminal repeat (LTR) promoter (Retro-LTR-effluc-thy1.1). Cells expressing both effluc and thy1.1 were sorted by magnetic cell sorting (MACS; Miltenyi Biotec, Auburn, CA) to isolate thy1.1 positive cells. After selecting the positive cells, RT-PCR and Western blot were used to confirm the expression of effluc at the mRNA and protein levels. To confirm the luciferase activity, D54 and D54/F cells were seeded at various cell densities in black and clear wells of 96 well plates. After 24 h incubation, 3 µl (3 mg/ml) of D-luciferin was added to each well and effluc activity was measured using the IVIS Lumina III imaging system (Perkin-Elmer, Wellesley, MA). The established stable cell lines expressing both effluc and thy1.1 gene were designated D54/F, MDA-MB-231/F, CAL-62/F and HepG2/F.
Animals
Specific pathogen-free, 6-week-old, female BALB/c nude mice (Hamamatsu, Shizuoka, Japan) were used in in vivo experiments. All animal protocols were conducted in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and approved by the Committee for the Handing and Use of Animals, Kyungpook National University.
Preparation of NK-exo and NK-EM
The EM were prepared as previously described with some modifications [Citation24,Citation26]. The cell suspensions were sequentially extruded eight times each through 5- and 1-μm polycarbonate membrane filters (Nuclepore; Whatman Inc., Clifton, NJ) using a mini-extruder (Avanti Polar Lipids, Birmingham, AL). The final extruded sample (1 ml) was placed at the top of an ultracentrifuge tube (Beckman Coulter, Brea, CA) overlaying 10% (2 ml) and 60% (2 ml) iodixanol (OptiPrep™, Sigma-Aldrich, St Louis, MO) and ultracentrifuged at 132,000 g for 3 h at 4 °C. NK-EM enriched in the interface between the 10% and 60% iodixanol were recovered, ultracentrifuged at 100,000 g for 1 h at 4 °C, and filtered through a 0.22-μm filter. The pellet was re-suspended in PBS and stored at −80 °C. Each sample was used within one month. The NK-Exo preparation was derived according to a previous protocol [Citation11].
Nanoparticle tracking analysis
The size, distribution and number of particles for exosomal formulations were assessed by nanoparticle tracking analysis using the NanoSight LM10 system equipped with a 405nm laser (Nanosight Ltd., Amesbury, UK) at 25 °C. NK-EMs and NK-Exos were each prepared at a concentration of 0.01 mg/mL and evaluated using an instrument equipped with NTA 2.0 analytical software. Furthermore, to compare NK-Exo and NK-EM productions, NK cells (5 × 106) were used to simultaneously prepare the NK-Exo and NK-EM preparations. The total protein and nanoparticle number were calculated. To measure the nanoparticle number, all experiments were carried out using a 1:1000 dilution, which produced particle concentrations of approximately 10 × 107/ml. Each experiment was carried out in triplicate.
Western blot
Western blot was performed as described previously [Citation2]. Whole cell lysates, NK-Exo and NK-EM were prepared in RIPA buffer (Thermo Fisher Scientific, Waltham, MA). Each lysate (50 µg protein/well) was separated using 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The resolved proteins from each preparation were transferred to a polyvinylidene fluoride membrane (Millipore) and probed sequentially with primary antibodies and horseradish peroxidase-conjugated secondary antibody. Proteins were detected using an enhanced chemiluminescence detection system (GE Healthcare, Milwaukee, WI) according to the manufacturer’s protocol. The antibodies used included CD63, GM130, Alix, cytochrome-c (all from Abcam, Cambridge, UK) and perforin and β-actin (both from Santa Cruz Biotechnology, Santa Cruz, CA). The FasL membrane protein component was harvested using the Mem-PER™ Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific) and detected by Western blot.
Transmission electron microscopy
Purified NK-Exos and NK-EMs were applied to glow-discharged carbon-coated copper grids (Electron Microscopy Sciences, Fort Washington, PA). After 20 min of incubation in a dry environment at room temperature (RT), the material on each grid was fixed using 2% paraformaldehyde (PFA) for 10 min, rinsed with droplets of deionized water, and negatively stained with 1% glutaraldehyde for 15 min to remove the negative background. TEM was done using a HT 7700 transmission electron microscope (Hitachi, Tokyo, Japan) operating at 100 kV.
Cell cytotoxicity assay
To evaluate the cytotoxicity of the NK-Exo and NK-EM preparations to cancer cells, bioluminescence imaging (BLI) and CCK-8 assay (Dojindo Molecular Technologies, Tokyo, Japan) were performed. For BLI, D54/F cells were cultured in serum-free medium containing 10, 20 and 30 µg/mL of NK-EM or NK-Exo concentrations (in 96 well black plates for 24, 48 and 72 h. For the CCK-8 assay, D54/F tumour cells were incubated with NK-Exo and NK-EM in 96-well plates for 24, 48 and 72 h in a NK-EM dose-dependent manner. Ten microliters of CCK-8 solution was added to each well and the plate was incubated in a humidified incubator. The absorbance at 450 nm was measured using a micro-plate reader. MDA-MB-231/F, CAL-62/F and HepG2 cells were co-incubated with NK-EM and NK-Exo for 72 h in a NK-EM dose-dependent manner and the CCK-8 assay was performed.
