
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Hepatocellular carcinoma (HCC) is one of the greatest public health problems worldwide, and chemotherapy remains the major approach for the HCC treatment. Doxorubicin (DOX) is one of the anthracycline antibiotics but its clinical use is limited due to its severe cardiotoxicity. In this study, novel hybrid nanoparticles by self-assembling based on pectin-doxorubicin conjugates (PDC-NPs) were fabricated for HCC treatment. The stabilized structure of the PDC-NPs was characterized by methylene blue absorption, the size, zeta potential and the morphology, which was investigated by Zetasizer nanoparticle analyzer and transmission electron microscope (TEM), of nanoparticles. The PDC-NPs achieved a sustained and prolonged release ability, which was illustrated with in vitro drug release profiles, anti-cell proliferation study, cellular uptake assay and in vivo pharmacokinetics analysis. Biocompatibility of the PDC-NPs was assessed with bovine serum albumin (BSA) adsorption test, hemolysis activity examination and viability evaluation of human umbilical vein endothelial cells. Importantly, in vivo studies of the PDC-NPs, which were performed in the athymic BALB/c nude mice, demonstrated that the PDC-NPs significantly reduced the lethal side effect of DOX. Additionally, the H&E staining and serum biochemistry study further confirmed the excellent biological security of the PDC-NPs.
Introduction
With the increases in incidence and mortality rates of hepatocellular carcinoma, HCC is gradually becoming a major public health problem and the leading cause of death in China now [Citation1,Citation2]. Surgery, chemotherapy, and radiotherapy are the main traditional methods for liver cancer treatment at present [Citation3]. For those patients with advanced HCC, chemotherapy is considered to be the predominant option because of its higher efficiency in healing compared with other therapies [Citation4].
Doxorubicin (DOX), a classical antineoplastic agent that has been widely used in HCC treatment, is one of the most effective chemotherapeutic agents approved by FDA [Citation5,Citation6]. However, therapeutic efficacy of DOX is challenged by its poor water solubility, rapid blood clearance, poor tumour selectivity and cardiotoxicity during chemotherapy [Citation7–9]. In order to conquer the defects of DOX, researchers have been investigating effective ways to reduce its clinical toxicity and improve its treatment efficacy [Citation10–16]. Macromolecular pro-drug delivery systems have been extensively studied for its attributes such as: chemical stability, sustained drug release, an increase in drug solubility and a reduced toxicity before metabolization occurs. Pectin is an anionic polysaccharide extracted from vegetable sources having many advantages, including good biocompatibility, hydrophilicity biodegradable properties and adjustable release properties [Citation17]. Moreover, the hydrophilic glucosyl-galactose in pectin allow the pectin-based drug carriers to specially interact with the asialoglycoprotein receptor (ASGPR) over-expressed on the surface of hepatoma cells [Citation18,Citation19]. In our previous study, we have prepared a macromolecular pro-drug based pectin-doxorubicin conjugates (PDC) via an amide condensation reaction [Citation20,Citation21]. Furthermore, the PDC macromolecular pro-drug self-assembles into a core-shell structure in water with DOX as a hydrophobic core and pectin as a hydrophilic shell. But the PDC macromolecular pro-drug still suffers from various drawbacks such as poor dispersibility, poor stability and irregular spherical shape [Citation20].
In the last two decades, nanoparticles (NPs) based drug delivery systems, have attracted a lot of public attention. When incorporated with suitable ligands, NPs as drug carriers could provide the site-specific delivery of anticancer agents as well as the controlled and sustained drug release [Citation22,Citation23]. Intravenous nanoparticles enter the liver via portal triads [Citation24]. In order to interact directly with hepatocytes, nanoparticles must be smaller than the diametre of liver sinusoidal fenestrations (up to 150–200 nm) [Citation25–28]. Since HCC is characterized as a typical hyper-vascular tumour, NPs could possess an enhanced in vivo performance for HCC treatment due to their enhanced permeability and retention (EPR) properties [Citation29].
According to our previous study, the presence of bound Ca2+ ions could decrease the electrostatic repulsion between the carboxyl (-COO-) groups in the pectin chains. Nucleation of inorganic compounds, CaCO3, could be used to prepare the mineralized cross-linked pectin-doxorubicin conjugates nanoparticles [Citation30,Citation31].
Herein, the aim of this research was to engineer a novel hybrid nanoparticle by solution self-assembling based on the pectin-doxorubicin conjugates for drug delivery. In this paper, PDC-NPs were prepared in aqueous media containing Ca2+ and CO3 2- ions by self-assembling based on macromolecular pro-drug with pectin as the hydrophilic shell and DOX as the hydrophobic core. As a result, the hydrophilic shell domains, the pectin chains in aqueous media containing Ca2+ and CO3 2- ions, became more condense. This method did not involve any toxic solvent and surfactant and could offer a good control over the morphology and the size of the nanoparticles. In vitro experiments showed that the PDC-NPs performed slow release and stable properties in all of the detection environments. Both in vitro biocompatibility experiments and in vivo animal studies clearly demonstrated the biological safety of PDC-NPs as an antitumour drug delivery system.
