
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
RNA interference shows a great strategy for biological studies; however, delivering of small interfering RNA (siRNA) remains challenging. Although several delivery vehicles, including cell-penetrating peptides, have been developed, their implementation is often restricted because of their endosomal entrapment. Herein, we report the formation of self-assembled nanostructures from rationally designed cyclic peptides and explore them for efficient delivery of functional biomacromolecules such as siRNA into mammalian cells. The newly obtained soft materials make stable complexes with siRNAs, thereby increasing their stability and deliver fluorescent labelled siRNA inside the cells as evident from confocal microscopy analysis. Flow cytometry analysis reveals that significant uptake of FAM-siRNA occurs in the presence of peptide nanostructures compared with siRNA alone. Peptide nanostructure-mediated delivery of very low concentration of siRNA causes significant knockdown of the target gene as observed at protein level by Western blot analysis, which is comparable to lipofectamine, commercially available transfection agent.
Background
Post-transcriptional gene silencing (PTGS) through RNA interference (RNAi) shows promising results towards the diagnosis of gene-related disease especially for cancer therapy [Citation1]. RNA interference is mediated by siRNA which binds and induces the degradation of target messenger RNA (mRNA) through RNA-induced silencing complex (RISC) [Citation2]. However, siRNA-based therapeutic strategy suffers from suitable delivery systems and poor biodistribution. Delivery of nucleic acids inside the cells is a challenging task due to the presence of negative charge on it [Citation3]. Moreover, blood nucleases are known to degrade naked siRNAs, and the generated biomolecules are quickly removed by kidneys [Citation4]. Furthermore, the hydrophilicity of siRNAs strongly hinders their cellular internalization [Citation5]. Thus, suitable delivery systems are needed to be developed to implicate siRNA therapy in the clinic. To address this issue, different types of gene delivery methods including viral and non-viral techniques have been developed to date. Viral vectors have several deep-rooted drawbacks including high acute inflammatory response, immune responses, high toxicity as well as difficulty in production of viral vector, and moreover, insertional mutagenesis is also a possible concern for some viral vectors [Citation6]. Therefore, physical non-viral approaches such as microneedle [Citation7] and hydrodynamic injections [Citation8] were exploited by researchers as alternative vehicles over to the existing viral vectors. In the last decade, nanotechnology-based non-viral siRNA delivery systems such as polymeric micelles [Citation9], liposomes [Citation10] and inorganic nanoparticles [Citation11] were explored to address the limitation of siRNA delivery and also tested in preclinical studies and human clinical trials. Even though therapeutic benefit has been noticed in animal models, efficiency of the non-viral approaches remains to be a major challenge for clinical applications owing to high toxicity, intracellular stability and resultant failure to release siRNA [Citation12], which renders them inadequate for in vivo application. In this context, peptides have drawn enormous attention to chemist, nanotechnologist and protein biologist because of their chemical and physical stability, diversity of amino acid sequence, simple structure/shapes and feasibility of synthesis in large amount. Among the diverse class of peptides, cell-penetrating peptides (CPPs) have been extensively utilized for delivery of therapeutics [Citation13]. Different CPPs including TP10, MPG, CADY, PepFect 6 (PF6), PF14, TAT, R9, R9-hLF, RV peptides, LAH4, RICK were reported for siRNA delivery [Citation14–24]. Among CPPs, RICK was reported to form self-assembled structure. Self-assembled nanostructure has several advantages including high drug loading capacities, protection of drug from external environment and stimuli responsive drug delivery [Citation25]. In order to improve the siRNA transfection efficiency, self-assembled peptide-based siRNA vectors were more commonly utilized in non-covalent formulations because covalent strategies remain less successful due to chemical modification of siRNA. Till date various self-assembled nanostructures including peptide nanotubes, nanovesicles, peptide nanofibres, peptide surfactants were reported for potential application in siRNA delivery [Citation26–28]. Compared to linear peptides cyclic peptides are much more stable as far as biological environment is concerned [Citation29]. Self-assembly of cyclic peptides towards nanotube formation using D-amino acids was first developed by Ghadiri et al. [Citation30, Citation31]. Recently, cyclic peptides L-AcC(FKFE)2CG-NH2, D-Ac-C(FKFE)2CG-NH2, L-AcC(WR)4CG-NH2 and RGD10-10R were reported for siRNA delivery and significant gene knockdown was observed in both in vitro and in vivo [Citation32,Citation33]. However, clinical implementation of these delivery systems is not successful yet. Hence, there is a need to develop more efficient self-assembled nanostructures for siRNA delivery. Very recently, tryptophan- and arginine-based cyclic peptides were reported to form self-assembled vesicle-like structures and they have been shown to act as molecular transporters. However, self-assembly process of these peptides was time-consuming (10–12 days) and frequency of nanostructures formation was found to be low [Citation34]. Moreover, they were not explored for macromolecular delivery. Based on this report, we hypothesized that tailoring of these peptide sequences might develop instant self-assembled nanostructures and potential carrier for siRNA delivery. To accomplish this, numbers of cyclic peptides containing L-tryptophan and L-arginine were synthesized using solid phase peptide methods where glycine was used as linker in all sequences. Herein, self-assembled nanostructure formation by cyclic peptides was demonstrated and characterized. Furthermore, their efficiency as delivery vehicles for siRNA was evaluated.
Materials
Fmoc-L-amino acid, 2-(1H−Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), Fmoc-amino acid, trityl chloride resins and coupling reagents were purchased from ChemPep (Miami, FL). Tris, glycine, SDS, agarose, Coomassie Brilliant Blue R250 were purchased from e, LLC. MTT, Na2EDTA, NaHPO4, KHPO4, KCl and NaCl were purchased from HiMedia (Mumbai, India). N,N-diisopropylethylamine (DIPEA), N,N-dimethylformamide (DMF), acetic acid, methanol, ethanol and other chemicals purchased from SRL Chemicals (India). Polypropylene columns were purchased from Bio-Rad (California, USA). GAPDH siRNA and FAM-GAPDH siRNA were purchased from Ambion. Monoclonal GAPDH antibody and polyclonal β-actin were purchased from Abgenex (Bhubaneswar, India).