Cellular uptake assay
The cellular uptake assay was performed using a modified chemical protocol [Citation12]. The fluorescent lipophilic tracer 2–(5-(1,3-dihydro-3, 3-dimethyl-1-octadecyl-2H-indol-2-ylidene)-1,3-pentadienyl)-3, 3-dimethyl-1-octadecyl-perchlorate (DiD; Invitrogen, Carlsbad, CA) was added into NK-Exo and NK-EM suspensions and incubated for 20 min at RT. Each plate was washed three times with PBS to eliminate unlabelled DiD. Thirty micrograms of DiD-labelled NK-Exo and NK-EM re-suspended in PBS were incubated with D54/F cells for 3 h at 37 °C. In one experiment, the chamber wells were washed five times with PBS and the adherent cells were fixed using 4% PFA for 20 min. After several washings using PBS, each sample was covered with the Vectashield mounting medium containing (4',6-diamidino-2-phenylindole, DAPI: Vector Laboratories, Burlingame, CA) and observed using confocal laser
Biodistribution and tumour targeting ability of NK-EM in a mouse model
The xenograft tumour mouse model was established according to a previous protocol [Citation11]. Briefly, 5 × 106 D54/F cells were subcutaneously injected into each mouse, and tumour development then occurred. After 28 days, NK-EM labelled with DiD were injected through the tail vein. Fluorescence imaging (FLI) of NK-EM was examined using an in vivo imaging system. The auto-fluorescence background was subtracted by imaging of control mice not injected with DiD. After 24 h, the organs (spleen, liver, kidney, lung, heart) and tumours of the mice were harvested, and ex vivo FLI was performed and quantified.
In vivo anti-tumour effect
After 10 days following injection of D54/F cells, the mice received 100 µg or 30 µg of the NK-EM suspension in PBS intravenously (I.V.) or intratumorally (I.T.), respectively, three times at intervals of 3 days. The anti-tumour effect of NK-EM was monitored using the BLI assay of the tumour every 2 days until day 14 using ultrasound three-dimensional (3-D) imaging (S-Sharp Corporation, New Taipei City, Taiwan). The mice were sacrificed after the final image acquisition on day 14, and BLI of the ex vivo tumour was obtained using the IVIS system. At the same time, the tumour weight was measured. Moreover, to assess NK-EM cytotoxicity, the body weight was measured during the treatment period.
Anti-tumour mechanism of NK-EM
Apoptosis and the cell survival signalling pathway in the NK-EM-treated cancer cells were assessed using Western blot. In addition, to identify the cell death pathway induced by the presence of FasL, an inhibition assay (AF-016, Kamiya Biomedical Company, Seattle, WA) was performed.
Statistical analyses
All quantitative data are expressed as mean ± standard deviation (SD). Student’s t-test or one-way analysis of variance (one-way ANOVA) followed by Scheffe’s post-hoc test was performed for normally distributed data, and Mann–Whitney test was performed for non-normal data using SPSS version 22.0 (SPSS, Chicago, IL). Statistical significance was set as p < .05.
Results
Production, characterization and cytotoxicity of NK-EM
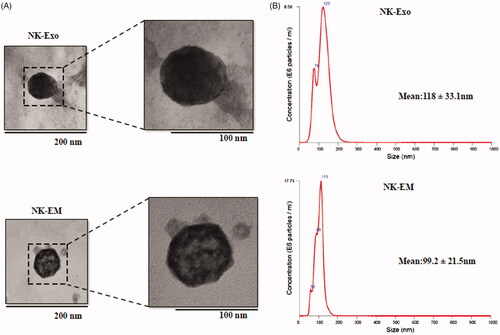
NK cells were extruded through polycarbonate membrane filters and the NK-EM were collected and purified by the density gradient ultracentrifugation (). NK-Exos derived from cultured NK cells were used as the control for the in vitro experiments. Characterization of NK-EM preparations by Western blot confirmed the presence of the typical CD63 and Alix exosomal proteins. However, different from NK-Exos, NK-EM also contained the cellular proteins GM130, cytochrome-c and β-actin) (). Moreover, using the same number of NK cells, we obtained 18.68 μg of NK-EM and 1.36 μg of NK-Exo (). TEM of NK-EM revealed 100–150 nm spherical particles with a complete membrane structure, fitting the recognized characteristics of exosomes (). Nanoparticle tracking analysis of the purified NK-EM and NK-Exo showed a size distribution with a peak diameter of approximately 100–120 nm (NK-EM: 99.2 ± 21.5 nm, NK-Exo: 118 ± 33.1 nm) similar to the TEM results (). Finally, the production of NK-Exo and NK-EM was measured by using NTA. Using 5 × 106 NK cells, 1.86 × 106 particles of NK-Exo were obtained, while 50-fold more NK-EM particles were obtained (9.59 × 107) (). The collective suggested a similar size and morphology of the NK-EM and NK-Exo but differing protein contents. In addition, more NK-EM particles were obtained than NK-Exo particles from the same number of NK cells.
Figure 1. Generation of NK-EM. (A) Schematic illustration of the procedure for the generation of NK-Exo and NK-EM. (B) Western blot analysis of exosomal marker proteins on NK-Exo and NK-EM (50 μg total protein). The yields of NK-Exo and NK-EM measured as the total protein (C) and particle number (D) from 5 × 106 NK cells. Experiments were performed in triplicate and mean ± SD values are plotted; ***p < .001.
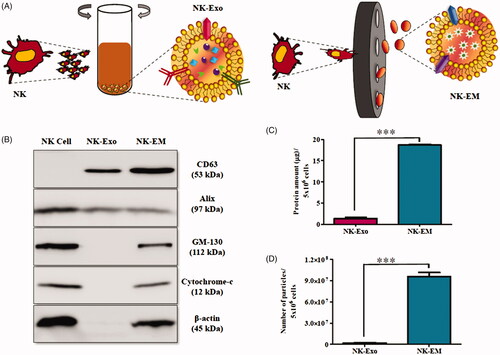
Figure 2. Characterization of NK-Exo and NK-EM. (A) Representative transmission electron micrograph of NK-Exo and NK-EM (scale bars denote 200 and 100 nm, respectively). (B) Size distributions of NK-Exo and NK-EM were measured by nanoparticle tracking analysis.