Materials and methods
Materials
Doxorubicin (DOX) were provided by the Milky Way Chemical Co., Ltd. Hubei. Pectin (esterification degree less than 50%, galacturonic acid more than 75.0%, Mw = 1.18 × 105 g/mol) were supplied by Quzhou Pectin Co., Ltd. Zhejiang. N-hydroxysuccinimide (NHS) was purchased from Shanghai Macklin Biochemical Co., Ltd. N-(3-Dimethyllaminopropyl)-N’-ethylcarbodllmlde hydrochlorlde (EDC) were obtained from Aladdin Industrial Corporation. Calcium hydroxide and Sodium bicarbonate were offered by Xilong Chemical Co., Ltd. [3–4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) was supplied by Amresco (Solon, OH). HepG2 cells and Human Umbilical Vein Endothelial Cells (HUVEC) were supplied by Shanghai Life Sciences Academe, Shanghai. BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Sprague-Dawley (SD) rats and New Zealand White rabbits were obtained from Laboratory Animal Centre, University of South China. All animals were raised at Laboratory Animal Centre in a specific pathogen-free (SPF) conditions. The use of animals and all procedures for care were licenced by the Ethics Committee of the University of South China, and were abided by the national guide and governmental regulations concerning the ethical use of animals. All solution was freshly prepared with deionized water and all other reagents were of analytical grade and used as received.
Fabrication of PDC and PDC-NPs
In our previous work, we had proposed the synthetic method of the PDC macromolecular pro-drug via amide condensation [Citation20,Citation21]. In a nutshell, 50 ml doxorubicin solution (1 mg/mL) was transfered into 50 ml pectin solution (2 mg/mL). Both NHS (30 mg) and EDC (100 mg) were added into the above mixture, and stirred for 8 h with 600 rpm at 50 °C. Then the PDC macromolecular pro-drug was generated. The targeted products were concentrated with rotary evaporator and washed with absolute alcohol, followed by a centrifugation and cleaning until the centrifugal liquid became colourless. The preparation of the prodrug was done here. In order to fabricate more condensed nanoparticles, 0.5 ml Ca(OH)2 (0.01 mol/l) solution was transferred into the 5 ml PDC macromolecular pro-drug solution system dropwise and was stirred (600 rpm) for 1 h at 50 °C. After that, 1.5 ml NaHCO3 (0.01 mol/l) solution was added to the above solution system dropwise and stirred for an additional 3 h at 50 °C. The resultant PDC macromolecular pro-drug (3 ml) was then put into a dialysis bag (MW: 14000) and dialyzed against 500 ml of distilled water at 20 °C for 24 h to obtain PDC-NPs.
Methylene blue absorption
The precise amount of the PDC macromolecular pro-drug (the control) and PDC-NPs was added to 4 ml MB (50 μg/mL) solutions and shaken for 2 min. The mixtures were separately incubated at 37 °C for 1 h, 2 h, 4 h and 6 h, followed by centrifugation at 15000 rpm for 10 min. The supernatant was aspirated after the centrifugation. The maximum absorbance of MB at the wavelength of 661 nm was selected to do the following analysis. The interaction of pectin with calcium ion was evaluated by the absorbance of MB aqueous solution.
Characterizations of PDC-NPs
In the preparation of the PDC-NPs, all of the supernatants were collected by a high-speed centrifugation (12000 rpms for 20 min). The remaining DOX in the solution was determined with a UV-vis spectrophotometer. The UV-vis values were converted into concentrations through a calibration standard curve (r2 = 0.9991), so the loading efficiency could be further determined. The drug loading content (%) and encapsulation efficiency (%) were calculated according to the following equations:
(1)
(1)
(2)
(2)
The mean particle size and Zeta-potential of the PDC-NPs in aqueous solutions were determined with a Zetasizer nanoparticle analyzer (Nano ZS-90, Malvern Instruments, Enigma Business Park Grovewood Road Malvern Worcestershire WR14 1XZ,UK). The morphological details of the PDC-NPs were evaluated by field transmission electron microscope (TEM, Hitachi S-4800, Hitachi Limited, Japan).
In vitro drug release study
In this study, the release of DOX from the PDC-NPs (5 ml) was determined by dialysis bag method. The dialysis bag was firstly immersed into 50 ml of the release media (Tris-HCl buffer) with different pH values (7.4, 6.8 and 5.0), and then shaken in a thermostatic oscillator at 37 °C. At predetermined intervals, samples from the dialysis solution (1 ml) were taken and replaced by the fresh media (1 ml).The cumulative release of DOX was subjected and investigated by a UV-Vis spectrophotometer with the absorbance at 480 nm.
