
Abstract
Osteoarthritis (OA) is a common debilitating disease primarily characterised by excessive loss of the articular ECM, which is composed of up to 95% type II collagen. Among the factors that contribute to the pathogenesis of OA, the natural process of aging is regarded as the most significant risk factor. AGEs, which are extremely resilient to degradation, are produced in the body naturally as a result of the Maillard process of nonenzymatic glycation and are also introduced through diet and tobacco smoke. AGEs have a high affinity for collagen and therefore accumulate in joint tissues, where they induce increased expression of proinflammatory cytokines, chemokines, and degradative enzymes. Additionally, AGEs induce oxidative stress, which further exacerbates the degradative process. Type II collagen is targeted for degradation by matrix metalloproteinases (MMPs), particularly MMP-3 and MMP-13, and AGEs have been shown to trigger increased expression of these MMPs. The role of retinoid and rexinoid receptors as specific treatment targets has been receiving increasing attention. Bexarotene is a retinoid X receptor (RXR) agonist used for the treatment of T-cell lymphoma and other cancers which has displayed a favourable safety profile. Here, we examined the roles of RXR agonism using bexarotene on AGE-induced markers of OA, including oxidative stress, inflammatory response, and MMP-mediated degradation of type II collagen. Furthermore, we demonstrate that bexarotene inhibited phosphorylation of IκBα, thereby suppressing activation of the proinflammatory NF-κB cellular signalling pathway. These findings present a basis for selective targeting of RXR by bexarotene as a potential treatment of OA induced by AGEs.
Introduction
Osteoarthritis (OA) [Citation1] is a major debilitating disease primarily characterised by excessive degradation of joint cartilage with abnormal bone growth and is considered the most prevalent joint disorder in the world [Citation2]. While OA can affect various joints throughout the body, symptomatic knee OA currently affects roughly 10% of males and 13% of females aged 60 years and older in the US alone. Furthermore, these numbers are expected to more than double by the year 2030 [Citation3]. While there are numerous factors known to contribute to the pathogenesis of OA, including idiosyncratic factors such as genetics, obesity, diabetes, joint injury, epigenetics, or excessive mechanical loading, and demographic factors including sex, age, and race, aging has been cited as the main risk factor for OA [Citation4,Citation5]. Aging contributes to the pathogenesis of OA in different ways, such as chronic age-related inflammation (“inflammaging”), cell senescence and the accompanying senescence-associated secretory phenotype (SASP), oxidative stress, and altered expression of proinflammatory cytokines and degradative enzymes [Citation6]. Another major contributing age-related factor is the accumulation of AGEs. In normal physiology, AGEs are produced over a period of weeks through the Maillard reaction, wherein glucose and other reducing sugars undergo a nonenzymatic reaction with protein, lipid, and amino acid amino groups, thereby forming Schiff bases and Amadori products. This process has been shown to be increased in diabetes, which is a prominent independent risk mediator for severe OA [Citation7,Citation8]. AGEs also come to accumulate in the body through tobacco smoke and diet. Increased plasma levels of AGEs have been shown to correlate with increased production of inflammatory markers such as cytokines and chemokines, including IL-6, MCP-1, and HMGB1, through activation of the surface receptor for AGEs (RAGE) [Citation9,Citation10,Citation11]. Additionally, studies have reported that activation of RAGE induces increased expression of matrix metalloproteinases (MMPs), which target the cartilage extracellular matrix (ECM) for degradation. Binding of HMGB1 to RAGE drives cartilage destruction by increasing MMP-3 and MMP-13 [Citation12,Citation13,Citation14]. Excessive degradation of the articular ECM is considered the main hallmark of OA. Type II collagen accounts for the major component of articular cartilage [Citation15]. Collagen molecules are especially susceptible to the accumulation of AGEs, and particularly the AGE pentosidine. Interestingly, the half-life of AGEs in articular collagen has been estimated to be 117 years, while the AGE half-life in skin collagen is only 15 years. This is due to the difference in the rates of cell turnover, as AGEs were shown to accumulate at approximately the same rate in both skin and articular collagen [Citation16]. While our understanding of how AGEs drive the development and progression of OA has increased significantly in recent years, the exact mechanisms driving AGE-induced degradation of the articular ECM remain to be fully elucidated. Thus, it is essential to seek out safe and effective therapies that can halt or alter the effects of AGE accumulation in cartilage.