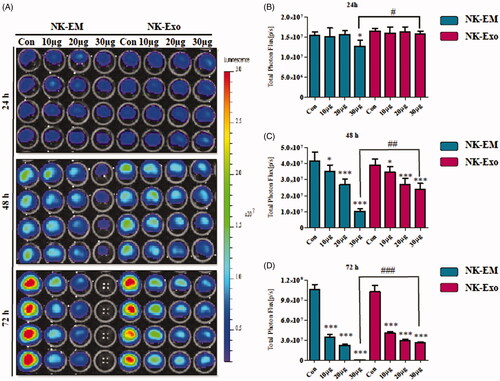
To check the cancer cell cytotoxicity of NK-EM, different concentrations of NK-EM and NK-Exo were cultured with glioblastoma (D54/F), breast carcinoma (MDA-MB-231/F), anaplastic thyroid cancer (CAL-62/F) and hepatic carcinoma (HepG2/F) cells, followed by BLI and CCK-8 assays. Time- and dose-dependent NK-EM cytotoxicity against D54/F cells was evident. The BLI signal strength of the cancer cells decreased with increasing NK-EM concentrations and incubation time (). Moreover, compared with NK-Exo treatment, NK-EM treatment was significantly more cytotoxic to cancer cells in the BLI assay () and CCK-8 assay (48 h: p < .001 at 48 and 72 h; Supplementary Figure 3(A)). The MDAMB-231/F, CAL-62/F and HepG2/F cancer cells also showed a decreased BLI signal intensity after 72 h co-incubation with 10, 20 and 30 µg NK-EM ((all p < .001; Supplementary Figure 2). The CCK-8 assay results (Supplementary Figure 3(B)) were consistent with the BLI results. These results indicated the cytotoxicity of NK-EM to various cancer cells.
Figure 3. In vitro cytotoxicity of NK-Exo against glioblastoma cells. (A) Concentration- and time-dependent cytotoxic effects of NK-EM and NK-Exo on D54/F cells. (B, C and D) Intensity of bioluminescence was quantified using an in vivo imaging system and live imaging software. Experiments were performed at least in triplicate and mean ± SD values are plotted; *p < .05, **p < .01, ***p < .001; ## p < .01, ### p < .001.
NK-EM biodistribution following systemic injection
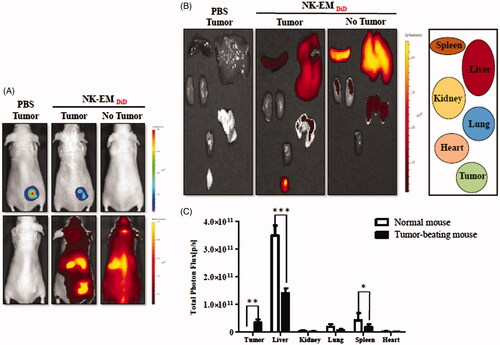
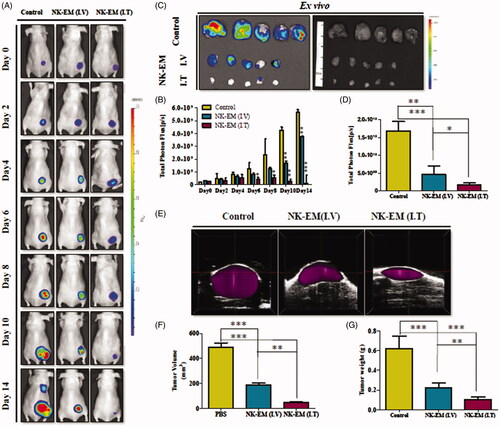
To investigate NK-EM biodistribution, DiD-labelled NK-EMs were I.V. administered to mice harbouring D54/F glioblastoma xenografts (Supplementary Figure 4). Fluorescence imaging revealed a strong signal in the region of the tumour. The distribution of NK-EM in different organs in mice with or without tumours is shown in . Ex vivo fluorescent imaging visualized NK-EM in the spleen, liver, kidney and lung. Mice with tumours showed clear tumour uptake of NK-EM and significantly less uptake of NK-EM in the liver (p < .001) and spleen (p < .05) compared with the control mice. Both the in vivo and ex vivo imaging confirmed the biodistribution of NK-EM and indicated their tumour targeting ability, which is crucial for oncologic application.
Figure 4. In vivo targeting of DiD-labelled NK-EM to tumours in mice. D54/F cells were injected subcutaneously into BALB/c nude mice. Four weeks later, the mice each received an intravenous injection of DiD-labelled NK-EM (100 μg total protein) or PBS. Normal mice without tumours were used as the control. After 24 h, the DiD fluorescence of the whole mouse body (A) or various tissues (B) was revealed in an IVIS spectrum. (C) Radiant efficiency was measured using living image 3.1 software. Data are presented as the mean ± SD values: *p < .05, ***p < .001.
In vivo cytotoxicity of NK-EM for the glioblastoma xenograft assessed by in vivo BLI
A schematic diagram of the in vivo experiments is presented in . Tumour cells were subcutaneously inoculated, and the mice were divided into three groups randomly at day 10 (n = 5 per group). NK-EM were administered at a dose of 100 µg (I.V.) 30 µg (I.T.), with control mice receiving PBS. The intensity of the BLI signal from tumours in the three groups did not show obvious differences until day 4. From day 6, the BLI signal intensity of the NK-EM treatment group (I.T) was significantly lower than that of the control group (p < .001, ). Of note, in the control group, a BLI signal was evident in the lung, which indicated metastasis to the lung that was not observed in the treatment groups. After harvesting the tumours from the mice, ex vivo measurements revealed a lower tumour weight in the NK-EM treatment group compared to the control group (p < .001; ). Ex vivo BLI of the tumours showed similar results to in vivo BLI; the control group exhibited a 4-fold increase in signal intensity compared with the NK-EM treatment group ().
