
Abstract
Adipose tissue (AT) homeostasis and expansion are dependent on complex crosstalk between resident adipose stromal/stem cells (ASCs) and AT extracellular matrix (ECM). Although adipose tissue ECM (atECM) is one of the key players in the stem cell niche, data on bidirectional interaction of ASCs and atECM are still scarce. Here, we investigated how atECM guides ASCs’ differentiation. atECM altered shape and cytoskeleton organization of ASCs without changing their proliferation, β-galactosidase activity and adhesion. Cytoskeleton modifications occurred due to fostered parallel organization of F-actin and elevated expression of Vimentin in ASCs. After seven-day cultivation, atECM impaired osteogenesis of ASCs, simultaneously decreasing expression of Runx2. In addition, atECM accelerated early adipogenesis concomitantly with altered Vimentin organization in ASCs, slightly increasing PPARγ, while elevated Adiponectin and Vimentin mRNA expression. Early adipogenesis triggered by atECM was followed by upregulated mitochondrial activity and Sirtuin 1 (SIRT1) expression in ASCs. Proadipogenic events induced by atECM were mediated by SIRT1, indicating the supportive role of atECM in adipogenesis-related metabolic state of ASCs. These results provide a closer look at the effects of atECM on ASC physiology and may support the advancement of engineering design in soft tissue reconstruction and fundamental research of AT.
Introduction
Among other adult tissues, human adipose tissue (AT) enriched with mesenchymal progenitors, referred as adipose-derived stromal/stem cells (ASCs), holds potential as a source of stem cells due to its ubiquitous localization and ease of accessibility [Citation1]. Considered as mesenchymal stem/stromal (MSC) population that contributes to the connective tissue homeostasis, multilineage differentiation as well as trophic, immunomodulatory and message-secreting activities of ASCs are under investigation [Citation2]. ASCs’ regenerative capacity on one side, and involvement in AT diseases with high frequency in human population on the other, draw additional attention to these progenitor cells. ASCs are surrounded by extracellular matrix (ECM) composed of many types of proteins and glycosaminoglycans that assemble into a highly complex network [Citation3]. The ECM is a fundamental component of specialized adipose niche and provides both architectural elements and non-structural molecules (such are growth factors) which influence progenitor cells and regulate the expandability of AT [Citation4]. However, the precise mechanisms whereby niche elements, such as cell–cell specific interactions or ECM constituents, can influence and modulate the differentiation potential of mesenchymal progenitors, remain mostly unclarified [Citation5]. Crosstalk of adipose tissue ECM (atECM) and ASCs underlies fat tissue homeostasis as well as abnormal expansion, and therefore its detailed understanding contributes to development of more efficient and safe strategies for AT engineering or adipose graft fate prediction. Autologous AT grafting has many disadvantages, such as high costs and unpredictable clinical outcome. Besides the lack of standardized procedures for AT harvest and preparation for transplantation [Citation6], major obstacles for successful adipose graft application are graft volume shrinkage and unpredictable rate of resorption. Therefore, many current studies are directed towards designing in situ injectable materials and porous 3D printed scaffolds for AT reconstruction [Citation7]. The focus of tissue engineering is set on the generation of biomimetic microenvironments which accurately recapitulate cell niches made by the cells themselves in vivo. Currently, two approaches are widely estimated, coating of growth surface with available ECM and in vitro matrices creation [Citation8–10].
Considered as an off-the-shelf natural biomaterial for soft tissue reconstruction, the atECM imparts chemical, physical and biological cues that affect cellular function as a result of signal transduction via cell surface receptors and activation of associated signalling pathways [Citation11]. In response to these cues, cells adjust their cytoskeleton tension following linkage of the actomyosin cytoskeleton with the surrounding ECM [Citation12]. Moreover, intracellular machinery engaged by ECM can interact with nuclear membrane and nucleoskeleton leading to cell transcriptome adaptation and epigenetic changes [Citation13]. Transcriptional cascade, triggered in crosstalk with extracellular cues is recognized as principal for the AT-resident stem/progenitor cell lineage commitment [Citation14], while mode of action of atECM in ASCs is still unknown. Therefore, elucidation of contribution of atECM in ASC behaviour is significant for understanding of ASC regenerative properties controlled by their proximate microenvironment.
Collagen or fibronectin coating of surface appeared to be rational strategy for in vitro cultivation of MSCs which ensure optimal cell growth [Citation15]. In order to analyse the role of native atECM in ASC fate, we compared the reactivity of ASCs cultured on standard tissue-culture plastic (TCP) and TCP coated with collagen or atECM, by evaluating the specific effects on ASC morphology, cytoskeleton organization, adhesion, proliferation, senescence and particularly differentiation into mesodermal lineages. We showed, for the first time, that atECM-derived external stimuli can modify mitochondrial activity and revealed that adipoinductive effects of atECM are mediated by metabolic sensor molecule Sirtuin 1 (SIRT1).
Materials and methods
Adipose tissue processing: atECM preparation and ASC isolation
Subcutaneous AT samples were obtained with informed consent from adult anonymous patients undergoing plastic surgery at National Cancer Research Center, University of Belgrade in accordance with ethical standards of local Ethical Committee and Declaration of Helsinki. The study protocol was approved by the Ethical Committee of the National Cancer Research Center (Decision No. 4428) and all patients signed a written consent for the collection of AT. All rights, security, physical and mental integrity of the patients are fully protected. For decellularization, AT samples were extensively washed with 1× phosphate-buffered saline (PBS, Capricorn Scientific, Ebsdorfergrund, Germany) for 2 h, dissected and minced by surgical scissors. Then, the AT samples were centrifuged at 1800×g for 5 min and supernatant with fat in the liquid phase was discarded. Viscous pellet containing tissue fragments, was incubated in 1 M NaCl (1:1 volume ratio) for 2 h at 37 °C. After centrifugation at 200×g for 5 min, pellet was incubated with 1% SDS for 1 h at room temperature. Thereafter, tissue fragments were extensively washed in extra pure water for 24 h at 37 °C. Between each step, tissue fragments were mechanically massaged with top of syringe. Finally, after incubation in 3% formaldehyde for 1 h, and thereafter with 0.2% DNase (2500 U/ml, Thermo Fisher, Waltham, MA) and 10 µg/ml RNase (Sigma-Aldrich, St. Louis, MO) for 1 h at 37 °C, tissue fragments were rinsed in extra pure water for 24 h. For scanning electron microscopy (SEM) analysis, atECM samples were fixed with 2.5% glutaraldehyde in 1× PBS for 1 h, and then washed twice in 1× PBS. Samples were post-fixed with 1% osmium tetroxide (Sigma-Aldrich, St. Louis, MO) for 1 h and rinsed in 1× PBS before dehydration with graded ethanol solutions [Citation16]. Final drying of samples was done using liquid CO2 critical point dryer (Bal-TecCPD030 Critical Point Dryer, Bal-TecAG, Balzers, Liechtenstein). SEM examination was performed using FE-SEM, a TESCAN MIRA 3 XMU, operated at 20 keV, after a deposition of gold/palladium thin layer on the samples surface.