Methods
Synthesis and purification of cyclic peptides
The synthesis of amphipathic cyclic peptides was performed according to the previously reported procedure [Citation35]. All reactions related to linear peptides were conducted in polypropylene columns by shaking and mixing using a vertical rotator at room temperature unless stated otherwise. Briefly, all linear peptides were made by SPPS strategy using Fmoc chemistry. HBTU and DIPEA in DMF were used as coupling and activating reagents, respectively. Fmoc deprotection at each step was done using piperidine in DMF (20% v/v). Side chain protected peptides were cleaved from the solid phase by shaking the resins with a mixture of trifluoroethanol (TFE)/acetic acid/dichloromethane (DCM) (2:2:6, v/v/v, 15 ml) for 2 h. The liquid was separated from resin by filtration and then evaporated by rotary evaporator to get dry side chain-protected linear peptides. The cyclization of the linear peptides was performed in the presence of HOAT and DIC in dry DMF (100 ml) and dry DCM (40 ml) for 24 h. Then, solvent was evaporated. The side chain deprotection was done with trifluoroacetic acid (TFA)/thioanisole/anisole/1,2-ethanedithiol (EDT) (90:5:2:3 v/v/v/v) for 2 h. The crude peptides were precipitated by the addition of cold diethyl ether (75 ml) and purified by Agilent 1260 infinity Quaternary LC system reversed-phase column (2.1 cm× 25 cm) using a gradient system.
Detection of molecular weight of cyclic peptides by mass spectrophotometer
The molecular mass of cyclic peptide was determined by matrix-assisted laser desorption/ionization-time of flight (MALDI TOF/TOF) mass spectrometry of AB SCIEX TOF/TOFTM 5800 System at central proteomics facility (Institute of life sciences, Bhubaneswar, India). Sample and matrix CHCA were mixed in 1:1 ratio and the sample was spotted in MALDI plate and allowed to dry. Molecular mass of the cyclic peptides was collected.
CP I: HR-MS (MALDI-TOF, m/z): [M]+ calcd for C76H100N28O12, 1597.79; found 1597.78
CP II: HR-MS (MALDI-TOF, m/z): [M]+ calcd for C59H78N22O10, 1255.39; found 1255.66
CP III: HR-MS (MALDI-TOF) (m/z): [M]+ calcd for C58H86N26O12, 1339.47; found 1339.7
CP IV: HR-MS (MALDI-TOF) (m/z): [M]+ calcd for C46H62N18O10, 1027.1; found 1027.47
CP V: HR-MS (MALDI-TOF) (m/z): [M]+ calcd for C46H62N18O10, 1027.1; found 1027.39
HR-TEM analysis
2-mM cyclic peptides were prepared in aqueous solution and incubated for 48 h at room temperature for formation of the self-assembled nanostructure. 10 µl samples were taken into the formvar-coated carbon grid of mesh size 300 and allowed for resting for 2 min. After drop casting of peptide solution, grids were then stained with uranyl acetate (20 μL, 2%) for 2 min. By using filter paper, excess stain was removed and grids were allowed to dry at room temperature overnight. The image was captured using a Field Emission Gun-Transmission Electron Microscope 300 Kv (HR-TEM 300 kV) at central facility, IIT Mumbai.
Dynamic light scattering
100 µM cyclic peptide, that is CP I, CP II, CP III, CP IV and CP V, was used to analyze the hydrodynamic size of the peptides in Zetasizer Nano series Nano ZS90 (Malvern Instruments, Malvern, UK). Particle sizes were measured in triplicate. 1 ml of sample was used in DLS. The intensity-based zeta-averaged hydrodynamic diameters were reported based on 11 scans and 3 repeated cycles.
Gel shift experiments to study siRNA/peptide binding
The binding assay was performed using 100 pmol siRNA and different concentrations of cyclic peptides ranging from 1:10 to 1:100 in 20 µl DMEM without serum and incubated for 30 min at room temperature. Then, samples were analyzed in agarose gel electrophoresis (Model: Bio-Rad, California, USA) using 2% agarose gel (w/v) impregnated with ethidium bromide for 30 min at 80 V. TAE buffer (5 mM Tris base, 2.5 mM acetic acids, 0.1 mM Na2EDTA) was used as an electrophoresis running buffer. Gels were scanned by Gel Doc™ EZ Bio-Rad imager. In order to investigate the stability of peptide–siRNA complex, the physical mixture was treated with 10% FBS, 1 N HCl,1N NaOH, 5 mM glutathione, 2 nmol proteinase K, 510 nmol trypsin, respectively, for 30 min. Effect of RNase A was investigated by incubating the mixture with RNase A for 5 min.
Self-assembly and thermal stability of self-assembled cyclic peptide
In order to evaluate the polymerization of self-assembled cyclic peptide, semi-native SDS-PAGE was performed according to previous reports [Citation36, Citation37]. In this study, cyclic peptides were dissolved separately in Milli-Q water and 1× PBS, respectively. Then, 20.7 µL of 100 µM self-assembled cyclic peptides were taken for the above-mentioned study which was performed on a Mini-PROTEAN 3 electrophoresis system (Bio-Rad, California, USA) using 15% gel. Sample buffer (200 mM Tris–HCl pH 6.8, 25% (v/v) glycerol, 8% (w/v) SDS, 0.2% (w/v) bromophenol blue, in Millipore H2O) required to load the sample was added in 1:3 ratio in electrophoresis at 80 V. The peptide bands were stained with Coomassie Brilliant Blue and gel was scanned in Gel Doc™ EZ Bio-Rad imager. In order to investigate thermal stability of self-assembled nanostructure, 20.7 µL of 100 µM CP I and CP III was heated at 90˚C for 15 min in thermomixer comfort (Eppendorf). Then, they were loaded in the gel and experiment was carried out following the above-mentioned methods.