Figure 5. Scheme of the in vivo treatment experiments.
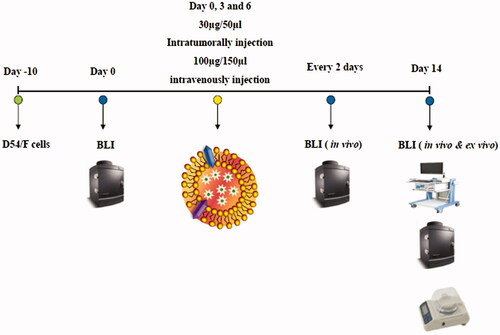
Figure 6. Immunotherapy with NK-EM in vivo. (A) BLI measurements of the D54/F activity in mice were performed in the control and treatment groups. In the treatment group, 100 µg or 30 µg of NK-EM was injected intravenously and intratumorally, respectively, three times every 72 h. (B) Quantitative BLI of D54/F activity. (C) BLI of the ex vivo tumour activity. (D) Quantitative BLI of ex vivo D54/F activity. (E) Ultrasound images were obtained in the B mode, and three-dimensional images were constructed. (F) Tumour volumes and tumour weight (G) were measured. Experiments were performed at least in triplicate and mean ± SD values are plotted; **p < .01 and ***p < .001.
3-D tumour model and in vivo tumour measurement
Ultrasound imaging has provided reliable tumour volume estimates in animal studies [Citation27,Citation28]. To confirm that the BLI results were correlated to tumour size, we also performed ultrasound imaging. In the quantitative analysis, the average tumour volume of the NK-EM treatment group was 189.235 (I.V.) and 45.896 (I.T.) mm3, in contrast to the average volume of 489.256 mm3 in the control group (p < .001, ). These results supported the reliability of the BLI results and demonstrated the in vivo therapeutic effect of NK-EM against glioblastoma cells. The final measurement of ex vivo tumour weight revealed a lower tumour weight in the treatment group (0.220 g by I.V and 0.105 by I.T) compared to the control group (0.621 g) (p < .001, ).
Tumour cell uptake of NK-EMs
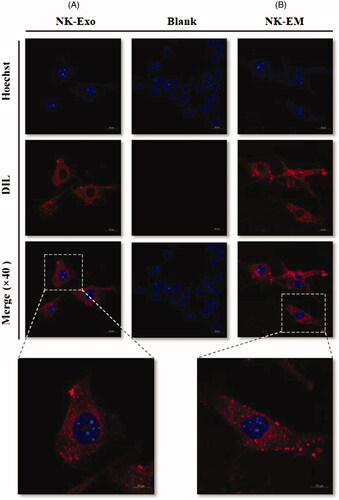
To explore the internalization of NK-EM into cancer cells, D54/F cells were treated with DiD-labelled NK-EM and NK-Exo for 3 h. Confocal laser microscopy imaging detected the labelled NK-EM and NK-Exo (red staining) in the cancer cells (). This result showed that both NK-EM and NK-Exo have the same biological behaviour and can interact with the target cells.
Figure 7. Trafficking of NK-Exo and NK-EM in cancer cells. DiD-labelled NK-Exo (A) and NK-EM (B) were used to treat D54/F cells, and confocal laser microscopy images were obtained. The scale bar denotes 20 and 10 µm, respectively.
Analysis of Fas ligand and perforin expression in NK-EMs
NK cells exert their cytotoxic activity through the release of cytotoxic effectors contained in their lytic granules [Citation10,Citation29]. At the same time, several transmembrane proteins, such as FasL and TRAIL [Citation2,Citation30], are exposed on the cell surface and dictate the fates of effector and target cells. Thus, we first established the presence of FasL and perforin in NK-EM by Western blot analysis. The results revealed the presence of FasL with a typical molecular mass of 42 kDa in NK-EM. Perforin was also detected in the NK-EM lysates (). Furthermore, in the quantitative assay, the levels of these two proteins were higher in NK-EM compared to that in NK cells (< .001). To confirm the membrane expression of FasL, the NK-EM envelope was collected using a membrane protein extraction kit and analyzed by Western blot, which revealed the expression of FasL ().
Figure 8. Identification of apoptosis-inducing proteins in NK-Exo and NK-EM. NK cells, NK-Exo, and NK-EM lysates were loaded into each lane (50 μg total protein; n = 3). (A) The apoptosis-inducing proteins FasL and perforin were detected in the lysates. Since FasL comprised a membrane associated form (FasL) and soluble homotrimeric form (sFasL), the membrane and cytoplasm were measured separately. (B) The abundance of FasL and perforin in the lysates was estimated using EvolutionCapt software. (C) To confirm the function of the FasL, the inhibitor was used and the BLI of the tumour was obtained. (D) CCK-8 assay was also performed as well. Experiments were performed at least in triplicate and values ± SD are plotted; *p < .05.