BSA adsorption
BSA (0.5000 g) was first dissolved in phosphate-buffered saline (PBS) (1000 ml, pH 7.4) buffer. Then, 5 ml of the solution was separately added to 500 μL DOX and different preparations of NPs with different DOX concentrations, which were followed by a vortex mixing for 5 min. Right after that, the mixtures were incubated at 37 °C in thermostatic oscillator for 1 h, 2 h, 4 h and 6 h, respectively. After the incubation, 1 ml of each sample was withdrawn and then centrifuged at 15000 rpm for 15 min. The BSA properties of the free DOX and PDC-NPs were examined by spectrophotometry at 280 nm.
Hemolysis activity examinations
Fresh blood was obtained from the auricular vein of six healthy New Zealand White rabbits and anticoagulated with potassium oxalate. The blood cells were then rinsed three times with PBS buffer, and centrifuged to concentrate the red blood cells until the supernatant was no longer red. Different concentrations of PDC-NPs (0.5 ml) and free DOX were mixed separately with 0.5 ml of 2% red blood cell suspension. After 3 h incubation at 37 °C, the mixtures were centrifuged at 3000 rpm for 10 min. Furthermore, the supernatant fluids were assembled, and their optical density values were measured at 540 nm by a microplate reader (Labsystems Multiskan, Bio-chromatic Labsystem, Osaka, Japan). PBS buffer and distilled water were used as the negative and positive controls, respectively. The hemolysis ratio (HR) was calculated as follows:
(3)
(3)
Cell viability assay
MTT assay was used to evaluate the in vitro cytotoxicity of the PDC-NPs and free DOX. Firstly, endothelial cells and HepG2 cells were seeded at 96-well plates with a density of 6 × 103 cells/well and grew overnight at 37 °C in 5% CO2. Then the medium was discarded and 200 µL of the culture medium with the PDC-NPs and free DOX was added. With 24 h, 48 h, 72 h incubation, the plates were washed three times with PBS buffer and were further incubated in 5 mg/mL MTT solution for another 4 h. Later the medium was discarded and dimethyl sulfoxide (DMSO) was then added to dissolve the formazan crystal formed by living cells. The absorbance was measured at 570 nm using a microplate reader. Finally, the relative cell viability was calculated as follows:
(4)
(4)
In EquationEquation (4)(4)
(4) , Asample and Acontrol were calculated as the absorbance of the sample and control groups, respectively.
Cellular uptake
HepG2 cells were cultured in a saturated humidity incubator (5% CO2, 37 °C) with DMEM supplemented with 1% penicillin-streptomycin solution and 10% fetal calf serum. After 1 h, 2 h, 4 h and 6 h incubation in six-well plates (5 × 103 cells/well) and then treated with the PDC-NPs and free DOX, the HepG2 cells were suspended in PBS and cellular uptake images were measured by a flow cytometer (Beckman, California, USA).
In vivo pharmacokinetics analysis
The pharmacokinetics (PK) of the PDC-NPs was evaluated in SD rats. Six SD rats were randomly divided into two groups and were injected intravenously with 30 mg/kg of DOX and the PDC-NPs. The blood was collected from the fossa orbitalis vein (at 0.5, 1, 2, 4, 8, 12, 24, 48, 72, 96 and 120 h) and centrifuged at 4000 rpm for 10 min to isolate plasma. The supernatant were stored at −20 °C prior to its quantitative analysis by a Agilent 1260 HPLC system (Santa Clara, CA, USA). For determining the concentrations of DOX in the plasma samples, 200 μL of plasma was mixed with 1 ml ethyl acetate, including 50 μL of the 0.5 mg/mL daunorubicin as an internal standard, and the mixture was centrifuged at 4000 rpm for 10 min. The supernatant was collected, and the solvent was nitrogen dried at room temperature. The mobile phase (the mixture of 95% water and 5% acetonitrile) was added to the dry residues and was mixed for 10 min. After centrifugation, the supernatant was injected into the HPLC system. The pharmacokinetic parameters of the PDC-NPs were obtained by running the software PK Solver 2.0.
In vivo antitumour efficacy
The antitumour efficacy of the PDC-NPs and free DOX in vivo was measured by the tumour volume changes within 14 days. BALB/c nude mice were subjected to a subcutaneous injection of approximately 2 × 106 HepG2 cells in the right flank. When the tumour volume reached approximately 200 mm3, the mice were divided into three groups randomly (six mice per group): (1) saline, (2) DOX, (3) the PDC-NPs. 0.2 ml of the above samples (DOX: 5 mg/kg) were injected into mice via the tail vein every other day, respectively. Animals were weighed and tumour volume was recorded with a digital caliper every other day. The tumour volume was calculated: tumour volume V (mm3) = a (long diametre, mm) × b2 (short diametre, mm2)/2. Animals were sacrificed after 14 days treatment and hearts and tumours were collected for the hematoxylin and eosin staining.