Cell culture
Human colon cancer cell line and HCT116 cells were obtained from ATCC (Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% foetal bovine serum (FBS), 3.5 mM glutamine, 1× antibiotic. Cells were cultured as exponentially growing sub-confluent monolayers in a humidified atmosphere containing 5% CO2.
Cellular uptake of FAM-siRNA: confocal microscopy
HCT 116 cells (2 × 105 cells in 1 ml media per well) were grown in six-well tissue culture plates on glass coverslips prior to the experiment. Control FAM-labelled siRNA (0.04 nmol) was mixed with 4 nmol peptides in 500 μl of DMEM and incubated at room temperature for 30 min, and then it was added to cells. The cells were incubated at 37 °C in 5% CO2 for 3 h and then rinsed twice with 1× PBS. Then, the cells were fixed for 5 min 4% paraformaldehyde in 1× PBS and nucleus staining was carried out using DAPI (1:5000) followed by washing with 1× PBS. The coverslips were mounted with mounting media. Finally, cells were observed under Leica TCS SP confocal laser scanning microscope. Images were acquired at excitation wavelength 488 nm argon laser (40%) and emission wavelength from 500 to 580 for FITC. DAPI images were collected at excitation wavelength 405 nm and emission at 410–490 nm. LAS AF software was used for image analysis.
Cellular uptake: FACS analysis
Human colon tumour (HCT116) cell lines were grown in six-well plates (2 × 105cells/well) in complete media overnight. Then, media was removed and cells were washed with PBS (1×) twice. FAM-labelled GAPDH siRNA (200 nM) was mixed with cyclic peptide (30 µM) in Opti-MEM and incubated for 30 min at room temperature. Then, the mixture was added into the cells and incubated for 4 h at 37 °C. After 4-h incubation, media containing peptide and siRNA was removed. The cells were digested with 0.25% trypsin/0.53 mM EDTA for 5 min to detach from the surface. Then, they were collected by centrifuge at the speed of 700 g for 5 min followed by washing with PBS twice and finally analyzed by BD FACSCanto™ II.
Mechanism of cellular uptake of FAM-siRNA in the presence of endocytic inhibitors: fluorescence microscopy
HCT 116 cells (2 × 105 cells in 1 ml media per well) were grown in six-well tissue culture plates on glass coverslips prior to the experiment. All wells containing cells were pretreated with endocytic inhibitors for 30 min before performing siRNA uptake. In order to check the pathway of siRNA uptake, several inhibitors such as methyl-β-cyclodextrin (2.5 mM), chloroquine (100 μM), nystatin (50 μg/mL), 5-(N-ethyl-N-isopropyl)amiloride (EIA, 50 μM) and chlorpromazine (30 μM) were used. FAM-labelled siRNA (0.04 nmol) was mixed with 4 nmol peptides in 500 μl of DMEM and incubated at room temperature for 30 min. Then, the mixture was added to the cells and incubated at 37 °C in 5% CO2 for 3 h. After incubation period, the cells were washed twice with 1× PBS and then they were fixed with 4% paraformaldehyde in 1× PBS for 5 min and DAPI (1:5000) was used for nucleus staining followed by washing with 1× PBS. The coverslips were put upside down on glass slide along with mounting media. Finally, cells were observed under Olympus BX61 fluorescence microscope with a 100× objective. Images were acquired with a Leica DFC365 FX camera and Image-Pro Express software.
GAPDH knockdown: Western blot analysis
HCT 116 cells were grown up to 80% confluence in six-well plates and washed three times with DMEM (without serum). Separately, cyclic peptide (24 μM) and GAPDH siRNA (600 nM; Ambion) were mixed with 500 μL of DMEM (without serum) and incubated for 30 min. Then, the cells were incubated with peptide–siRNA complex for 3 h at 37 °C in 5% CO2. Finally, complete media was added to the cells and incubated for 48 h in the above-mentioned condition. Collected cells were lysed in RIPA lysis buffer for protein isolation. Bradford assay was carried out to estimate the proteins. Samples were loaded in electrophoresis gel and proteins were separated by SDS-PAGE. Then, protein was transferred from SDS-PAGE to PVDF membrane by Western blotting apparatus (Bio-Rad, California, USA). 7.5% skim milk in 1× TBST was used as a blocking reagent in membrane for 1 h. The membrane was further incubated overnight with monoclonal GAPDH (1:7000) anti-mouse antibody (Abgenex, Bhubaneswar, India) at 4 °C. The blots were washed with 1× TBST followed by addition of alkaline phosphatase (AP) conjugated secondary antibody (Abgenex, Bhubaneswar, India). Then, the blot was washed with 1× TBST and developed using AP buffer and BCIP/NBT (Promega, Wisconsin, USA). β-actin (Abgenex, Bhubaneswar, India) polyclonal (1:5000) was used as loading control. The blot was scanned and images were quantified using ImageJ software.