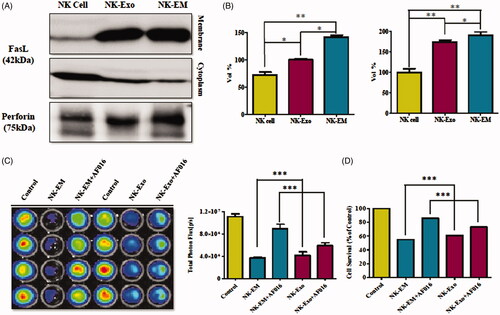
To explore the predominant cell death-mediated pathway triggered by FasL by NK-EM, we assessed the inhibitory effect of a FasL inhibitor (AF-016) on the cytotoxicity of NK-EM using BLI and CCK-8 assays. The intensity of the BLI signal from cancer cells in the absence of AF-016 was significantly lower compared with cells treated with AF-016 (p < .001). However, the FasL inhibitor was not able to completely block the cytotoxic effect of NK-EM (p < .001, ). The CCK-8 assay displayed similar trends to the BLI analysis. These results confirmed that NK-EM-mediated cell death is partly related to the presence of FasL.
Mechanism of the anti-tumour effect of NK-EM on D54/F cells
Previous studies revealed that NK cell-mediated death s is regulated by two major mechanisms: aggregation of target cell death receptors (FasL or tumour necrosis factor-related apoptosis-inducing ligand [TRAIL]-related receptor) and perforin/granzymes [Citation30,Citation31].
The combination of perforin and granzyme leads to the intrinsic apoptosis pathway involving cytochrome-c, while, at the same time, death receptor-mediated apoptosis leads to cell death through the extrinsic apoptosis pathway, resulting in activation of caspase-8, caspase-3 and poly ADP ribose polymerase (PARP) and cell death [Citation4,Citation13,Citation14,Citation23]. To confirm the cell death mechanism, we assessed apoptosis-related proteins by Western blot analysis. As shown in the schematic diagram () of the apoptosis signalling pathway, the levels of caspase-3, cleaved caspase-3, cleaved PARP and cytochrome-c were detected. Cleaved caspase-3, cleaved PARP and cytochrome-c were increased after 24 h of NK-EM treatment (). These results indicated that NK-EM could induce the apoptosis of the cancer cells by both the extrinsic and intrinsic apoptotic pathways, which was consistent with the Western blot results that the NK-EM particles contained the FasL and perforin proteins.
Figure 9. Effect of NK-EM on the apoptosis and cell proliferation pathways. After 24 h of co-incubation of D54/F with NK-EM, proteins were collected from the whole cell lysates and Western blotting was performed. Marker protein in the apoptosis (A) and proliferation (B) signalling pathways were checked. (C) Levels of apoptosis proteins, cleaved-caspase 3, cleaved-PARP, and cytochrome-C, were increased. (D) Coincidentally, levels of cell proliferation proteins p-ERK and p-AKT were decreased after NK-EM treatment.
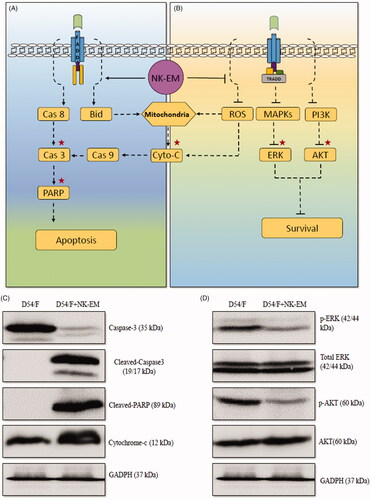
In addition to the apoptosis signalling pathway, the mitogen-activated protein kinase (MAPK) and phosphoinositol-3-kinase (PI3K) signalling pathways related to cell proliferation [Citation32–34] were also examined. The protein levels of p-ERK and p-AKT in cancer cells were decreased after 24 h NK-EM treatment (), which demonstrated that NK-EMs could inhibit the tumour proliferation and exert an anti-tumour effect. The results indicate two functional aspects of the anti-tumour ability of NK-EM. NK-EMs can induce cancer cell apoptosis, which leads to cell death. As well, NK-EM can decrease cancer cell proliferation, which leads to growth inhibition of the tumour.
Discussion
In the current study, the extrusion method was used to efficiently isolate the EM fraction from the Exo fraction from the same number of NK cells. The size distribution, morphology and contents of the NK-EMs were confirmed. The tumoricidal effect of NK-Exo has been proven. This study provides the first evidence of the tumoricidal effect of NK-EM both in vitro with cell cultures and in vivo using a xenograft mouse tumour model. In addition, the in vivo tumour targetability of NK-EM was proven.
Bioengineered biological agents, including cells, provide a novel approach to cancer treatment that differs from chemotherapy or radiotherapy. Compared with other cells, NK cells, which are lymphocytes of the innate immune system, are important for defense against infectious pathogens and various cancers [Citation1,Citation3,Citation16,Citation30], and have additional therapeutic advantages due to their ability to kill the cells they target without prior stimulation [Citation35,Citation36]. However, clinical responses have so far been limited. A potential limiting factor in patients with solid tumours is the defective homing of the infused NK cells to the tumour site [Citation37,Citation38]. As well, some cancers can evade or dampen the cytotoxic effect of NK cells by downregulating the expression of recognition receptors or by actively suppressing or diverting NK cell functions [Citation39]. Exosomes of NK cells (NK-Exo), which can communicate cell-to-cell and cell-to-environment, have attracted attention as a potential therapeutic agent [Citation12,Citation40,Citation41]. Since exosomes have the characteristics of both nano-sized and cell-based delivery systems, they are regarded as promising anti-tumour tools for both endogenous and exogenous cargos. Furthermore, exosomes may retain the natural properties of NK cells by maintaining a topology of plasma membrane proteins [Citation12,Citation24,Citation41,Citation42].