Organ damage assays
The SD rats were randomly divided into three groups and injected intravenously with saline, DOX and the PDC-NPs (DOX: 5 mg/kg) every other day. Blood was daily collected for 7 days after the treatment. Blood samples were incubated at 25 °C for 3 h for coagulation and then centrifuged for 10 min at 4000 rpm to obtain serum inside. With a fully automatic clinical chemistry and immunoturbidimetric analyzer (Liasys, AMS Alliance), serum biochemical analysis was carried out. Creatine kinase (CK), creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) were measured to evaluate the cardiac toxicity. Serum aminotransferase (AST) and alanine transaminase (ALT) were used as the markers for the diagnosis of liver functions. Moreover, blood urea nitrogen (BUN) and creatinine (Cr) were measured to assess the kidney functions.
Statistical analysis
All values were presented as the mean ± standard deviation (SD). Statistical analysis was performed with unpaired Student’s t-test. *p < .05, **p < .01 and ***p < .001 was considered to be statistically significant.
Results and discussion
Synthesis and characterization of PDC-NPs
As shown in scheme 1, a novel nanoparticle was fabricated. DOX shows vigorous hydrophobic property while pectin exhibits strong hydrophilicity, therefore they can easily form a core-shelled structure in which DOX is the hydrophobic core and pectin is the hydrophilic shell.
Scheme 1. Schematic illustration of the PDC-NPs.
The structural characterization of the PDC-NPs was confirmed by methylene blue absorption in our previous research [Citation20]. In aqueous medium, calcium cations were bound to ionic carboxylic groups of pectin by electrostatic force, which resulted in a 3 D structure known as “egg-box” in order to reduce the absorption of MB cations onto the carboxyl groups of pectin [Citation32,Citation33]. As illustrated in , each concentration of PDC-NPs presented a lower MB cation adsorption rate than that of the corresponding control, which could be ascribed to an interaction of the carboxylic groups of the pectin structure with calcium cations and the resultant reduced the adsorption of MB onto pectin. Furthermore, adsorption rate of MB was unchanged in different incubation time, indicating PDC-NPs were stable.
Figure 1. Characterization of the PDC-NPs. (A) Methylene blue absorption (B) Size distribution and (C) Zeta potential of the PDC-NPs. (D) TEM images of the PDC-NPs. (E) In vitro cumulative release profiles of the PDC-NPs suspended in PBS at pH 5.0, 6.8, 7.4 and 37 °C.
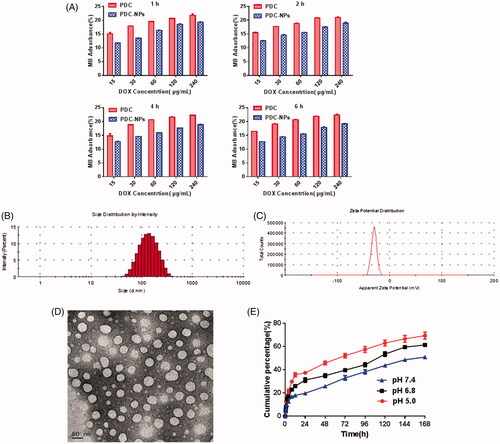
The size and size distribution of the PDC-NPs were measured by Zetasizer. From , we can see that the mean particle size of water-soaked nanoparticles was about 123.5 nm, in accordance with the standard of nanoparticle size. The TEM images of the dried PDC-NPs were presented in . The diametre of nanoparticles was about 50 nm with a regular spherical shape. Compared with the size of water-soaked nanoparticles, the size of dried nanoparticles from TEM is much smaller. The same regularity can be seen in the previous studies [Citation31]. The zeta potential of the PDC-NPs was −29.3 mV as shown in . The negative charge on the surface of the nanoparticles is favorable for a long circulation in vivo and an accumulation into the tumours via EPR effects.
According to our previous study [Citation20,Citation30,Citation31], the aqueous media used to prepare the nanoparticles are basic through the addition of Ca(OH)2 and NaHCO3. And the Ca2+ and CO3 2− play a role in stabilizing the structures of nanoparticles. In an aqueous medium containing Ca2+, pectin can bind Ca2+ cations for a formation of the “egg-box” structure. Due to the repulsive forces between the deprotonated carboxyl groups, pectin chains exist in a stretched conformation. Therefore, the PDC-NPs were prepared in aqueous solutions containing Ca2+ and CO3 2- ions by the self-assembling of the macromolecular pro-drugs with pectin as the hydrophilic shell and DOX as the hydrophobic core. The presence of the bound Ca2+ ions could decrease the electrostatic repulsion between carboxyl (–COO-) groups in the pectin chains. Hydrophilic shell domains, the pectin chains in aqueous media containing Ca2+ and CO3 2− ions became more condense, which led to a significant decrease of the mean size of the nanoparticles. Compared with our previous research [Citation21], the good dispersity, superior stability, smooth surface and uniform size of the nanoparticles indicated that we proposed method in this work could offer a good control over the size of the nanoparticles.
UV analyses showed that the encapsulation efficiency and drug loading for the PDC-NPs was 56.96% ± 2.8% (n = 3) and 22.07% ± 2.6% (n = 3), respectively.