MTT assay
Cytotoxicity of CPs was determined by using 3-(4,5-(dimethyl thiazol-2-yl) 2,5-diphenyltetrazolium bromide) (MTT) assay. Briefly, cells were plated (104cells/well) in 100 µl media/well in 96-well tissue culture plates and incubated for 24 h at 37 °C in a humidified atmosphere of 5% CO2. Then, cells were treated with cyclic peptides (10 µM) for 24 h at the same condition. 100 µl of MTT solution (0.5 mg/ml) was added to each well followed by incubation for 4 h. After the incubation period, the MTT solution was removed cautiously. Then, DMSO was added to dissolve generated formazan crystals. To find out the cell viability, the coloured solution was measured using microplate reader (Epoch BioTek ELISA reader) at 570 nm. Negative control experiment was performed by using untreated cells. All experiments were carried out in triplicate and the cell viability was calculated as follows:
Results
Self-assembly of peptides
Numbers of cyclic peptides namely Cyclo[WWWWGGRRRRGG] (CP I), Cyclo [WWWGGRRRGG] (CP II), Cyclo[WWGGGRRRRGGG] (CP III), Cyclo [WWGGGRRGGG] (CP IV) and Cyclo[WRGGGWRGGG] (CP V) containing L-tryptophan, L-arginine and L-glycine were synthesized () using solid phase peptides synthesis. These peptide sequences () were selected based on our previous report where cyclic amphipathic peptides were made consisting of hydrophobic (Trp) and positive charge (Arg) residues [Citation34]. Glycine was incorporated in the sequence to add more flexibility in the peptide. The sequences were designed in such a way that they might generate self-assembled structures. It was hypothesized that proper ratio of hydrophobic and positively charged residues could produce self-assembled nanostructures and entrapped biomacromolecules.
Figure 1. Chemical structures of synthesized cyclic peptides (A = CP I, B = CP II, C = CP III, D = CP IV, E = CP V).
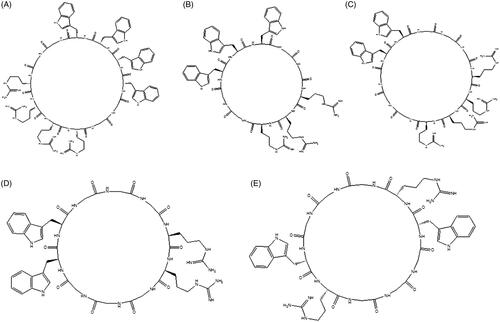
Table 1. Sequences of cyclic peptides.
In order to evaluate the structural morphology, all cyclic peptides (2 mM) were dissolved in aqueous solution and structural patterns were checked by high-resolution transmission electron microscopy (HR-TEM). HR-TEM images () indicated that all cyclic peptides form the self-assembled structure in aqueous solution.
Figure 2. HR-TEM images of self-assembled structures of cyclic peptides (A:CP I, B:CP II, C:CP III, D:CP IV, E:CP V).

Dynamic light scattering study
To determine the size distribution of nanostructures generated by cyclic peptide (100 µM), dynamic light scattering (DLS) was analysis was performed. DLS results showed that the majority (80%) of nanostructures were found in the size range of 100–250 nm. Detail hydrodynamic diameter is mentioned in .
Table 2. Hydrodynamic size of cyclic peptide measured by DLS.
Binding study of cyclic peptide with siRNA
The affinity of siRNA towards cyclic peptides was evaluated by using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) siRNA as model system. Different ratio of siRNA/cyclic peptide was incubated and their complex formation was monitored in agarose gel-shift assay. As shown in , formation of CP/siRNA complexes was started at a higher mole ratio of CP/siRNA (100/1); however, at a mole ratio of 40/1 and 20/1 siRNA molecules were found to be entirely associated with CP I and CP III, respectively (). No clear band corresponding to free siRNA was observed on the agarose gel in the above experiment, indicating strong complex formation between CP and siRNA.
Figure 3. Gel retardation assay: study of complex formation between cyclic peptide and GAPDH siRNA. (A) The preformed CP/siRNA complexes were analyzed by agarose gel electrophoresis. GAPDH gene was incubated with CP at molar ratio 1:100. (B) siRNA was incubated with different concentrations of CP I to a molar ratio ranging between 1/20, 1/40, 1/60, 1/80 and 1/100, respectively. (C) siRNA was incubated with different concentrations of CP III to a molar ratio ranging between 1/20, 1/40, 1/60, 1/80 and 1/100, respectively. (D) siRNA release from CP I and CP III in the presence of glutathione. (E) siRNA release from CP I and CP III in the presence of RNase A. (F) siRNA release from CP I and CP III in the presence of proteinase K. (G) siRNA release from CP I and CP III in the presence trypsin (H). siRNA release from CP I and CP III in different pH condition. (I) siRNA release from CP I and CP III in the presence of FBS.
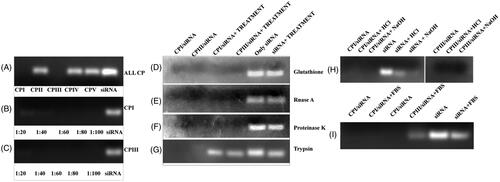
In order to mimic the in vivo system, the stability of siRNA/CP complex was evaluated in the presence of enzymes and different pH environment (). Serum RNase also plays a crucial role in reducing siRNA as the half-life of siRNA is less than 30 min [Citation38]. To accomplish the stability issue, siRNA/CP complexes were incubated with different biomolecules such as glutathione, RNase A, proteinase K, trypsin and serum, respectively. The stability of the above complex also assessed in different pH. One of the major requirements of siRNA vector is to provide protection to siRNA. Remarkably, complexes were found to be stable under all above-mentioned conditions. In contrast, in the presence of trypsin, both the complexes were found to be unstable and released siRNA from the complex. While evaluating the effect of serum towards CP/siRNA complex, siRNA was found to be partially released from CPIII as observed in the gel. The above results indicate that CP I and CP III can efficiently protect the naked siRNA from external environment.