After decellularization, atECM was lyophilized using BETA 1-8 LD plus lyophilizator (Martin Christ, Osterode am Harz, Germany). After cooling to –70 °C, the primary drying was conducted at shelf temperature of –60 °C for 24 h followed by –65 °C for 2 h. Obtained lyophilized samples were hermetically sealed and stored at –20 °C. To solubilize atECM, minced lyophilized atECM was further digested with pepsin solution (Sigma-Aldrich, St. Louis, MO) for 48 h at room temperature under constant stirring. In detail, pepsin was solubilized in 0.1 M HCl and added to the atECM at a ratio of 1 mg pepsin for every 10 mg of lyophilized atECM. The pH was raised to 7.4 using 1 M NaOH and then the matrix was diluted to 15 mg/ml using 10× PBS so that the final solution contained 1× PBS [Citation17]. Solubilized atECM was stored at 4 °C and used for coating of tissue culture plastic after previous dilution with 1× PBS to adjust concentration to 5 mg/ml. For determination of solubilized atECM protein concentration, BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL) was used according to the manufacturer’s protocol.
For isolation of ASCs, our recently reported protocol was applied and modified [Citation18]. Pieces of AT were collected and kept in growth medium (GM), consisting of Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, St. Louis, MO), Hepes (1%), penicillin/streptomycin (1%), and 10% foetal bovine serum (FBS) (all bought from Capricorn Scientific, Ebsdorfergrund, Germany) at 4 °C for no more than 24 h after surgery. Upon reception in the laboratory, unidentified raw samples were washed extensively using 1× PBS. The specimens were minced using surgical scissors and tissue fragments were digested in a solution of 1 mg/ml collagenase type 2 supplemented with 3.5% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO), 0.5 mM CaCl2, 1% penicillin/streptomycin, for 2 h at 37 °C. The pellets were re-suspended in GM and cells were counted and seeded in 25 cm2 plastic tissue culture flasks (Greiner Bio-One, Kremsmünster, Austria) in GM. The medium was replaced twice a week, and after reaching 90% of confluence, adherent cells were detached using 0.25% solution of trypsin/EDTA (Gibco, Carlsbad, CA). All experiments were performed using ASCs between passages 2 and 6.
Preparation of tissue culture plastic: coating with atECM and collagen
Solutions of atECM and rat tail vein-derived collagen (concentration 3.5 mg/ml) were stored at 4 °C. Tissue culture plastic flasks, plates and coverslips were coated with solubilized atECM or collagen for 24 h at 4 °C. atECM or collagen was added to cover growth surface of dishes in volumes 30 µl/0.34 cm2, 100 µl/1.9 cm2, 500 µl/9.6 cm2 and 3 ml/25 cm2. Further, atECM and collagen were discarded, and surfaces (including TCP) were washed with 1× PBS and 75% ethanol. Then, plates were treated with 3% BSA in 1× PBS for 1 h at 4 °C. After discarding the BSA solution, growth surfaces were washed with 1× PBS for three times. These pre-coated surfaces were used for ASC cultivation.
Phenotype analysis of cultivated human ASCs
ASCs were maintained in standard conditions and after reaching confluence were harvested using 1 mM EDTA and washed in cold PBS supplemented with 0.5% BSA. Cells were incubated (30 min in the dark at 4 °C) with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated antibodies against human antigens CD44H, CD73, CD90, (all from R&D Systems, Minneapolis, MN), CD105 (from Invitrogen, Carlsbad, CA) and CD34 (Dako Cytomation, Glostrup, Denmark). The percentage of non-specific binding was determined by using the appropriate FITC- and PE-conjugated isotype control antibodies (R&D Systems, Minneapolis, MN). Flow cytometry was performed using flow cytometer Partec (Munster, Germany).
Cell proliferation and viability
ASCs were seeded at concentration 2 × 104 cells/well in 24 well-plates in GM and cultivated in standard conditions. After 24, 48 and 120 h, exclusion test was performed and cell number was counted using Trypan Blue (0.4% solution).
To evaluate the viability of ASCs, 5 × 103 cells/well were seeded in 96 well-plates in GM. After 24, 48, and 120 h, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/ml) (Sigma-Aldrich, St. Louis, MO) was added to each well and incubated for the next 2 h. Then, culture media were discarded and formazan crystals were dissolved in isopropanol. Optical density was measured at 540 nm using automatic reader for microtiter plates (Labsystems Multiskan PLUS, Vantaa, Finland).
Adhesion, cell shape and area: single cell image analysis
Cells were seeded at concentration 2000 cells/well in 96 well plates in GM and cultivated in standard conditions. After 2, 10 and 24 h, adhered cells were stained by 0.1% crystal violet. Three separate fields were photographed by phase contrast microscope and the number of spread attached cells was estimated by microscopic cell counting. For cell area and shape analysis, aspect ratio and circularity were determined by single cell analysis using NIH-Image J program. Aspect ratio was calculated using formula AR = major axis/minor axis of the cell, while circularity was calculated by formula 4π × Area/Perimeter2. Twenty five cells on five separated fields were analysed in each group.
Differentiation assays in vitro
Cells were cultivated in GM or corresponding differentiation medium in standard conditions until reach 80% confluency. Osteogenic differentiation medium contained DMEM supplemented with 5% FBS, 1% penicillin/streptomycin, 50 μM ascorbic acid-2-phosphate (Sigma-Aldrich, St. Louis, MO), 10 mM β-glycerophosphate (Sigma-Aldrich, St. Louis, MO) and 10 nM dexamethasone. After seven days, ASCs were fixed with 3.7% formaldehyde in 96% ethanol and enzyme activity of ALP was quantified by adding p-nitrophenyl-phosphate (pNPP) and measuring absorbance at 405 nm using automatic reader for microtiter plates (Labsystems Multiskan PLUS, Vantaa, Finland). For visualization of ALP activity, cells were washed with 0.9% NaCl and NBT/BCIP substrate solution was added. The presence of calcium-containing deposits and mineralization of ECM was visualized with 0.5% Alizarin Red staining after 21-day cultivation.
Cartilage-associated glycosaminoglycan formation was detected with Safranin O after 3 weeks of cultivation in chondrogenic medium containing DMEM with 5% FBS, 1% penicillin/streptomycin, 50 μM ascorbic acid-2-phosphate, 10 nM dexamethasone and 5 ng/ml of transforming growth factor-β1 (TGF-β, R&D Systems, Minneapolis, MN). After staining protocols, ASCs were examined using a light microscope (Olympus, Tokyo, Japan) and differentiation was semi-quantified by densitometry, using NIH-Image J software.