In vitro drug release
The PDC-NPs release profile at different pHs were evaluated as shown in . pH 5.0, 6.8 and 7.4 were used to simulate the intracellular acidic, lysosome environment and physiological condition. In the experiments, the PDC-NPs performed a prolonged release ability under different pHs. There were significant differences between pH 7.4 and pH 5.0 in terms of the rate of DOX release. The rate of DOX release at pH 5.0 was notably higher than that at pH 7.4, and the cumulative percentage of DOX released at pH 7.4 was approximately 30% within 72 h but the same percentage in 8 h at pH 5.0. The results manifested that the resulting nanoparticles possessed injectable properties as a sustained and prolonged drug release system.
Biocompatibilities of PDC-NPs
As now well known, the good biocompatibility is essential for applications of most drug delivery. In this work, the protein adsorption test was carried out with BSA as a model protein to examine the stability of PDC-NPs in blood circulation and its potential applications in vivo. As shown in , the PDC-NPs indicated a slight BSA adsorption for all the concentrations during the incubation while the free DOX interacted strongly with BSA.
Figure 2. (A) BSA adsorption on free DOX and the PDC-NPs at 1 h, 2 h, 4 h and 6 h. (B) Percentage of red blood cell hemolysis incubated with free DOX and the PDC-NPs. (C) In vitro cell viability toward endothelial cells incubated with free DOX and the PDC-NPs at 24 h, 48 h and 72 h (*p < .05, **p < .01, ***p < .001, n = 3).
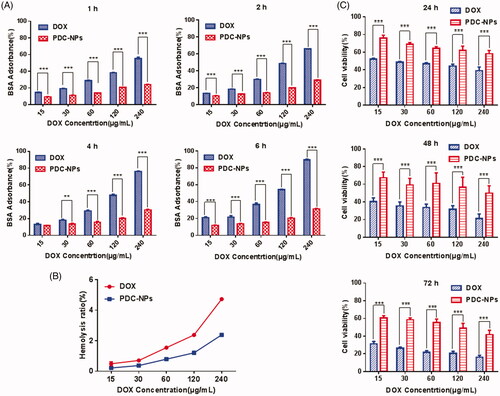
In addition, the haemolysis rate (HR) of the PDC-NPs was assessed by a hemolysis assay. The exposure of rabbit blood cells to different concentrations of the PDC-NPs was investigated (). The PDC-NPs resulted in a less haemolysis destruction to red blood cells than the free DOX even at high concentration, which appeared be due to its egg-box shaped structure between carboxyl groups on the pectin and calcium ion. These results indicated that the PDC-NPs possesses good suitability and good haemocompatibility for intravenous injection.
The viability of endothelial cells was assessed by MTT to investigate the cytotoxicity of the PDC-NPs. demonstrated the viabilities of cells treated with the different concentrated PDC-NPs for 24 h, 48 h and 72 h, respectively. As seen in , the PDC-NPs exhibited a less cytotoxicity than that of DOX. Furthermore, cell viability was decreased either when the concentration of PDC-NPs was higher or when the incubation time was longer, suggesting a concentration-dependent and time-dependent cytotoxic effect of PDC-NPs on cell viability. All results indicated that the PDC-NPs were low toxic, highly biocompatible and hemocompatible, which were the essential characteristics for drug delivery.
Cell viability assay
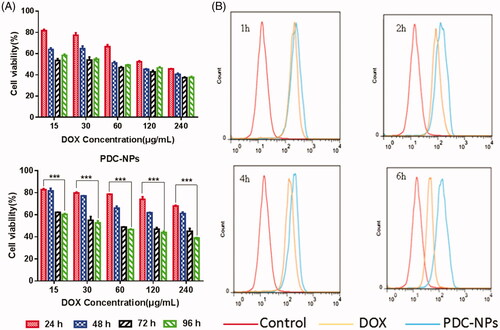
The effect of the PDC-NPs on the viability of HepG2 cells was evaluated with a MTT assay. As shown in , the viability of HepG2 cells decreased as the concentration of the PDC-NPs or the incubation time increased. The result revealed a PDC-NPs concentration-dependent and incubation time-dependent manner for cell viability. Similar results were found in the drug release profiles, which were attributed to the sustained and prolonged release of the PDC-NPs.
Figure 3. (A) In vitro cell viability toward HepG2 cells incubated with free DOX and the PDC-NPs with various DOX concentrations for 24 h, 48h, 72h and 96 h. (B) Flow cytometry of HepG2 cells incubated with free DOX and PDC-NPs for 1 h, 2 h, 4 h and 6 h (***p < .001, n = 3).
Cellular uptake
In order to assay the cell uptake process, the uptake of the PDC-NPs and free DOX by HepG2 cells was measured with flow cytometry. As presented in , the PDC-NPs were ingested by cells significantly more than the free DOX at all incubation periods (1 h, 2 h, 4 h and 6 h). Moreover, the cell uptake of the PDC-NPs finally reached a plateau at the end of the measurement. These results further proved that the PDC-NPs exhibited the sustained and prolonged release, which was also in consistence with the drug release profiles in vitro.