The potential clinical use of extracellular vesicles as a therapeutic agent and drug delivery system [Citation11,Citation12,Citation43–45] has been tempered by major drawbacks and hurdles. The complexity of extracellular vesicles makes their short- and long-term adverse clinical effects difficult to gauge, since harmful and as yet unidentified components could be present. Moreover, since there is currently no “gold standard protocol” for preparation of pure exosome preparations, it is likely that exosomes produced in different facilities using different preparation methods will not have the same characteristics and functions. The main isolation methods for exosomes include ultracentrifugation, buoyant density ultracentrifugation, filtration, use of commercially available agglutinating agents and chromatography-based methods [Citation11,Citation14,Citation46]. These methods differ in their experimental principles. Furthermore, extracellular vesicles derived from biological fluid comprise of a mixture of diverse extracellular vesicles originating from various cell types [Citation17]. This makes it difficult to isolate a signal vesicle population in order to determine the vesicle type that best induces the function of interest. Lastly, the low production yield of natural secreted extracellular vesicles is insufficient for scaled-up production, and remains one of the biggest challenges for their clinical application [Citation24,Citation26,Citation40,Citation43].
The presently reported development of the NK-EM could overcome these aforementioned limitations of exosomes. The NK-EM combines the characteristics of cells and exosomes. The novel NK-EM were prepared by disrupting NK cells through serial extrusion using nano-sized filters with diminishing pore sizes followed by density gradient centrifugation to obtain the NK-EM at the interface between the 10 and 60% iodixanol layers (). NK-EM displayed similar properties to exosomes. The size distribution of the NK-Exo and NK-EM fractions was confirmed by NTA, which revealed a size of approximately 100 nm for both nano-particles (). This size is consistent with that reported by previous studies [Citation12,Citation15,Citation47–49]. TEM also confirmed that the NK-EM are lipid bilayer membrane vesicles with diameters of approximately 100 nm (). The yield of the exosomes-mimetic nanovesicles was more than 50-fold higher than that of exosomes derived from the same number of cultured NK cells (). The collective results indicate the similar physical characteristics of NK-EM and exosomes, with NK-EM capable of being generated in markedly higher amounts through a simple and affordable procedure.
In addition to their higher production, the novel exosome mimetics from NK cells displayed more potent cytotoxicity against various cancer cells compared with NK-Exo, the exosomes directly derived from the culture medium of NK cells ( and Supplementary Figures 2 and 3). In this study, the NK-EM cytotoxicity was superior against four different human cancer cell lines (glioblastoma, breast cancer, anaplastic thyroid cancer and hepatic carcinoma) than a low or high dose of NK-Exo both in the low and high dose. Strong anti-tumour activity of NK-EM might be due to their biological contents inherited from the parental NK cells. Perforin and granzyme B are the core molecules required for NK cell-mediated tumour killing and the death-receptor pathways (involving FasL and TRAIL) are commonly used for the killing [Citation2,Citation36,Citation50]. Perforin triggers the intrinsic pathway and elevates the release of cytochrome-c [Citation36,Citation51]. FasL triggers the extrinsic apoptotic pathway by activating caspase-8, caspase-3 and PARP [Citation31]. In this study, the expression levels of the FasL and perforin in the NK cells and the NK-EM were compared using the Western blot. The protein content was greater in the NK-EM fraction (), which indicates that the NK-EM could be a suitable therapeutic candidate for cancer treatment. Moreover, NK-EM displayed increased contents of cleaved caspase-3, cleaved-PARP and cytochrome-c, with the simultaneous decreases of the cell proliferation markers p-ERK and p-AKT (. The collective results demonstrate that NK-EM could induce cell apoptosis and exert an anti-tumour effect.
Even with the in vitro demonstration of NK-EM-mediated cytotoxicity, the in vivo biodistribution of this potential therapeutic agent to tumours are necessary before their use in cancer treatment can be contemplated. We confirmed the tumour targetability of NK-EM by exploring their biodistribution in a mouse model featuring tumour xenograft. Previous studies have reported the biodistribution of exosomes from different cells in vivo. Wen et al. showed that breast cancer cell-derived exosomes were predominantly distributed in the lungs, which is a frequent site of metastasis [Citation21]. In contrast, dendritic cell-derived exosomes mainly accumulated in the liver. The exosomes from cancer cells, macrophages and even mesenchymal stromal cells [Citation21,Citation26,Citation52–56] have been intensively studied. However, NK cells exosomes have not been fully explored. In this study, the NK-EM were injected through the mouse tail vein and subsequently non-invasively monitored by FLI (. The results are the first to establish the tumour targetability of the NK-EM. The results of this study should be useful as a basis for developing NK cells based on therapeutic strategies. Several studies have proved that NK cells are capable of eliminating tumour in vitro and in vivo conditions, and the result suggested that the tumour targeting dependents on the over-express of NK cell receptor ligands like NKp30 ligands in tumour cells [Citation29,Citation30,Citation57]. Recently, Pitchaimani et al. developed a membrane camouflage fusogenic liposomal delivery system “NKsome” for targeted tumour therapy using NK cell-ghost [Citation58]. NK cells attacking tumour cells are mediated by active receptors including NKG2D, NKp44, NKP46, NKp30 and DNAM-1. The ligands may use as a detector system to target inflamed and malignant cells and play a crucial role in anti-tumour effect [Citation26]. In this study, we also confirmed the FasL expression on the NK-EM witch was consist to the previously study and demonstrated the ability of the tumour homing.