To induce adipogenic differentiation, ASCs were cultivated in adipogenic medium (AM) containing 5% FBS in DMEM, 1% penicillin/streptomycin, 100 μg/ml isobutyl-methylxanthine (IBMX; Sigma-Aldrich, St. Louis, MO), 1 μM dexamethasone and 10 μg/ml insulin (Sigma-Aldrich, St. Louis, MO). The appearance of intracellular lipid droplets and their accumulation was confirmed by Oil Red O (Merck Chemicals, Darmstadt, Germany), which labels cholesteryl esters and triacylglycerols. ASCs were washed with water and the stained fat droplets in the cells were visualized by light microscopy and photographed. The percentage of differentiated cells was determined by counting cells positive for Oil Red O staining in the lipid vacuoles. Otherwise, to follow adipogenic differentiation, ASCs were seeded and cultivated on cover slips in AM, while lipid droplets were stained with fluorescent hydrophobic probe 9-(diethylamino)-5H-benzo[a]phenoxazin-5-one, Nile Red (Santa Cruz Biotechnology, Santa Cruz, CA) at final concentration 0.05 μg/ml (Santa Cruz Biotechnology, Santa Cruz, CA) for detection of neutral lipids (preferentially unesterified cholesterol) and 1 ng/ml of DAPI (Sigma-Aldrich, St. Louis, MO) for nucleus labelling for 20 min at room temperature. Cells were examined using epi-fluorescent microscope (Olympus, Tokyo, Japan).
β-Galactosidase staining
Cells were seeded in a 96-well plate and cultivated for 3, 7 and 14 days in standard conditions. Afterwards, plates were washed with 1× PBS, fixed for 5 min at room temperature, and incubated at 37 °C overnight with 1 mg/ml X-gal buffered solution prepared according to the manufacturer’s instructions (Sigma Aldrich, St. Louis, MO). Histochemical staining for β-galactosidase activity was documented by light microscope (Olympus, Tokyo, Japan). Microphotographs of four random fields for each sample were taken. Altered (flattened) shape and staining intensity of senescent cells was applied as discrimination strategy when number of senescent-like cells per visual field was determined.
Immunofluorescence
ASCs (5 × 103 cells/coverslip) were seeded and cultivated on coated coverslips in standard conditions. For visualization of cytoskeletal proteins and SIRT1, at certain time points (experimental conditions indicated in figure legend), cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton-X100 (Sigma-Aldrich, St. Louis, MO) in 1× PBS and blocked with 3% BSA in PBS for 30 min. Then, samples were incubated with primary antibodies: phalloidin-tetramethylrhodamine B isothiocyanate (Sigma Aldrich, St. Louis, MO, dilution 1:1000), anti-mouse Vimentin (Santa Cruz Biotechnology, Santa Cruz, CA, dilution 1:100), mouse anti-α-smooth muscle actin (SMA) (dilution 1:100, Santa Cruz Biotechnology, Santa Cruz, CA), anti-rabbit SIRT1 (Cell Signaling Technology, Danvers, MA, dilution 1:100). Samples incubated in 1% BSA in PBS served as negative control. After appropriate incubation, cells were washed three times with PBS and incubated with the corresponding FITC/TRITC-coupled secondary antibodies (dilution 1:100, both from Sigma-Aldrich, St. Louis, MO) at room temperature for 1 h. The mitochondria of the viable ASCs were visualized with membrane potential sensitive dye [Citation19], MitoTracker Orange CMTMRos (MTO) (Invitrogen, Thermo Fisher Scientific, Waltham, MA). After appropriate treatment, 10 nM MTO was added during the least 30 min of incubation, in GM without FBS. For nucleus labelling, 1 ng/ml of DAPI (Sigma-Aldrich, St. Louis, MO) was added. Mounted cells were analysed using an epi-fluorescent microscope (Olympus, Tokyo, Japan).
Semi-quantitative RT-PCR
ASCs seeded at concentration 3 × 105 cells/well in six-well-plate in GM/AM were cultivated in standard conditions for seven days. Afterwards, ASCs were washed with 1× PBS and total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA). RNA was precipitated in isopropanol, resuspended in nuclease-free water and 200 ng of RNA was reverse transcribed into cDNA by RevertAidTM H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA), using oligo (dT) as a primer. PCR products were obtained after 33 cycles of amplification with adjusted annealing temperature ranging from 47 to 56 °C. Primer sets were both from Invitrogen (Carlsbad, CA). Applied annealing temperatures are indicated in . As a housekeeping gene control, GAPDH was amplified for the amount of cDNA control in each sample. Amplicons were resolved in 1.5% agarose gel and stained with ethidium bromide.
Table 1. Primer sets used in experiments.
Statistics
Statistical analysis was performed with the use of GraphPad Prism 6 (La Jolla, CA). Results are presented as a mean value ± SEM calculated from data of at least three independent experiments. Differences between groups were tested for statistical significance by Student’s t-test. Statistical significance is reached for p<.05.
Results
atECM does not affect viability, proliferation and β-galactosidase activity of cultivated ASCs
As a confirmation of delipidization and decellularization, no cellular or lipid components were observed in atECM after processing, in comparison to native human AT, while structural and fibre-morphological shape with different size bundles of varying thickness in atECM was observed by SEM. After lyophilization and solubilization, protein concentration of neutralized solution of atECM (15 mg/ml) was 4.053 ± 0.160 mg/ml ().
Figure 1. atECM preparation and testing of cell viability, proliferation, β-galactosidase activity of cultivated ASCs. (a) Adipose tissue before and after processing. For fluorescence imaging, lipids are stained by Nile Red and nuclei by DAPI (DNA). For light microscopy, lipids are stained by Oil Red O. Representative photos of decellularized and delipidized atECM observed by scanning electron microscopy (SEM) and lyophilized and solubilized form used for coating. Scale bar 200 and 10 μm. (b) Representative histograms of FACS analysis showing expression of CD44, CD90, CD73, CD105 and CD34 (shaded histograms: isotype control; empty histograms: antigen expression). Cell number and viability were determined after 24, 72 and 120 h. (c) By Trypan blue exclusion test and (d) by MTT assay, respectively. (e) Activity of β-galactosidase in ASCs expressed as percentage of β-gal positive cells per visual field. Photos obtained after cultivation of ASCs for 3, 7 and 14 days in standard conditions are shown. Representative images of obtained by light, fluorescent and SEM are shown.
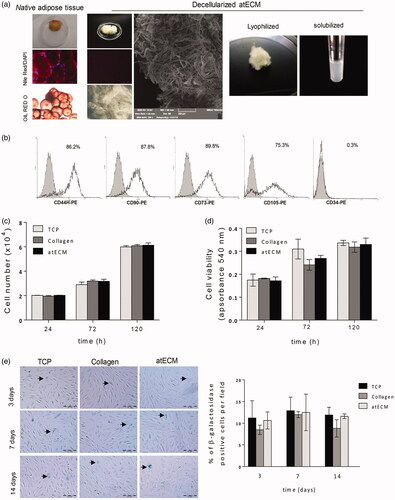
Isolated ASCs showed positive expression of CD73, CD44, CD90 and CD105, without expression of hematopoietic and endothelial progenitor marker CD34 (), thus satisfying minimal criteria recommended for mesenchymal stromal cell (MSC) characterization [Citation20].