In vivo pharmacokinetics analysis
The mean plasma concentration-time curves of DOX and PDC-NPs were shown in . It indicated that the PDC-NPs exhibited a significant pharmacokinetic advantage compared to the free DOX. Twenty-four h after the tail vein injection, the free DOX was undetected while the DOX released by the PDC-NPs was still detectable after 120 h. This meant that the PDC-NPs displayed prolonged PK properties over the free DOX in vivo. The corresponding pharmacokinetic parameters were presented in . The half-life (T1/2) of the PDC-NPs was 147.91 h and was 8 times longer than the T1/2 of the free DOX (18.28 h). The area under curve from time zero extrapolated to infinity (AUC0-∞) of the PDC-NPs was 6.3 times higher than that of the free DOX. Furthermore, AUC from time zero to the sampling time of the last measurable concentration (AUC0–t) values of the PDC-NPs were 11233.93 μg·mL−1·h−1 higher than that of the free DOX. This result indicated that the bioavailability of the PDC-NPs was dramatically improved compared to that of the free DOX. The steady-state volume of distribution (Vss) of the PDC-NPs was 6.60 mg/(μg/mL), which was 1.3 times higher than that of the free DOX (Vss = 5.09 mg/(μg/mL)). These results demonstrated that the PDC-NPs performed as a proper and reliable drug delivery system for a sustained drug release, which was in consistence with the findings of the in vitro drug release profiles.
Figure 4. Mean plasma DOX concentration–time curves after tail vein i.v. injection of free DOX and PDC-NPs to SD rats (n = 3).
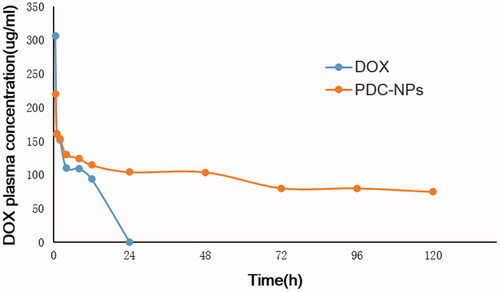
Table 1. Pharmacokinetic parameters of DOX and the PDC-NPs administration to SD rats (n = 3).
In vivo antitumour efficacy
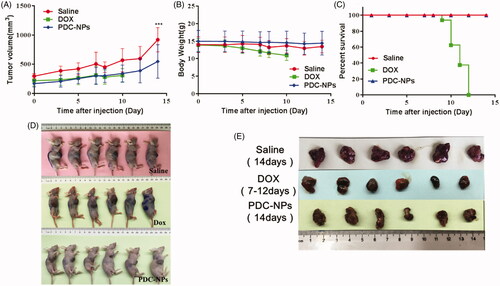
The in vivo anti-cancer efficacy of the PDC-NPs was assessed with the HepG2 xenograft BALB/c nude mice model. Within 14 days of observations, the tumour growth was partially inhibited by both the PDC-NPs and free DOX (). Although the group of mice treated with the free DOX showed a remarkable inhibition of tumour growth, the body weight of this group decreased gradually since day 3, indicating the appreciable toxicity of DOX (). The body weight of mice injected with the PDC-NPs remained stable, which suggested a low systemic toxicity of the PDC-NPs. Following the free DOX injection, the weak mice were euthanized and their tumours were collected (). Other investigators also showed that the terminated animals in free DOX group in the treatment of 6 mg/kg on day 7 due to the severe systemic toxicity [Citation34,Citation35]. In addition, the tumour volumes was investigated by Digital SLRs and given in . The tumour size from the control (saline) group was obviously larger than that from the PDC-NPs group, which was a strong evidence for the tumour-inhibiting effect of the PDC-NPs. Moreover, the PDC-NPs showed a higher in vivo safety in the nude mice models compared to the free DOX.
Figure 5. Antitumour efficacy of DOX and the PDC-NPs in tumour-bearing athymic BALB/c nude mice including (A) tumour growth curve, (B) body weight change, (C) survival rate, (D and E) photos of tumour tissues and mice treated with saline, DOX and the PDC-NPs at the end of the study. (***p < .001, n = 6).
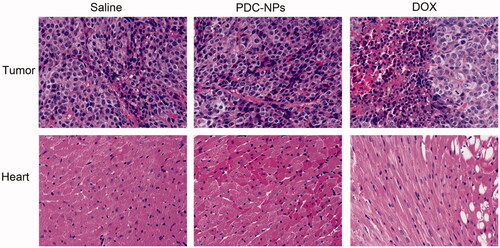
The morphological changes of tumour and heart were evaluated by the H&E staining. As shown in , the heart morphology of the PDC-NPs group appeared to be normal and significantly indifferent from the control (saline) group. However the nude mice treated with the free DOX suffered from heart muscle cracks, myocardial distortions and the emergence of empty structure. On another hand, we observed that the dissolution and necrotic tumour cells existed in the tumour tissues of the animals that received either the PDC-NPs or the free DOX, indicating that the tumour cells underwent apoptosis. Therefore, these results supported that the PDC-NPs were much safer in vivo when compared to the free DOX.