Oligomerization and thermal stability of self-assembled peptide nanostructures
The aggregation behaviour of all cyclic peptides was investigated in aqueous solution as well as in the presence of ions. The extent of oligomerization may vary in different solvents including water and PBS. In order to compare the polymerization of peptides, they were dissolved in water and 1× PBS, respectively. The results indicated that bands corresponding to CP I and CP III in water as well as in 1× PBS were observed near 10 kDa which is much higher than their molecular weights, whereas no band was observed for CP II in aqueous solution (). In contrast, CP II in 1× PBS produced clear bands in the gel. Moreover, band intensity of CP I and CP III in 1× PBS was found to be 2.16- and 1.5-fold greater than their aqueous solution, respectively, suggesting enhanced oligomerization of CP I and CP III in the presence of salt ions.
Figure 4. Semi-native SDS-PAGE to study (A) oligomerization of CP I and CP III in water as well as in 1× PBS, and (B) thermal stability of the CP I and CP III.
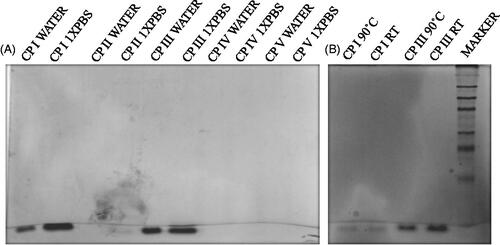
To evaluate the stability of self-assembled peptide nanostructure at high temperature, thermal stability assay was performed by semi-native SDS-PAGE [Citation36, Citation37]. To accomplish this issue, CP I and CP III were heated at 90˚C for 15 min. Bands corresponding to CP I and CP III were observed in the gel with the same intensity in both control and treated peptides. Thus, this result confirmed the thermal stability of self-assembled peptide nanostructures even at 90 °C.
Cellular uptake study: confocal microscopy
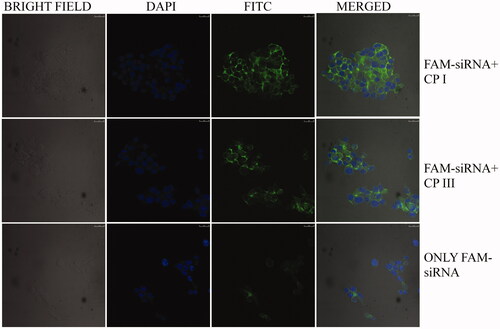
To evaluate the efficiency of carrier peptides to deliver siRNA to the cells, FAM-labelled GAPDH siRNA was taken as model siRNA and confocal microscopy was performed. For this purpose, HCT 116 cells were treated with cyclic peptides along with FAM-labelled GAPDH siRNA. As shown in , it was found that FAM-GAPDH siRNA without peptide did not enter the cells to any appreciable degree, whereas significant cellular uptake of FAM-siRNA was observed in the presence of CP I. Modest degree of uptake was noticed in the presence of CP III. Other three cyclic peptides (CP II, CP IV and CP V) did not significantly influence siRNA uptake (data not shown here).
Figure 5. Confocal microscope images of FAM-GAPDH siRNA uptake in HCT 116 cells in the presence of CP I and CP III after 3-h incubation (DAPI: 4',6-diamidino-2-phenylindole, FITC: fluorescein isothiocyanate).
Cellular uptake of FAM-siRNA by flow cytometry
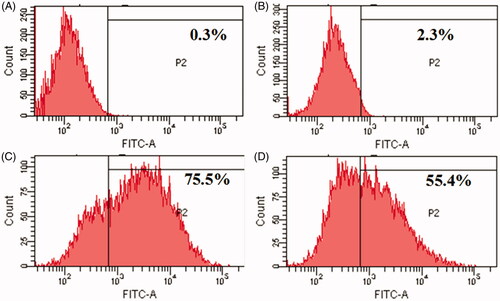
To evaluate cyclic peptides CP I and CP III as siRNA carrier, a model experiment with non-targeting fluorescent siRNA, FAM-siRNA as a cargo molecule was performed. Intracellular uptake of FAM-siRNA was measured in HCT 116 cells using fluorescence-activated cell sorter (FACS). FACS showed higher fluorescence signal in cells treated with FAM-siRNA-loaded CP I and CP III compared to that of FAM-siRNA alone. For instance, the cellular internalization of FAM-siRNA mediated by CP I and CP III was found to be approximately 75.5% and 55.4%, respectively, which is much higher than uptake of FAM-siRNA alone (2.3%) (). The above results further confirm the confocal microscopy results where bright green fluorescence of FAM-siRNA facilitated by the carriers is clearly visible inside the cells.
Figure 6. Flow cytometry analysis for cellular uptake of siRNA-loaded cyclic peptide in HCT 116 cells: (A) control cells, (B) only siRNA, (C) CP I with siRNA and (D) CP III with siRNA.