Clinically, the predominant method of NK cell-based immunotherapy is still the transfer of unmodified autologous or allogeneic NK cells [Citation36]. However, the clinical effectiveness of NK cell-based immunotherapy for cancerous diseases is quite limited mainly due to the limited cancer cell targetability of NK cells. For example, the blood-brain barrier is a tightly-regulated interface between the bloodstream and the central nervous system that creates an immune-privileged site that evades immune surveillance [Citation57]. In this regard, extracellular vesicles are the ideal immunotherapy for brain disease, as these vesicles can cross the blood-brain barrier. Exosomes isolated from three brain tumour cell lines (U-87, PFSK-1, A-712 and bEND.3) have been detected in the brains of zebrafish embryos [Citation58]. Thus, exosomes and EM have the advantages of cell-mediated therapeutic activity, while avoiding the inherent limitations associated with cellular therapeutics. The generation of EM is a well-established and relatively simple process that requires only minimal laboratory effort and reagents. In this study, we successfully generated NK-EM using the same simple procedure. The previous finding that injection of nanovesicles produced from both human and mouse cells show similar activities without systemic side effects in an immunocompetent mouse tumour model [Citation26,Citation59] suggests that NK-EM or even non-autologous NK cell-derived EM can be used for cancer treatments.
Various types of nano-sized particles ranging from 10 to 1000 nm in diameter have been fabricated. These include liposomes, nanoparticles and polymeric particles [Citation57]. However, synthetic nanoparticles require complicated and sophisticated chemical crosslinking processes that have a myriad of hurdles, such as various safety issues including immunogenicity concerns [Citation26]. NK-EM might be a potent and useful anti-tumour agent because they have ability to target tumours with functional protein, are highly cytotoxic based on their contents, and are feasible to produce. This is primarily a consequence of the adsorption of plasma proteins to the nanoparticle surface, leading to uptake and activation of immune cells, reduced circulation time and limited tumour uptake or targeting [Citation25,Citation49,Citation59]. In addition, NK cells used to produce EM can be engineered to express tumour targeting molecules. A previous study examined the use of chimeric antigen receptor-modified NK cells to target human epidermal growth factor receptor (EGFR)-positive brain cancer [Citation60]. Another study showed that chimeric antigen receptor-modified NK cells target ERBB2, EGFR, or mutant EGFRvIII, which are expressed in many patients with glioblastoma [Citation61]. Exosomes generated from immature DC cells with rabies virus glycoprotein can bind to nicotinic acetylcholine receptor expressed on the blood-brain barrier to increase the brain targeting ability [Citation62]. Various targeting moieties, including antibodies, have been conjugated onto the surfaces of nanocarriers to improve their targeting efficacy [Citation25,Citation38,Citation49,Citation63]. Furthermore, anti-cancer drugs such as curcumin, doxorubicin and paclitaxel can be spontaneously packaged into exosomes under ambient conditions [Citation53]. Compared to standard liposomes, exosomes exhibit higher loading efficiency and, as the components of the exosome membrane are derived from the donor cell, the particles are non-immunogenic, allowing them to resist fast clearance from circulation and increase the efficiency of drug delivery to target tissues [Citation64]. All these attributes can potentially be incorporated in EM to enable the targeting of tumour cells and could be combined with many other personalized treatments [Citation60,Citation65].
Conclusions
The results of the current study suggest that NK-EM exert stronger killing effects to cancer cells compared with the traditional NK-Exo and are able to target cancers in vivo. NK-EM might be a promising immunotherapeutic agent for cancerous diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Lee SJ, Kang WY, Yoon Y, et al. Nam, natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15:1011.
- Zhu L, Li XJ, Kalimuthu S, et al. Ahn, natural killer cell (NK-92MI)-based therapy for pulmonary metastasis of anaplastic thyroid cancer in a nude mouse model. Front Immunol. 2017;8:824
- Zamai L, Ponti C, Mirandola P, et al. NK cells and cancer. J Immunol. 2007;178:4011–4016.
- Shoae-Hassani A, Hamidieh AA, Behfar M, et al. NK cell-derived exosomes from NK cells previously exposed to neuroblastoma cells augment the antitumor activity of cytokine-activated NK cells. J Immunother. 2017;40:265–276.
- Moore TC, Kumm PM, Brown DM, et al. Interferon response factor 3 is crucial to poly-I:C induced NK cell activity and control of B16 melanoma growth. Cancer Lett. 2014;346:122–128.
- Zhang G, Liu R, Zhu X, et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol Cell Biol. 2013;91:615–624.
- Hwang MH, Li XJ, Kim JE, et al. Potential therapeutic effect of natural killer cells on doxorubicin-resistant breast cancer cells in vitro. PloS One. 2015;10:e0136209.
- Kim HW, Kim JE, Hwang MH, et al. Enhancement of natural killer cell cytotoxicity by sodium/iodide symporter gene-mediated radioiodine pretreatment in breast cancer cells. PloS One. 2013;8:e70194.
- Lee HW, Singh TD, Lee SW, et al. Evaluation of therapeutic effects of natural killer (NK) cell-based immunotherapy in mice using in vivo apoptosis bioimaging with a caspase-3 sensor. FASEB J. 2014;28:2932–2941.
- Michel T, Poli A, Cuapio A, et al. Human CD56 bright NK cells: an update. J Immunol. 2016;196:2923–2931.
- Zhu L, Kalimuthu S, Gangadaran P, et al. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics. 2017;7:2732–2745.
- Kalimuthu S, Gangadaran P, Li XJ, et al. In vivo therapeutic potential of mesenchymal stem cell-derived extracellular vesicles with optical imaging reporter in tumor mice model. Sci Rep. 2016;6:30418.
- Fais S. NK cell-released exosomes: natural nanobullets against tumors. Oncoimmunology. 2013;2:e22337.
- Jong AY, Wu CH, Li J, et al. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracell Vesicles. 2017;6:1294368.
- Sobo-Vujanovic A, Munich S, Vujanovic NL. Dendritic-cell exosomes cross-present Toll-like receptor-ligands and activate bystander dendritic cells. Cell Immunol. 2014;289:119–127.
- Kim OY, Lee J, Gho YS. Extracellular vesicle mimetics: novel alternatives to extracellular vesicle-based theranostics, drug delivery, and vaccines. Semin Cell Dev Biol. 2017;67:74–82.