Figure 6. Tumours and hearts sections of mice stained with hematoxylin and eosin (HandE) after treatments with saline, the free DOX, and the PDC-NPs for 14 days.Original magnifications are 200×.
Organ damage assays
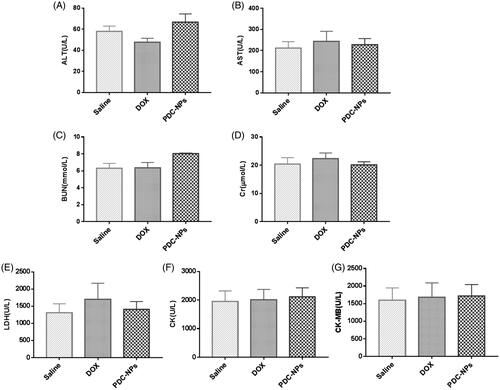
In vivo toxicity of the prepared PDC-NPs was assessed through biochemical measurement of various functional indexes of heart (CK, CK-MB and LDH), liver (ALT, AST) and kidney (BUN and Cr). As shown in , like the saline control group, all of these clinical indexes remained normal in the PDC-NPs treated animals, suggesting that the PDC-NPs did not cause severe damages to the heart, kidney or liver of treated mice.
Figure 7. Plasma levels of biochemical markers in mice after treatment with saline, the free DOX, and the PDC-NPs (A) ALT, (B) AST, (C) BUN, (D) Cr, (E) LDH, (F) CK, (G) CK-MB (n = 6).
Conclusions
In summary, we developed a novel strategy to fabricate the hybrid PDC-NPs in aqueous media containing Ca2+ and CO3 2- ions by self-assembling of the macromolecular pro-drugs with pectin as the hydrophilic shell and DOX as the hydrophobic core. In an aqueous medium containing Ca2+, the hydrophilic shell domains become more condense. The good dispersity, superior stability, smooth surface and uniform size of the nanoparticles indicated that this method could offer good controls over the morphology and the size of the resulting PDC-NPs. Furthermore, in vitro drug release profile, cell viability study, cellular uptake assay and in vivo pharmacokinetics analysis indicated that the resulting PDC-NPs performed a slow release and stable properties in all detection environments. Both in vitro cellular experiments and in vivo animal studies revealed that these PDC-NPs possessed a good biological safety. In a word, the resulting PDC-NPs demonstrated great potential as a sustained drug delivery system for the HCC therapy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. Ca Cancer J Clin. 2016;66:115–132.
- Zamani P, Riahi MM, Momtaziborojeni AA, et al. Gankyrin: a novel promising therapeutic target for hepatocellular carcinoma. Artifi Cell Nanomed Biotechnol. 2017;1:1–13.
- Huang X, Zhang W, Guan G, et al. Design and functionalization of the nir-responsive photothermal semiconductor nanomaterials for cancer theranostics. Acc Chem Res. 2017;50:2529–2538.
- Li Y, Lin J, Liu G, et al. Dual-acting, function-responsive, and high drug payload nanospheres for combining simplicity and efficacy in both self-targeted multi-drug co-delivery and synergistic anticancer effect. Int J Pharm. 2016;512:194–203.
- Zhu W, Li Y, Liu L, et al. Supramolecular hydrogels as a universal scaffold for stepwise delivering Dox and Dox/cisplatin loaded block copolymer micelles. Int J Pharm. 2012;437:11–19.
- Pan JX, Chen G, Li JJ, et al. Isocorydine suppresses doxorubicin-induced epithelial-mesenchymal transition via inhibition of ERK signaling pathways in hepatocellular carcinoma. Am J Cancer Res. 2018;8:154–164.
- Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16:3267–3285.
- Gabizon AA. Liposome circulation time and tumor targeting: implications for cancer chemotherapy. Adv Drug Deliv Rev. 1995;16:285–294.
- Ak G, Yɪlmaz H, Sanlɪer SH. Preparation of magnetically responsive albumin nanospheres and in vitro drug release studies. Artifi Cell Nanomed Biotechnol. 2014;42:18–26.
- Li M, Bu W, Ren J, et al. Enhanced synergism of thermo-chemotherapy for liver cancer with magnetothermally responsive nanocarriers. Theranostics. 2018;8:693–709.
- Hu R, Zheng H, Cao J, et al. Synthesis and in vitro characterization of carboxymethyl chitosan-cba-doxorubicin conjugate nanoparticles as pH-sensitive drug delivery systems. J Biomed Nanotechnol. 2017;13:1097–1105.
- Hu R, Zheng H, Cao J, et al. Self-assembled hyaluronic acid nanoparticles for pH-sensitive release of doxorubicin: synthesis and in vitro characterization. J Biomed Nanotechnol. 2017;13:1058–1068.