To analyse the response of ASCs cultivated on atECM, we pre-coated common used plastic dishes with collagen or solubilized atECM. Results demonstrated that atECM did not affect proliferation () nor viability of ASC after 24, 72 and 120 h of cultivation in GM (), in comparison to TCP or collagen. Additionally, we showed that atECM did not change the frequency of β-galactosidase positive cells after 3, 7 and 14 days of cultivation in comparison to TCP and collagen (). Taken together, our results confirm absence of cytotoxic or senescence-associated changes in ASCs during cultivation on surface coated with solubilized atECM.
atECM modifies the morphology and cytoskeleton organization of cultivated ASCs
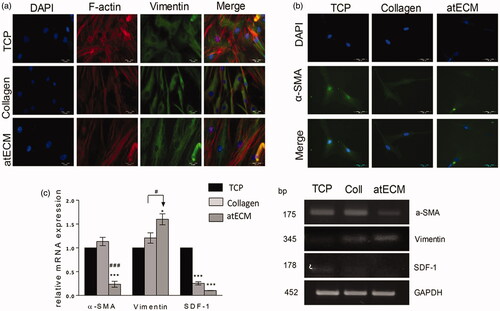
Although atECM did not alter adhesion level of ASCs after 2, 10 and 24 h, differences in cell morphology were observed after 24 h (). The indicator of cell morphology, aspect ratio was significantly higher in collagen and atECM groups (), while cultivating ASCs on atECM significantly decreased cell circularity in comparison with TCP and collagen group (). Thus, we concluded that TCP supported flattened or round shape, while collagen and atECM favoured elongated morphology of ASCs. This state was confirmed by immunofluorescence staining of cytoskeleton molecules, where was shown that atECM fostered spindle-shape extended morphology of ASCs after seven-day cultivation more efficiently than collagen, in comparison with TCP (). We revealed that morphological changes were followed by the change in filamentous (F)-actin and intermediate filament, Vimentin organization. While after cultivation on TCP for seven days, ASCs acquired flattened morphology with criss-cross organized F-actin and net-like organized Vimentin, collagen and more efficiently, atECM, promoted parallel organization of F-actin and Vimentin in ASCs, with fibres elongating through the length of the cell contributing to preservation of their spindle-shape morphology (). In addition, atECM and collagen stimulated Vimentin mRNA expression, in comparison to TCP. Similar to collagen, atECM attenuated stromal-derived factor 1 (SDF-1) mRNA expression in ASCs after seven-day cultivation, in comparison with TCP (). Also, obtained results indicated that atECM strongly decreased α-smooth muscle actin (α-SMA) at protein and gene level () in comparison with TCP and collagen.
Figure 2. Adhesion and cell morphology of ASCs. (a) Number of adherent ASCs was counted after 2, 10 and 24 h after seeding. Photos of adherent cells stained after 24 h are shown. Percentage of spread cells/field was calculated, while three separate fields were observed per well. (b) Aspect ratio and (c) circularity were quantified for 25 cells observed in five different fields for each group. Results are presented as means ± SEM from three independent experiments in three or four replicates. Statistically significant differences: *p<.05; ***p<.001, ****p<.0001.
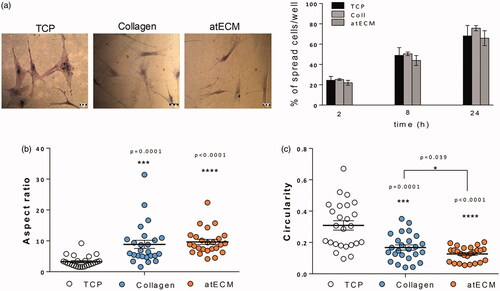
Figure 3. Cytoskeleton organization in ASCs. For protein and gene expression analysis, cells were cultivated on TCP, collagen and atECM for seven days in GM in standard conditions. Immunofluorescence staining was performed for: (a) for F-actin (phalloidin), Vimentin and (b) α-SMA (c) Relative gene expression of α-SMA, SDF-1 and Vimentin was determined by RT-PCR. GAPDH was used as gel loading control. Molecular weight of PCR products is indicated in base pairs (bp). Representative photos and gels are shown. Results are presented as means ± SEM from three independent experiments in three or four replicates. Statistically significant differences in comparison to the TCP (set as 1): *p<.05; ***p<.001, and in comparison with collagen: #p<.05; ###p<.001.
Effect of atECM on differentiation in vitro: accelerated early adipogenesis and delayed/impaired osteogenesis of ASCs
To evaluate the potential of atECM to direct cell differentiation, we estimated its effects on osteogenesis, chondrogenesis and adipogenesis of ASCs. Results indicated that atECM, although it did not affect mineralization, attenuated early phase of osteogenesis of ASCs, decreasing activity of ALP in both GM and osteogenic medium (OM), which was in accordance with decreased expression of Runt-related transcription factor 2 (Runx2) mRNA (). However, atECM did not affect chondrogenic capacity of ASCs nor expression of SRY (sex determining region Y)-box 9 (Sox9) mRNA which was undetectable in our conditions (), while Safranin O staining was similar as in TCP and collagen group.
Figure 4. Differentiation potential of ASCs cultivated on atECM. (a, b) Osteogenic differentiation determined by ALP after seven days and Alizarin Red after 21 days. Determination of ALP level (c, d) chondrogenic differentiation of ASCs determined by Safranin O and content of glycosaminogycans. (e) Relative gene expression of Runx2 and Sox9 was determined by RT-PCR. GAPDH was used as gel loading control. Molecular weight of PCR products is indicated in bp. Results are shown as mean ± SEM from three independent experiments. Statistically significant differences in comparison to the TCP (set as 1): *p<.05; **p<.01; ***p<.001 and in comparison with collagen: ##<.01; ###<.001. (f) Adipogenic differentiation of ASCs; lipid droplets are stained by Oil Red O staining after 7, 14 and 21 days. Percentage of Oil Red positive cells per field was calculated, while three separate fields were observed per well. Representative photos and gels are shown. All results are shown as mean ± SEM from three independent experiments in three or four replicates. Statistically significant differences in comparison to the TCP: **p<.01; and in comparison with collagen: ##<.01.
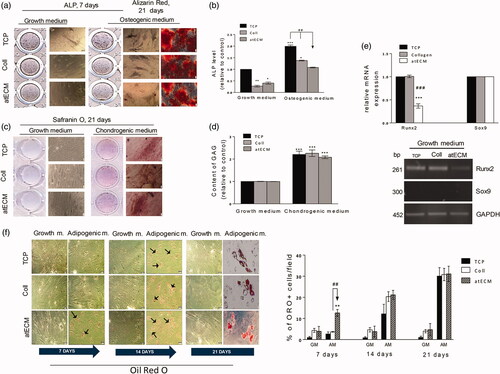
Additionally, our results revealed that although atECM did not alter adipogenic capacity of ASCs, it accelerated lipid droplets formation. Oil Red O positive cells were observed in ASCs after only seven days of cultivation in the presence of adipogenic stimuli when cells were cultivated on atECM-coated surface (). However, the percentage of Oil Red positive cells after 21 days of cultivation in the presence of atECM was not different from TCP and collagen. Simultaneously, atECM did not change the level of weak spontaneous adipogenic differentiation of ASCs which could be detected by Oil Red staining. Thus, we hypothesized that atECM can affect the commitment of ASCs in favour of adipogenic lineage in early phase (seven days) of differentiation induction. Additionally, regardless of adipogenic stimuli, atECM decreased peroxisome proliferator-activated receptor gamma (PPAR-γ) mRNA expression in comparison to TCP, while slightly stimulating it in comparison with collagen in the presence of adipogenic stimuli. Also, Adiponectin mRNA expression was elevated in the presence of atECM in comparison with TCP in the presence of AM, while it did not differ from the collagen group ().