Mechanism of cellular uptake
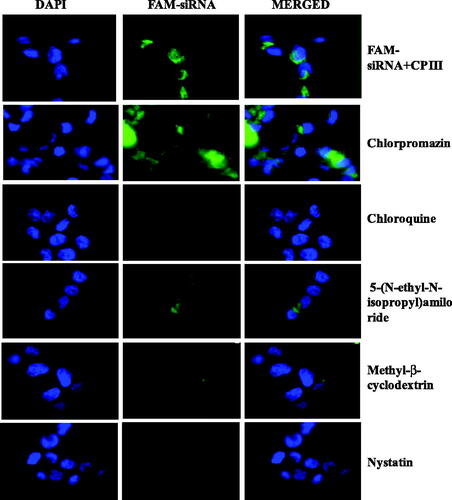
Transport of siRNA into cells involves a number of selective pathways; thus, in order to develop efficient carriers, it is imperative to understand the underlying mechanisms in each step and the fate of carrier/siRNA complexes within the cells. In order to understand the cellular uptake mechanism involved in the current study, transfection experiments were performed in the presence of several endocytic inhibitors including chloroquine, methyl-β-cyclodextrin, nystatin, 5-(N-ethyl-N-isopropyl)amiloride (EIA) and chlorpromazine that interfere with the endosomal pathway. Chloroquine inhibits the maturation of the transport vesicles into late endosomes and neutralizes the pH of the later [Citation39]. Methyl-β-cyclodextrin is known to interact with cholesterol present in cell membrane and therefore has an effect on clathrin- and caveolae-mediated uptake [Citation40]. Nystatin is also known to interact with cholesterol and influence on caveolae-mediated uptake [Citation41]. The function of EIA is identified to inhibit lipid-raft-dependent micropinocytosis and phagocytosis pathways [Citation42]. Clathrin-mediated endocytosis is inhibited by the presence of chlorpromazine [Citation43]. As depicted in , the transfection efficiency of CP I was inhibited by chlorpromazine, nystatin and partially affected by methyl-β-cyclodextrin, suggesting that the cellular uptake of CP I/siRNA complexes is dependent of the classical endosomal pathway such as caveolae-mediated uptake and clathrin-mediated endocytosis, whereas the transfection efficiency of CP III affected by chloroquine, methyl-β-cyclodextrin and nystatin, suggesting that clathrin- and caveolae-mediated uptake as well as late endosome stage is responsible as shown in .
Figure 7. Fluorescence microscopy images of CP I-mediated FAM-GAPDH siRNA uptake in HCT 116 cells in the presence of different inhibitor.
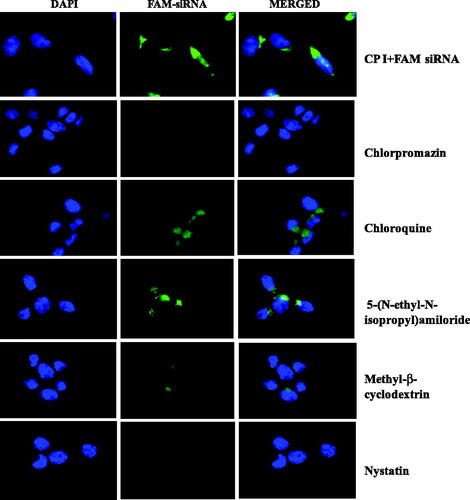
Figure 8. Fluorescence microscopy images of CP III-mediated FAM-GAPDH siRNA uptake in HCT 116 cells in the presence of different inhibitor.
GAPDH gene silencing study
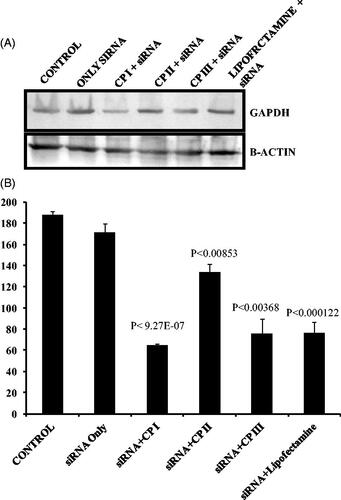
In order to evaluate the gene silencing efficiency of peptide nanostructures, functional GAPDH gene was delivered using CP I, CP II and CP III. GAPDH gene knockdown was assessed in HCT 116 cell. Western blot analysis indicated that around 2.8- and 2.4-fold gene knockdown was observed in case of CPI and CPIII, respectively, and also the data set was statistically significant (). Gene knockdown efficiency was found to be comparable with commercially available transfection agent lipofectamine.
Figure 9. GAPDH gene silencing efficiency of CP I and CP III and comparison with lipofectamine by measuring the protein level. (A) Western blots of GAPDH expression relative to β-actin expression. (B) Quantitative analysis of GAPDH expression with respect to beta actin (n = 3) in ImageJ software.
Cytotoxicity study
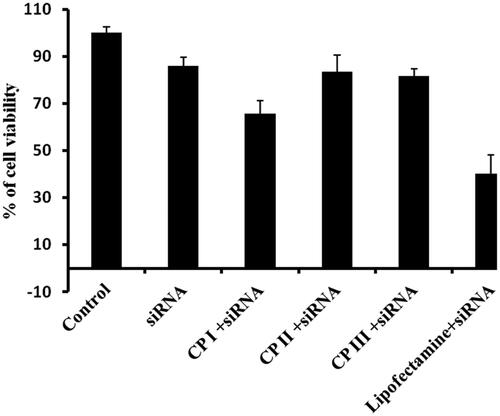
In order to exploit any drug delivery vehicle, the primary criteria, that is non-toxicity of the carrier, should be evaluated. In order to assess the cytotoxicity, cyclic peptides were incubated with cancer cells (HCT 116). The cytotoxicity of all cyclic peptides was evaluated at 10 µM concentration for 24 h (). More than 90% cell viability was observed in all cyclic peptides. Furthermore, cytotoxicity of siRNA/carrier mixture was evaluated by MTT assay. No significant cytotoxicity was observed in case of siRNA/CP complex except CP I (); however, lipofectamine 2000 exhibited significant cytotoxicity, suggesting the acceptability of cyclic peptides as delivery system for siRNA. Therefore, accounting all the above results, it be concluded that CP I and CP III are promising delivery vehicles in siRNA therapy.
Figure 10. Cytotoxicity of cyclic peptide (10 µM) in HCT 116 cancer cell line.
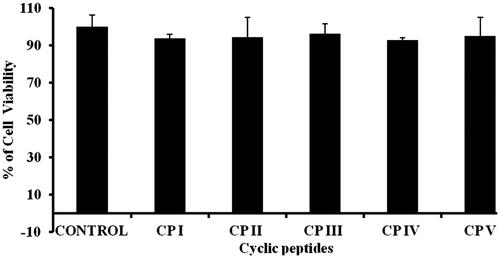
Figure 11. Cytotoxicity of CP/siRNA complex in HCT 116 cancer cell line.