- Li Y, Yang DD, Zhang Y, et al. Novel DSPE-PEG-cholic acid-modified liposomes with hepatic targeting properties improve the anti-tumor efficacy of oral doxorubicin hydrochloride for liver tumor-bearing mice. J Biomed Nanotechnol. 2017;13:727–736.
- Hu DR, Chen LJ, Qu Y, et al. Oxygen-generating hybrid polymeric nanoparticles with encapsulated doxorubicin and chlorin e6 for trimodal imaging-guided combined chemo-photodynamic therapy. Theranostics. 2018;8:1558–1574.
- Qu Y, Niu T, Liu ZG, et al. Recent progress of doxorubicin nanomedicine in hematologic malignancies. Nanosci Nanotechnol Lett. 2017;9:1861–1874.
- Yao C, Tian J, Wang H, et al. Loading-free supramolecular organic framework drug delivery systems (sof-DDSs) for doxorubicin: normal plasm and multidrug resistant cancer cell-adaptive delivery and release. Chinese Chem Lett. 2017;28:893–899.
- Ji F, Li J, Qin Z, et al. Engineering pectin-based hollow nanocapsules for delivery of anticancer drug. Carbohydr Polym. 2017;177:86–96.
- Zheng G, Zhao R, Xu A, et al. Co-delivery of sorafenib and siVEGF based on mesoporous silica nanoparticles for ASGPR mediated targeted HCC therapy. Eur J Pharm Sci. 2018;111:492–502.
- Zhao R, Li T, Zheng G, et al. Simultaneous inhibition of growth and metastasis of hepatocellular carcinoma by co-delivery of ursolic acid and sorafenib using lactobionic acid modified and pH-sensitive chitosan-conjugated mesoporous silica nanocomplex. Biomaterials. 2017;143:1–16.
- Yu CY, Yang S, Li ZP, et al. The in-situ one-step synthesis of a PDC macromolecular pro-drug and the fabrication of a novel core-shell micell. Curr Pharm Des. 2016;22:506–513.
- Li ZP, Jiang MC, Chen B, et al. Fabrication and characterization of a novel self-assembling micelle based on chitosan cross-linked pectin–doxorubicin conjugates macromolecular pro-drug for targeted cancer therapy. Rsc Adv. 2018;8:12004–12016.
- Nafee N, Forier K, Braeckmans K, et al. Mucus-penetrating solid lipid nanoparticles for the treatment of cystic fibrosis: Proof of concept, challenges and pitfalls. Eur J Pharm Biopharm. 2018;124:125–137.
- Zhou Y, Peng Z, Seven ES, et al. Crossing the blood-brain barrier with nanoparticles. J Control Release. 2018;270:290–303.
- Zhang YN, Poon W, Tavares AJ, et al. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348.
- Wang H, Thorling CA, Liang X, et al. Diagnostic imaging and therapeutic application of nanoparticles targeting the liver. J Mater Chem B. 2015;3:939–958.
- Gershwin ME, Vierling JM, Manns MP. Liver immunology: principles and practice. 2nd ed. Germany: Springer; 2013.
- Takakura Y, Mahato RI, Hashida M. Extravasation of macromolecules. Adv Drug Deliv Rev. 1998;34:93–108.
- Gaumet M, Vargas A, Gurny R, et al. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69:1–9.
- Xu L, Xu S, Wang H, et al. Enhancing the Efficacy and Safety of Doxorubicin against Hepatocellular Carcinoma through a Modular Assembly Approach: the Combination of Polymeric Prodrug Design, Nanoparticle Encapsulation, and Cancer Cell-Specific Drug Targeting. Acs Appl Mater Interfaces. 2018;10:3229–3240.
- Yu CY, Cao H, Zhang XC, et al. Hybrid nanospheres and vesicles based on pectin as drug carriers. Langmuir. 2009;25:11720–11726.
- Yu CY, Wang YM, Li NM, et al. In vitro and in vivo evaluation of pectin-based nanoparticles for hepatocellular carcinoma drug chemotherapy. Mol Pharmaceutics. 2014;11:638–644.
- Lessa EF, Gularte MS, Garcia ES, et al. Orange waste: a valuable carbohydrate source for the development of beads with enhanced adsorption properties for cationic dyes. Carbohydr Polym. 2017;157:660–668.
- Chambin O, Dupuis G, Champion D, et al. Colon-specific drug delivery: influence of solution reticulation properties upon pectin beads performance. Int J Pharm. 2006;321:86–93.
- Kiziltepe T, Ashley JD, Stefanick JF, et al. Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacyin vivo. Blood Cancer J. 2012;2:e64.
- Tran TH, Chi TN, Gonzalez-Fajardo L, et al. Long circulating self-assembled nanoparticles from cholesterol-containing brush-like block copolymers for improved drug delivery to tumors. Biomacromolecules. 2014;15:4363–4375.