Figure 5. Effects of atECM on early adipogenesis: Vimentin, mitochondria and SIRT1 distribution and expression in ACSs. ASCs are cultivated for seven days in GM/AM on TCP, collagen and atECM. (a) Gene expression of PPARγ, Adiponectin and Vimentin. Relative mRNA expression was determined by RT-PCR. (b) Adipogenic differentiation of ASCs after seven-day cultivation on TCP or atECM in the presence of adipogenic stimuli. Immunofluorescence staining of Vimentin and lipid droplets (Nile red). Representative photos of at least three independent experiments performed in duplicate are shown. (c) MitoTracker Orange was added into the media for fluorescence staining of mitochondria, followed by staining of nuclei (DAPI). Representative photos of two separate experiments are shown. (d) Immunofluorescence staining of SIRT1 and nuclei (DAPI). (e) Relative mRNA expression was determined by RT-PCR. Representative photos and gels are shown. GAPDH was used as gel loading control. Molecular weight of PCR products is indicated in bp. Results are presented as means ± SEM from at least three independent experiments. For PCR analysis, statistically significant differences in comparison to the TCP in GM (set as 1): *p<.05; **p<.01; ***p<.001; and in comparison to collagen: #<.05.
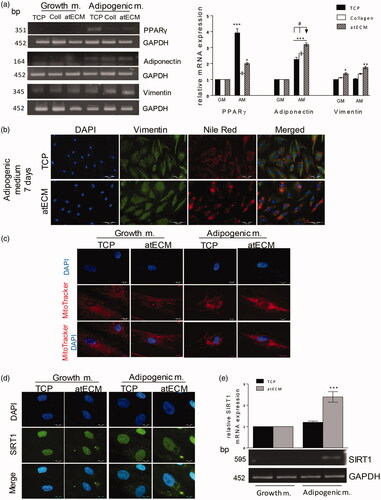
atECM-induced early adipogenesis is associated with increased Vimentin expression in ASCs
We found that pro-adipogenic events in ASCs were followed by alteration of Vimentin expression after seven days of cultivation. atECM elevated Vimentin mRNA expression both in absence and presence of adipogenic differentiation stimuli in ASCs in comparison to TCP and collagen (). These observations are in accordance with fluorescence staining of Vimentin protein and lipid droplets by Nile Red. It has been observed that increased lipid droplet presence induced by atECM, was followed by alteration of Vimentin subcellular organization. In comparison to TCP, where during adipogenic stimuli, Vimentin tended to form cage-like structure (), in the presence of atECM, Vimentin expression was more diffuse-like, while its nuclear localization was also observed. Immunofluorescence labelling results indicated that atECM, concomitantly with adipogenic stimuli, increased lipid droplet formation and accumulation in ASCs leading to state of preadipocyte-like cells after seven days ().
atECM alters mitochondrial distribution and expression of SIRT1 in ASCs in early phase of adipogenesis
Adipogenic stimuli increased mitochondrial activity detected by mitochondrial membrane potential sensitive probe (MTO) staining and merged mitochondrial mass away from perinuclear region. In addition, atECM supported these changes, additionally increasing mitochondrial activity and their shuttling in cytosol space (). Immunofluorescence analysis revealed that adipogenic stimuli increased Silent mating type information regulation 2 homolog 1 (SIRT1) protein expression in perinuclear compartment of ASCs cultivated on TCP, while this event was more clearly observed in the atECM group (). In accordance, atECM elevated SIRT1 mRNA expression in ASCs, in comparison to TCP in the presence of adipogenic stimuli. In the absence of adipogenic stimuli, SIRT1 was not affected by either TCP or atECM in ASCs ().
atECM-induced pro-adipogenic events are dependent on SIRT1 activity
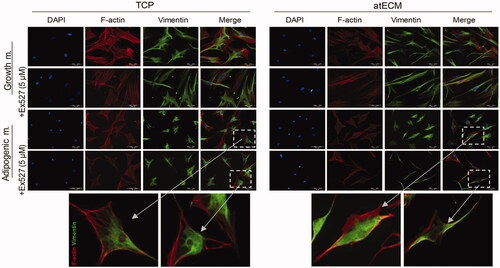
First, nontoxic concentration of inhibitor of SIRT1 class III histone deacetylase enzyme, Ex-527 (5 µM, Santa Cruz Biotechnology, Santa Cruz, CA) for ASCs was determined by MTT test (data not shown). To avoid undesired interference with cell functions, total volume of DMSO (0.001%, Santa Cruz Biotechnology, Santa Cruz, CA) was tested as a vehicle and showed no harmful effect on cells (data not shown). Our results demonstrated that after seven-day cultivation in GM on TCP, presence of Ex-527 inhibitor slightly attenuated F-actin expression, without changing Vimentin expression and organization and therefore modification of ASC shape was not observed. On the other hand, after seven-day cultivation in GM on atECM, SIRT1 inhibition supported parallel organization F-actin and Vimentin, thus contributing to spindle shape maintenance (). In addition, after seven-day cultivation in AM on TCP, presence of Ex-527 inhibitor did not change F-actin and cage-like Vimentin organization and expression. On the contrary, SIRT1 inhibitor diminished effects of AM when ASCs were cultured on atECM, predominantly affecting Vimentin expression and distribution. Namely, presence of Ex-527 in AM during seven days, fostered the spindle shape of ASCs which was observed in GM, and diminished AM-associated Vimentin distribution in nucleus region in ASCs cultivated on atECM ().
Figure 6. Cytoskeleton rearrangement in ASCs in dependence of SIRT1. ASCs are cultivated for seven days in GM/AM on TCP or atECM in presence or absence of Ex-527 (Santa Cruz Biotechnology, Santa Cruz, CA) at concentration 5 µM. Immunofluorescence staining of F-actin (phalloidin), Vimentin and nuclei (DAPI). Representative photos of at least three independent experiments performed in triplicate are shown.