Discussion
There is an urgent need to develop suitable delivery system for siRNA in order to implement in the clinic. The objective of this work was to design non-viral and non-toxic peptide-based vector to address this issue. For this purpose, few cyclic peptides were designed and synthesized to achieve self-assembled nanostructures. Among them, CP I, CP II, CP IV and CP V generated nicely organized bunch of vesicle-like nanostructures. CP III formed micelle-like structure in aqueous solution. It is pertinent to mention that all the cyclic peptides were constituted of hydrophobic tryptophan and hydrophilic arginine. It is already reported that hydrophobicity generated by tryptophan plays a major role in the self-assembly process [Citation34]. Additionally, guanidine group of arginine residues in all cyclic peptides could take part in hydrogen bonding interactions. Hence, it is presumed that hydrophobic and/or hydrogen bonds are the driving force for the self-assembly process.
Binding study indicated that selective peptides formed strong complex with siRNA. This may be due to the greater number of arginine present in those sequences compared to others. siRNA release study suggested that peptide–siRNA complexes were very much stable in the presence of several enzymes and serum; however, in the presence of trypsin, siRNA was found to be released from the peptide matrix, indicating trypsin triggered release of siRNA inside the cells.
To understand the extent of self-assembly, oligomerization of peptides should be evaluated. SDS-PAGE (sodium dodecyl sulphate–polyacrylamide gel electrophoresis) is commonly used electrophoretic technique to detect oligomerization in protein aggregation studies. SDS-PAGE depends on the capability of SDS, a negatively charged detergent, to connect to the protein of interest. Herein, SDS-PAGE technique was used to detect peptide aggregation. As a consequence of binding between negative charge of SDS and positive charge on peptide, secondary structure of the peptide is compromised. The different self-assembly behaviour of the peptides in aqueous solution as well as in PBS buffer can be attributed to the change in non-covalent interaction among the peptide monomers in the presence of different ions in the surrounding environment. Among all the cyclic peptides, CP IV and CP V did not produce any band either in water or PBS, indicating the absence of any significant oligomerization in these peptides as reflected in the gel.
In order to evaluate the efficiency of peptide nanostructures to deliver siRNA inside the cells, fluorescent labelled GAPDH siRNA was taken as model gene and observed under confocal microscope. The results clearly suggested that among the designed cyclic peptides, CP I and CP III exhibited promising results in delivering siRNA efficiently inside the cytoplasm of the cells. In order to assess the quantitative cellular uptake, FACS analysis was performed, where significant uptake in the presence of CP I and CP III was observed compared to control experiment. The results confirm that CP I and CP III can be used as siRNA carriers. Mechanism of uptake was found to be caveolae- and clathrin-mediated endocytosis. Based on the cellular uptake study, in order to evaluate the efficacy of peptide nanostructures to deliver functional gene, gene silencing study was performed by using GAPDH gene. The above study indicated significant gene knockdown in HCT 116 cells, which confirmed that delivered GAPDH gene was functional inside the cells. The newly developed delivery system was found to be equally efficient like lipofectamine, a commercially available reagent. Moreover, unlike lipofectamine cytotoxicity of cyclic peptides was found to be negligible which adds advantage for the newly developed delivery system compared to lipofectamine.
In conclusion, formation of self-assembled vesicle-like nanostructures was demonstrated from rationally designed cyclic peptides containing tryptophan and arginine residues. Furthermore, they were found to form stable non-covalent complex with siRNA. To the best of our knowledge, this is the first report on cyclic peptide-based nanostructures which allow the formation of stable complexes with siRNA via non-covalent strategy and that efficiently promote cellular uptake of siRNA. The mechanism of siRNA uptake is found to be caveolae- and clathrin-mediated endocytosis. Accounting all the results, with the fact that designed cyclic peptides are not toxic and gene knockdown efficiency at low nanomolar concentration of siRNA, suggests that these cyclic peptide carriers are extremely efficient for silencing different target genes. Therefore, peptide-based nanostructures comprise a promising technology for siRNA delivery for therapeutic purpose. In summary, water soluble efficient self-assembled peptide nanostructures were developed for siRNA delivery.
Acknowledgments
The authors thank Dr Bikash Ranjan Sahu, KIIT School of Biotechnology for assisting in FACS analysis. The authors are also thankful to Dr Amol Suryawanshi and Mr R. Rajendra Kumar Reddy, ILS, Bhubaneswar, for assisting in MALDI analysis.
Disclosure statement
The authors have no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Lam JK, Chow MY, Zhang Y, et al. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids. 2015;4:e252.
- Agrawal N, Dasaradhi PVN, Mohmmed A, et al. RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev. 2003;67:657–685.
- Sokolova V, Epple M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew Chem Int Ed Engl. 2008;47:1382–1395.
- Moreira JN, Santos A, Moura V, et al. Non-viral lipid-based nanoparticles for targeted cancer systemic gene silencing. J Nanosci Nanotechnol. 2008;8:2187–2204.
- Ly S, Navaroli DM, Didiot MC, et al. Visualization of self-delivering hydrophobically modified siRNA cellular internalization. Nucleic Acids Res. 2017;45:15–25.
- Al-Dosari MS, Gao X. Nonviral gene delivery: principle, limitations, and recent progress. AAPS J. 2009;11:671.
- Tang T, Deng Y, Chen J, et al. Local administration of siRNA through microneedle: optimization, bio-distribution, tumor suppression and toxicity. Sci Rep. 2016;6:30430.
- Bonamassa B, Hai L, Liu D. Hydrodynamic gene delivery and its applications in pharmaceutical research. Pharm Res. 2011;28:694–701.