- Bretz NP, Ridinger J, Rupp AK, et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J Biol Chem. 2013;288:36691–36702.
- Watson DC, Bayik D, Srivatsan A, et al. Efficient production and enhanced tumor delivery of engineered extracellular vesicles. Biomaterials. 2016;105:195–205.
- Munoz JL, Bliss SA, Greco SJ, et al. Delivery of functional anti-miR-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol Ther Nucleic Acids. 2013;2:e126.
- Katakowski M, Buller B, Zheng X, et al. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013;335:201–204.
- Wen SW, Sceneay J, Lima LG, et al. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res. 2016;76:6816–6827.
- Yu DD, Wu Y, Shen HY, et al. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015;106:959–964.
- Lugini L, Cecchetti S, Huber V, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. 2012;189:2833–2842.
- Oh K, Kim SR, Kim DK, et al. In vivo differentiation of therapeutic insulin-producing cells from bone marrow cells via extracellular vesicle-mimetic nanovesicles. ACS Nano. 2015;9:11718–11727.
- Jang SC, Gho YS. Could bioengineered exosome-mimetic nanovesicles be an efficient strategy for the delivery of chemotherapeutics? Nanomedicine (Lond). 2014;9:177–180.
- Jang SC, Kim OY, Yoon CM, et al. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano. 2013;7:7698–7710.
- Cheung AM, Brown AS, Hastie LA, et al. Three-dimensional ultrasound biomicroscopy for xenograft growth analysis. Ultrasound Med Biol. 2005;31:865–870.
- Wirtzfeld LA, Wu G, Bygrave M, et al. A new three-dimensional ultrasound microimaging technology for preclinical studies using a transgenic prostate cancer mouse model. Cancer Res. 2005;65:6337–6345.
- Vivier E, Tomasello E, Baratin M, et al. Functions of natural killer cells. Nat Immunol. 2008;9:503–510.
- Dubois S, Conlon KC, Muller JR, et al. IL15 infusion of cancer patients expands the subpopulation of cytotoxic CD56bright NK cells and increases NK-cell cytokine release capabilities. Cancer Immunol Res. 2017;5:929–938.
- Augstein P, Heinke P, Schober C, et al. Impact of cytokine- and FasL-induced apoptosis in the beta-cell line NIT-1. Horm Metab Res. 2009;41:207–212.
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344.
- Gaestel M. MAPK-activated protein kinases (MKs): novel insights and challenges. Front Cell Dev Biol. 2016;3:88.
- Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83.
- Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91.
- Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–1036.
- Kremer V, Ligtenberg M, Zendehdel R, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunotherapy Cancer. 2017;5:73.
- Roth JC, Curiel DT, Pereboeva L. Cell vehicle targeting strategies. Gene Ther. 2008;15:716–729.
- Lam PY, Nissen MD, Mattarollo SR. Invariant natural killer T cells in immune regulation of blood cancers: harnessing their potential in immunotherapies. Front Immunol. 2017;8:1355.
- Lunavat TR, Jang SC, Nilsson L, et al. RNAi delivery by exosome-mimetic nanovesicles – implications for targeting c-Myc in cancer. Biomaterials. 2016;102:231–238.
- Yuan D, Zhao Y, Banks WA, et al. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials. 2017;142:1–12.
- Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med. 2005;3:10.
- Kim OY, Choi SJ, Jang SC, et al. Bacterial protoplast-derived nanovesicles as vaccine delivery system against bacterial infection. Nano Lett. 2015;15:266–274.
- Gangadaran P, Rajendran RL, Lee HW, et al. Extracellular vesicles from mesenchymal stem cells activates VEGF receptors and accelerates recovery of hindlimb ischemia. J Control Release. 2017;264:112–126.
- Imai T, Takahashi Y, Nishikawa M, et al. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles. 2015;4:26238.
- Lobb RJ, Becker M, Wen SW, et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles. 2015;4:27031.
- Vashisht M, Rani P, Onteru SK, et al. Curcumin encapsulated in milk exosomes resists human digestion and possesses enhanced intestinal permeability in vitro. Appl Biochem Biotechnol. 2017;183:993–1007.
- Pitt JM, Andre F, Amigorena S, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest. 2016;126:1224–1232.
- Gilligan KE, Dwyer RM. Engineering exosomes for cancer therapy. Int J Mol Sci. 2017;18:1122.
- Gangadaran P, Ahn BC. Molecular imaging: a useful tool for the development of natural killer cell-based immunotherapies. Front Immunol. 2017;8:1090.
- Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–747.
- Smyth T, Kullberg M, Malik N, et al. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145–155.
- Munagala R, Aqil F, Jeyabalan J, et al. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016;371:48–61.
- Gangadaran P, Hong CM, Ahn BC. Current perspectives on in vivo noninvasive tracking of extracellular vesicles with molecular imaging. BioMed Res Int. 2017;2017:1.
- Xin H, Li Y, Buller B, et al. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells. 2012;30:1556–1564.
- McDonald MK, Tian Y, Qureshi RA, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain. 2014;155:1527–1539.
- Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30.
- Yang T, Martin P, Fogarty B, et al. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res. 2015;32:2003–2014.
- Tan S, Wu T, Zhang D, et al. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5:863–881.
- Chen X, Han J, Chu J, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7:27764–27777.
- Zhang C, Oberoi P, Oelsner S, et al. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533.
- Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345.
- Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays. 2011;33:737–741.
- Kim SM, Kim HS. Engineering of extracellular vesicles as drug delivery vehicles. Stem Cell Investig. 2017;4:74.
- Muller N, Michen S, Tietze S, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother. 2015;38:197–210.