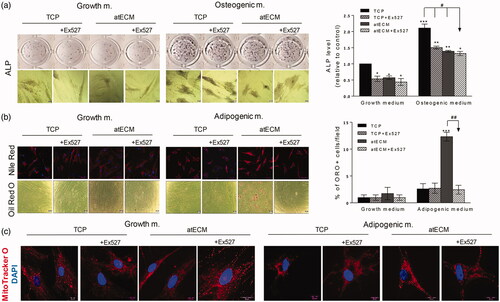
Concomitantly, we examined whether SIRT1 could be involved in atECM-affected differentiation of ASCs, observed in vitro after seven-day cultivation. Our results demonstrated that Ex-527 inhibited spontaneous ALP activity in ASCs cultivated on TCP, while it did not change the decreased ALP activity in ASCs cultivated on atECM in comparison with TCP. Similarly, in the presence of osteogenic stimuli, Ex-527 attenuated ALP activity in ASCs on TCP, while it did not affect impaired ALP activity in ASCs cultivated on atECM in comparison with TCP. These results suggest that SIRT1 activity is not involved in the regulation of osteogenic potential of ASCs cultivated on atECM (). In contrary, SIRT1 inhibitor decreased stimulated adipogenic differentiation observed after cultivation of ASCs on atECM after seven days in AM, not affecting invisible adipogenic differentiation in ASCs on TCP (). In comparison to ASCs cultivated on TCP, presence of SIRT1 inhibitor impaired the distribution of mitochondria away from perinuclear region and their increased activity observed in ASCs on atECM in the presence of adipogenic stimuli, without changing mitochondrial distribution in ASCs cultivated on TCP and atECM in GM ().
Figure 7. Effects of SIRT1 inhibition on differentiation and mitochondria in ACSs. To estimate their differentiation, ASCs are cultivated for seven days in GM/OM/AM on TCP or atECM in presence or absence of Ex-527 at concentration 5 µM. (a) Osteogenic differentiation determined by ALP after seven days. Results are shown as mean ± SEM from three independent experiments. Statistically significant differences in comparison to the TCP (set as 1): *p<.05; **p<.01; ***p<.001 and in comparison with TCP (OM): #<.001. (b) Adipogenic differentiation of ASCs; lipid droplets are stained by Oil Red O for light microscopy while Nile Red was applied for immunofluorescence. Percentage of Oil Red positive cells/field was calculated, while three separate fields were observed per well. All results are shown as mean ± SEM from three independent experiments. Statistically significant differences in comparison to the TCP: ***p<.001; or within AM group: ##<.01. (c) MitoTracker Orange was added into the media for fluorescence staining of mitochondria, followed by staining of nuclei (DAPI). Representative photos of three independent experiments are shown.
Discussion
Our study pointed toward elucidation how atECM affects differentiation process in human ASCs. We found atECM lead to stimulation of ASC adipogenesis-associated lipid droplet formation, which was accompanied by decreased osteogenic capacity, cytoskeleton rearrangement and increased mitochondria and SIRT1 activity.
While ECM-mimicking materials, such as chitosan or tendon-derived ECM [Citation21,Citation22] were found to promote proliferation of MSCs, fibronectin, laminin, collagen [Citation23] or ECM produced by ASCs cultured in monolayer [Citation24] did not significantly changed bone marrow (BM)-MSC and ASC growth, which is in accordance with our results. Recent study reported that cell-deposited ECM proteins protected umbilical cord MSCs from oxidative stress-induced senescence [Citation25], while we demonstrated that atECM had no effect on activity β-galactosidase in ACSs. The inequality of ECM effect on MSC reported in the studies, might be the consequence of different ECM present in different anatomical locations, unequal properties of MSCs isolated from different adult human tissues [Citation26] and finally, variations of cell-type-determined “cell–matrix interface” [Citation27]. However, we can conclude that presence of atECM did not interfere with ASC proliferation capacity or senescence-associated phenotype appearance.
Although adhesion stayed intact, atECM significantly increased aspect ratio and decreased circularity of ASCs, which was followed by cytoskeletal rearrangement. Control of ASC shape and gene expression by biomaterial such as atECM, can be an important tool in directing the cell fate, and particularly differentiation [Citation28] contributing to the fields of biomaterials, biomechanics, and tissue engineering [Citation29]. Importantly, our analysis of spreading and morphology at single cell level indicate intra-population variability within ASCs, which is in accordance with previous studies where similar heterogeneity was reported [Citation11,Citation23,Citation30]. Apart from donor-to-donor heterogeneity, it is worth to mention that heterogeneity among MSC population, whether constitutive or achieved during in vitro cultivation, can significantly complicate analysis of their functional properties and still represents great challenge for MSC clinical manufacturing protocols [Citation31]. Our findings showed that atECM has important effects on cytoskeletal arrangement which has been described as crucial event in cell polarization, cytokinesis, migration and differentiation [Citation32]. Results suggest that atECM can modify F-actin and Vimentin organization and expression. After seven-day cultivation of ASCs on atECM or collagen, parallel organization of F-actin and Vimentin filament was observed, while expression of α-SMA was diminished. It could be speculated that atECM, as well as collagen, represent soft cell culture substrate which can attenuate F-actin and α-SMA arrangement associated with multipotency maintenance in MSCs, which could be truncated in standard cell culture conditions [Citation33]. It is possible that the increase of Vimentin expression in ASCs cultivated on atECM is related to weakened actin element expression, as previously shown in osteosarcoma cells and dermal fibroblast [Citation34], where interplay of F-actin and Vimentin controls cytoskeleton dynamics and cell morphogenesis. Although often recognized as marker of cultivated MSCs and pericytes, α-SMA was not expressed in native isolated ACSs [Citation35]. It might be speculated that use of atECM improve in vitro conditions providing native-like environment to ASCs, thus allowing us to understand their physiology and response more properly, which is particularly important for the development of ASC-based cell therapies. Decreased stromal derived factor 1 (SDF-1) expression in our study may be associated with the ability of atECM to regulate ASC adipogenesis, as low SDF-1 expression has been correlated to ASCs already committed to adipocyte differentiation [Citation36]. Thus, the elucidation of atECM role in the regulation of ASC differentiation state could be of the great importance for the planning strategies for AT augmentation, repair and long-term maintenance.
In accordance with data revealing the crucial role of ECM in AT physiology [Citation37], we demonstrated that atECM can alter differentiation of ASCs. Previous reports showing that atECM coating stimulates osteogenesis of BM-MSCs [Citation38] and that atECM-based scaffold stimulates chondrogenic differentiation of ASCs [Citation39] are partly in contrast with our results as we showed that atECM inhibit osteogenic without change of chondrogenic capacity in ASCs. atECM stimulated lipid droplet formation and Adiponectin expression in ASCs, accelerating adipogenic differentiation in comparison with collagen and TCP. Weak increase of PPARγ mRNA expression in atECM group might be related to duration of applied treatment (seven days), as it has been indicated that PPARγ is activated soon after adipogenic stimuli and is further controlled by crosstalk with numerous positive and negative regulators [Citation40]. However, observed atECM-accelerated adipogenesis of ASCs in our study is in accordance with previous report where was demonstrated that decellularized AT hydrogel enhanced adipogenesis in porcine ASCs in vitro and in vivo [Citation41].