- Gary DJ, Puri N, Won YY. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Control Release. 2007;121:64–73.
- Kim HK, Davaa E, Myung CS, et al. Enhanced siRNA delivery using cationic liposomes with new polyarginine-conjugated PEG-lipid. Int J Pharm. 2010;392:141–147.
- Jiang Y, Huo S, Hardie J, et al. Progress and perspective of inorganic nanoparticle-based siRNA delivery systems. Expert Opin Drug Deliv. 2016;13:547–559.
- Ozpolat B, Sood AK, Lopez, ‐et al. Nanomedicine based approaches for the delivery of siRNA in cancer. J Intern Med. 2010;267:44–53.
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell Res. 1988;55:1189–1193.
- Arukuusk P, Pärnaste L, Oskolkov N, et al. New generation of efficient peptide-based vectors, NickFects, for the delivery of nucleic acids. Biochim Biophys Acta. 2013;1828:1365–1373.
- Simeoni F, Morris MC, Heitz F, et al. Insight into the mechanism of the peptide‐based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724.
- Crombez L, Aldrian-Herrada G, Konate K, et al. A new potent secondary amphipathic cell–penetrating peptide for siRNA delivery into mammalian cells. Mol Ther. 2009;17:95–103.
- EL Andaloussi S, Lehto T, Mäger I, et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011;39:3972–3987.
- Ezzat K, EL Andaloussi S, Zaghloul EM, et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011;39:5284–5298.
- Chiu YL, Ali A, Chu CY, et al. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. ACS Chem Biol. 2004;11:1165–1175.
- Yamashita H, Kato T, Oba M, et al. Development of a cell-penetrating peptide that exhibits responsive changes in its secondary structure in the cellular environment. Sci Rep. 2016;6:33003
- Van Asbeck AH, Beyerle A, McNeill H, et al. Molecular parameters of siRNA-cell penetrating peptide nanocomplexes for efficient cellular delivery. ACS Nano. 2013;7:3797–3807.
- Ryu DW, Kim HA, Ryu JH, et al. Amphiphilic peptides with arginine and valine residues as siRNA carriers. J Cell Biochem. 2012;113:619–628.
- Langlet-Bertin B, Leborgne C, Scherman D, et al. Design and evaluation of histidine-rich amphipathic peptides for siRNA delivery. Pharm Res. 2010;27:1426–1436.
- Vaissière A, Aldrian G, Konate K, et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J Nanobiotechnology. 2017;15:34.
- Yan CY, Gu JW, Hou DP, et al. Synthesis of Tat tagged and folate modified N-succinyl-chitosan self-assembly nanoparticles as a novel gene vector. Int J Biol Macromol. 2015;72:751–756.
- Yan X, He Q, Wang K, et al. Transition of cationic dipeptide nanotubes into vesicles and oligonucleotide delivery. Angew Chem Int Ed Engl. 2007;46:2431–2434.
- Mazza M, Hadjidemetriou M, de Lázaro I, et al. Peptide nanofiber complexes with siRNA for deep brain gene silencing by stereotactic neurosurgery. ACS Nano. 2015;9:1137–1149.
- Qiu Y, Chow MY, Liang W, et al. From pulmonary surfactant–synthetic KL4 peptide as effective siRNA delivery vector for pulmonary delivery. Mol Pharmaceutics. 2017;14:4606–4617.
- Kwon YU, Kodadek T. Quantitative comparison of the relative cell permeability of cyclic and linear peptides. Chem Biol. 2007;14:671–677.
- Ghadiri MR, Granja JR, Buehler LK. Artificial transmembrane ion channels from self-assembling peptide nanotubes. Nature. 1994;369:301.
- Ghadiri MR, Granja JR, Milligan RA, et al. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature. 1993;366:324.
- Welch JJ, Swanekamp RJ, King C, et al. Functional delivery of siRNA by disulfide-constrained cyclic amphipathic peptides. ACS Med Chem Lett. 2016;7:584–589.
- Huang Y, Wang X, Huang W, et al. Systemic administration of siRNA via cRGD-containing peptide. Sci Rep. 2015;5:12458.
- Mandal D, Tiwari RK, Shirazi AN, et al. Self-assembled surfactant cyclic peptide nanostructures as stabilizing agents. Soft Matter. 2013;9:9465–9475.
- Mandal D, Shirazi AN, Parang K. Cell‐penetrating homochiral cyclic peptides as nuclear‐targeting molecular transporters. Angew Chem Int Ed. 2011;50:9633–9637.
- Petkau KM, Sonntag MH, Colditz A, et al. Multivalent protein assembly using monovalent self-assembling building blocks. Int J Mol Sci. 2013;14:21189–21201.
- Jung JP, Jones JL, Cronier SA, et al. Modulating the mechanical properties of self-assembled peptide hydrogels via native chemical ligation. Biomaterials 2008;29:2143–2151.
- Raemdonck K, Vandenbroucke RE, Demeester J, et al. Maintaining the silence: reflections on long-term RNAi. Drug Discov Today. 2008;13:917–931.
- Maxfield FR. (1982). Weak bases and ionophores rapidly and reversibly raise the pH of endocytic vesicles in cultured mouse fibroblasts. J Cell Biol. 1982;95:676–681.
- Kilsdonk EP, Yancey PG, Stoudt GW, et al. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–17256.
- Rothberg KG, Heuser JE, Donzell WC, et al. (1992). Caveolin, a protein component of caveolae membrane coats. Cell 1992;68:673–682.
- Gold S, Monaghan P, Mertens P, et al. A clathrin independent macropinocytosis-like entry mechanism used by bluetongue virus-1 during infection of BHK cells. PLoS One. 2010;5:e11360.
- Rejman J, Oberle V, Zuhorn IS, et al. Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169.