Our results indicate that atECM coating provides stimuli which modulates cytoskeleton during early phase of adipogenesis. Decreasing of F-actin polymerization in ASCs might be associated with spherical shape acquisition, osteogenesis inhibition [Citation42] and improvement of adipogenic differentiation of MSCs [Citation43]. Moreover, our results showed that only atECM could increase Vimentin mRNA expression in ASCs. As Vimentin participates in cholesterol transport [Citation44] and interacts with PPARγ in the cytosol, regulating its turnover rate [Citation45], it is plausible that upregulated Vimentin might be involved in atECM-triggered adipogenesis of ASCs. Importantly, nuclear localization of Vimentin might indicate potential implication of Vimentin in DNA-based nuclear events, like DNA replication, transcription and repair [Citation46], as well as intracellular position and physiological activity of mitochondria, thus controlling cell differentiation [Citation47].
We revealed possibility that changes in mitochondria properties are related to the initiation of adipogenic differentiation in ASCs observed in atECM group. Increased mitochondrial activity and conspicuous perinuclear position in ASCs, were also demonstrated in recent report where ASCs were cultivated in AM for seven days, and this time was suggested to be enough to establish adipogenic lineage commitment of ASCs [Citation48]. While mitochondria in different tissues have distinctive ultrastructural features consistent with differential bioenergetic demands, it is suggested that mitochondrial respiration is required for adipogenic differentiation of BM-MSCs [Citation49]. Involvement of mitochondria in MSC differentiation indicates adjustment of cell metabolic pathways to microenvironment conditions such as high glucose level, insulin or serum [Citation50], while here we demonstrated for the first time that atECM stimuli may affect mitochondrial distribution in ASCs. The mitochondrial processes are dependent on the proper nicotinamide adenine dinucleotide (NAD)+/NADH redox balance and the key regulators of mitochondrial metabolism, sirtuins (SIRTs) [Citation51], where SIRT1 regulatory factor, acting as NAD + dependent deacetylase, regulates metabolism and stress responses, lipid homeostasis and maintenance of mitochondrial function [Citation52]. As presence of SIRT1 activity inhibitor abolished atECM-stimulated Vimentin protein expression in nucleus region of ASCs in the presence of adipogenic stimuli, it is possible that SIRT1 regulates atECM-supported cytoskeleton arrangement in ASCs. Inhibitor strategy indicated an involvement of SIRT1 molecule in atECM-associated adipogenesis of ASCs without its implication in ASC osteogenesis. These results are not in complete accordance with previous findings showing that SIRT1 deletion causes weak adipogenic differentiation of mouse BM-MSCs [Citation53]. On the other hand, SIRT1 was involved in positive regulation of Adiponectin expression in preadipocyte cell line 3T3-L1 [Citation54], which suggested that role of SIRT1 in differentiation may be dependent on tissue-controlled lineage commitment and cell differentiation state. Moreover, SIRT1 inhibitor abolished increased activity of mitochondria in ASCs cultivated on atECM in adipogenic media, thus indicating that SIRT1 may be involved in the regulation of atECM induced metabolic changes in ASCs, which have been reported as crucial in the first phase of ASC adipogenesis [Citation48]. Since it has been revealed that ECM microfibrils have protective role against metabolic stress in adipocytes [Citation55], our finding might imply a supporting role of atECM in the maintenance of adipose cell metabolic state.
Conclusions
Our results demonstrated that atECM does not change proliferation, senescence or adhesion of ASCs, while fosters extended morphology of ACSs followed by increased Vimentin and weakened F-actin expression. atECM attenuates the early phase of osteogenesis of ASCs, accelerating lipid droplet formation in ASCs. This stimulation of early adipogenesis is followed by increased Vimentin expression and mitochondrial activity. We revealed that cytoskeletal changes and adipoinductive effect of atECM in ASCs were mediated by SIRT1. Regarding the use of atECM, obtained results emphasize the importance of improving biomimetic cell environment when optimizing in vitro settings. Taken together, revealing adipose stem/progenitor niche cues through the clarification of atECM influence on ASC differentiation can contribute to the better cell engineering design for soft tissue reconstruction as well as fundamental research of AT in homeostasis and disease.
Acknowledgements
We owe special gratitude to Dr. S. Nikolić, MD at National Cancer Research Center, for providing subcutaneous adipose tissue samples. We thank Mrs. S. Marković for her technical assistance. In addition, we are thankful to Dr. V. Vukić, Faculty of Technology, University of Novi Sad, Serbia; Dr. Dj. Veljović and Dr. K. Trifković, Faculty of Technology and Metallurgy, University of Belgrade, and Dr. M. Herrmann, University of Würzburg, Germany.
Disclosure statement
There are no conflicts to declare.
Additional information
Funding
References
- Dykstra JA, Facile T, Patrick RJ, et al. Concise review: fat and furious: harnessing the full potential of adipose-derived stromal vascular fraction. Stem Cells Transl Med. 2017;6:1096–1108.
- Kariminekoo S, Movassaghpour A, Rahimzadeh A, et al. Implications of mesenchymal stem cells in regenerative medicine. Artif Cells Nanomed Biotechnol. 2016;44:749–757.
- Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840:2506–2519.
- Spencer M, Unal R, Zhu B, et al. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. Clin Endocrinol Metab. 2011;96:E1990–E1998.
- Pope BD, Warren CR, Parker KK, et al. Microenvironmental control of adipocyte fate and function. Trends Cell Biol. 2016;26:745–755.
- Suszynski TM, Sieber DA, Van Beek AL, et al. Characterization of adipose tissue for autologous fat grafting. Aesthet Surg J. 2015;35:194–203.
- Van Nieuwenhove I, Tytgat L, Ryx M, et al. Soft tissue fillers for adipose tissue regeneration: from hydrogel development toward clinical applications. Acta Biomater. 2017;63:37–49.
- Benny P, Raghunath M. Making microenvironments: a look into incorporating macromolecular crowding into in vitro experiments, to generate biomimetic microenvironments which are capable of directing cell function for tissue engineering applications. J Tissue Eng. 2017;8:1–8.
- Saei Arezoumand K, Alizadeh E, Pilehvar-Soltanahmadi Y, et al. An overview on different strategies for the stemness maintenance of MSCs. Artif Cells Nanomed Biotechnol. 2017;45:1255–1271.
- Casagrande S, Tiribuzi R, Cassetti E, et al. Biodegradable composite porous poly(dl-lactide-co-glycolide) scaffold supports mesenchymal stem cell differentiation and calcium phosphate deposition. Artif Cells Nanomed Biotechnol. 2017;21:1–11.
- Lee J, Abdeen AA, Tang X, et al. Matrix directed adipogenesis and neurogenesis of mesenchymal stem cells derived from adipose tissue and bone marrow. Acta Biomater. 2016;42:46–55.
- Schiller ZA, Schiele NR, Sims JK, et al. Adipogenesis of adipose-derived stem cells may be regulated via the cytoskeleton at physiological oxygen levels in vitro. Stem Cell Res Ther. 2013;4:79.
- Crowder SW, Leonardo V, Whittaker T, et al. Material cues as potent regulators of epigenetics and stem cell function. Cell Stem Cell. 2016;18:39–52.
- Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896.
- Somaiah C, Kumar A, Mawrie D, et al. Collagen promotes higher adhesion, survival and proliferation of mesenchymal stem cells. PLoS One. 2015;10:e0145068.
- Kostić IT, Ilić VLj, Đorđević VB, et al. Erythrocyte membranes from slaughterhouse blood as potential drug vehicles: isolation by gradual hypotonic hemolysis and biochemical and morphological characterization. Colloids Surf B Biointerfaces. 2014;122:250–259.
- Young DA, Ibrahim DO, Hu D, et al. Injectable hydrogel scaffold from decellularized human lipoaspirate. Acta Biomater. 2011;7:1040–1049.
- Trivanović D, Nikolić S, Krstić J, et al. Characteristics of human adipose mesenchymal stem cells isolated from healthy and cancer affected people and their interactions with human breast cancer cell line MCF-7 in vitro. Cell Biol Int. 2014;38:254–265.
- Cottet-Rousselle C, Ronot X, Leverve X, et al. Cytometric assessment of mitochondria using fluorescent probes. Cytometry. 2011;79A:405–425.
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317.
- Debnath T, Ghosh S, Potlapuvu US, et al. Proliferation and differentiation potential of human adipose-derived stem cells grown on chitosan hydrogel. PLoS One. 2015;10:e0120803.
- Yang G, Rothrauff BB, Lin H, et al. Enhancement of tenogenic differentiation of human adipose stem cells by tendon-derived extracellular matrix. Biomaterials. 2013;34:9295–9306.
- Salzig D, Leber J, Merkewitz K, et al. Attachment, growth, and detachment of human mesenchymal stem cells in a chemically defined medium. Stem Cells Int. 2016;2016:5246584.
- Guneta V, Zhou Z, Tan NS, et al. Recellularization of decellularized adipose tissue-derived stem cells: role of the cell-secreted extracellular matrix in cellular differentiation. Biomater Sci. 2018;6:168–178.
- Zhou L, Chen X, Liu T, et al. SIRT1-dependent anti-senescence effects of cell-deposited matrix on human umbilical cord mesenchymal stem cells. J Tissue Eng Regen Med. 2018;12:e1008–e1021.
- Trivanović D, Jauković A, Branka Popović B, et al. Mesenchymal stem cells of different origin: comparative evaluation of proliferative capacity, telomere length and pluripotency marker expression. Life Sci. 2015;141:61–73.
- Holley RJ, Tai G, Williamson AJ, et al. Comparative quantification of the surfaceome of human multipotent mesenchymal progenitor cells. Stem Cell Rep. 2015;4:473–488.
- Lee J, Abdeen AA, Kilian KA. Rewiring mesenchymal stem cell lineage specification by switching the biophysical microenvironment. Sci Rep. 2014;4:5188.
- Uynuk-Ool T, Rothdiener M, Walters B, et al. The geometrical shape of mesenchymal stromal cells measured by quantitative shape descriptors is determined by the stiffness of the biomaterial and by cyclic tensile forces. J Tissue Eng Regen Med. 2017;11:3508–3522.
- Pärssinen J, Hammarén H, Rahikainen R, et al. Enhancement of adhesion and promotion of osteogenic differentiation of human adipose stem cells by poled electroactive poly(vinylidene fluoride). J Biomed Mater Res. 2015;103:919–928.
- Phinney DG. Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem. 2012;113:2806–2812.
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–643.
- Talele NP, Fradette J, Davies JE, et al. Expression of α-smooth muscle actin determines the fate of mesenchymal stromal cells. Stem Cell Rep. 2015;4:1016–1030.
- Jiu Y, Peränen J, Schaible N, et al. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J Cell Sci. 2017;130:892–902.
- Maumus M, Peyrafitte JA, D'Angelo R, et al. Native human adipose stromal cells: localization, morphology and phenotype. Int J Obes. 2011;35:1141–1153.
- Oñate B, Vilahur G, Camino-López S, et al. Stem cells isolated from adipose tissue of obese patients show changes in their transcriptomic profile that indicate loss in stemcellness and increased commitment to an adipocyte-like phenotype. BMC Genomics. 2013;14:625.
- Levin MC, Borén J. The extracellular matrix protein MAGP1 is a key regulator of adipose tissue remodeling during obesity. Diabetes. 2014;63:1858–1859.
- Wei W, Li J, Chen S, et al. In vitro osteogenic induction of bone marrow mesenchymal stem cells with a decellularized matrix derived from human adipose stem cells and in vivo implantation for bone regeneration. J Mater Chem B. 2017;5:2468–2482.
- Choi JS, Kim BS, Kim JD, et al. In vitro cartilage tissue engineering using adipose-derived extracellular matrix scaffolds seeded with adipose-derived stem cells. Tissue Eng Part A. 2012;18:80–92.
- Lee JE, Ge K. Transcriptional and epigenetic regulation of PPARγ expression during adipogenesis. Cell Biosci. 2014;4:29.
- Tan QW, Zhang Y, Luo JC, et al. Hydrogel derived from decellularized porcine adipose tissue as a promising biomaterial for soft tissue augmentation. J Biomed Mater Res. 2017;105:1756–1764.
- Müller P, Langenbach A, Kaminski A, et al. Modulating the actin cytoskeleton affects mechanically induced signal transduction and differentiation in mesenchymal stem cells. PLoS One. 2013;8:e71283.
- Mathieu PS, Loboa EG. Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways. Tissue Eng B Rev. 2012;18:436–444.
- Leung CL, Pang Y, Shu C, et al. Alterations in lipid metabolism gene expression and abnormal lipid accumulation in fibroblast explants from giant axonal neuropathy patients. BMC Genet. 2007;8:6.
- Tsai YC, Tsai SH, Chang EY, et al. Cytoskeletal protein vimentin interacts with and regulates peroxisome proliferator-activated receptor gamma via a proteasomal degradation process. J Cell Biochem. 2013;114:1559–1567.
- Hartig R, Shoeman RL, Janetzko A, et al. DNA-mediated transport of the intermediate filament protein vimentin into the nucleus of cultured cells. J Cell Sci. 1998;111:3573–3584.
- Chernoivanenko IS, Matveeva EA, Gelfand VI, et al. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015;29:820–827.
- Drehmer DL, de Aguiar AM, Brandt AP, et al. Metabolic switches during the first steps of adipogenic stem cells differentiation. Stem Cell Res. 2016;17:413–421.
- Zhang Y, Marsboom G, Toth PT, et al. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS One. 2013;8:e77077.
- Li Q, Gao Z, Chen Y, et al. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell. 2017;8:439–445.
- Jokinen R, Pirnes-Karhu S, Pietiläinen KH, et al. Adipose tissue NAD+-homeostasis, sirtuins and poly(ADP-ribose) polymerases-important players in mitochondrial metabolism and metabolic health. Redox Biol. 2017;12:246–263.
- Ou X, Lee MR, Huang X, et al. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells. 2014;32:1183–1194.
- Simic P, Zainabadi K, Bell E, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating β-catenin. EMBO Mol Med. 2013;5:430–440.
- Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J Biol Chem. 2006;281:39915–39924.
- Craft CS, Pietka TA, Schappe T, et al. The extracellular matrix protein MAGP1 supports thermogenesis and protects against obesity and diabetes through regulation of TGF-β. Diabetes. 2014;63:1920–1932.