Bexarotene (trade name Targretin) is a selective retinoid X receptor (RXR) agonist, and third generation retinoid produced by Ligand Pharmaceuticals Inc. Bexarotene was first sold in the US in 2000 and was initially marketed as a treatment against T-cell lymphoma, with later clinical trials investigating its efficacy for breast and lung cancers [Citation17,Citation18]. Additionally, bexarotene is considered to be well-tolerated and has good efficacy [Citation19]. There are three types of RXRs: RXR-α, RXR-β, and RXR-Γ. While the retinoic acid form of vitamin A triggers the activation of both retinoic acid receptors (RARs) and RXRs, these classes of receptors differ in that RARs regulate cell differentiation and proliferation, while RXRs regulate cell apoptosis. Hence, the anti-cancer capacity of bexarotene is primarily mediated through apoptosis of tumour cells [Citation20]. Recent research has demonstrated the potential for bexarotene as a treatment against Alzheimer’s disease. Bexarotene was found to reduce neuroinflammation, clear amyloid-β plaques, and improve cognition in an Alzheimer’s disease mouse model [Citation21]. However, there has been considerable debate regarding these findings [Citation22]. Presently, research is limited regarding the potential role of RXR agonism in different diseases and cell types. Here, we assessed the functions of RXR agonism by bexarotene against AGE-induced impairment of articular ECM using the SW1353 human chondrosarcoma cell line. Our findings demonstrate a promising role for RXR agonism in preventing AGE-induced loss of ECM by inhibiting oxidative stress, suppressing the release of proinflammatory factors and chemokines, and ameliorating degradation of type II collagen via downregulating MMP expression.
Materials and methods
Cell culture and treatment
SW1353 cells from ATCC, USA, were stored in DMEM/HAM’s 12 containing 10% FBS with 2 mM L-glutamine and 1% penicillin/streptomycin (P/S). Cells were cultured with 100 µg/mL AGEs with 30 and 60 nM bexarotene for 24 h.
Dihydroethidium (DHE) staining
The fluorescent dye DHE was used to determine the production of ROS induced by AGEs [Citation23]. After stimulation, cells were loaded with 2 μM DHE at 37 °C for 1 h. Fluorescence was captured using a fluorescence microscope (Nikon, Japan) (Excitation at 510 nm, emission at 580 nm). Image J software was used to quantify the fluorescent density of DHE staining. Regions of interest (ROI) were defined. Then, the number of cells in the ROI was quantified and the integrated density value (IDV) was calculated. Intracelluar levels of ROS equal IDV divied by cell number.
Levels of reduced glutathione (GSH)
A fluorometric assay was used to assess reduced GSH in SW1353 chondrocytes [Citation24]. Cells were collected and lysed with cell lysis buffer. After careful centrifugation at 15,000 × g for 15 min, the supernatant was mixed with reaction buffer containing OPAME and incubated for 30 min, and the resulting fluorescence was read (Excitation at 350 nm, emission at 420 nm).
Real-time PCR
After the indicated treatment, Qiazol was used to isolate total intracellular RNA from chondrocytes. Then, 1 μg RNA from each group was applied to generate cDNA via RT-PCR using a RT-PCR kit (Bio-Rad, USA). Real-time PCR experiment was carried out to assess target gene expressions using a SYBR Green PCR Master Mix kit (Bio-Rad, USA) [Citation25]. Results are normalised to GAPDH using the 2-ΔΔCt method.
Enzyme-linked immunosorbent assay (ELISA)
Commerical ELISA kits were used to determine the expressions of target protein [Citation26]. Briefly, primary antibodies were added to 96-well plates and incubated overnight in a cold room. After blocked with a blocking buffer for 1 h, equal amounts (50 μL) of samples from each group were added and incubated overnight at 4 °C. Reactions were stopped with a stop buffer. Absorbance was measured and recorded at 490 nm.
Western blot analysis
SW1353 human chondrocytes were lysed. A hypotonic buffer was used to obtain nuclear extracts via lysis, and the cytoplasm was removed. 10% SDS-PAGE was used to immobilise cell lysates [Citation27,Citation28]. Then, the separated proteins were transferred to PVDF membranes and sequentially blotted against specific antibodies and a secondary antibody. Pierce™ ECL Plus substrate (Catalog # 32132) was used to visualise the blots. The following antibodies from Cell Signalling Technology were used: iNOS (#13120, 1:2000); Lamin B (#12255, 1:3000); IκBα (1:1000, #4814); β-actin (#3700, 1:5000); p-IκBα (#2859, 1:1000); p65 (#4764, 1:2000). Type II collagen was from Chemicon (#MAB8887, 1:1000).
Nitric oxide (NO) measurement
Intracellular NO in SW1353 cells was determined with the dye DAF-FM [Citation29]. After necessary treatment, SW1353 cells were loaded with 0.5 μM DAF-FM in HBSS for 45 min at RT. The green fluorescence was monitored with a microscope (excitation/emission: 480 nm/540 nm). Image J software was used to quantify the fluorescent density of DAF-FM staining. Regions of interest (ROI) were defined. Then, the number of cells in the ROI was quantified and the integrated density value (IDV) of the ROI was calculated. Levels of NO = IDV/cell number.
Luciferase activity of NF-κB reporter
Briefly, Lipofectamine 2000 (Invitrogen, USA) was used to transfect NF-κB promoter-luciferase and β-galactosidase plasmids (Clontech, USA) into SW1353 chondrocytes. Cells were then lysed using cell lysis buffer. A luminescence assay kit (Gene Copoeia, MD) was used to examine the activity of the luciferase and β-galactosidase.
Statistical analysis
All the experiments have been repeated for 3 times. The results of experiments are expressed as means ± SEM (standard error of measurement). ANOVA followed by Bonferroni’s test was used to perform statistical analysis. A p value <.05 was considered statistically significant.
Results
Bexarotene ameliorates AGE-induced oxidative stress
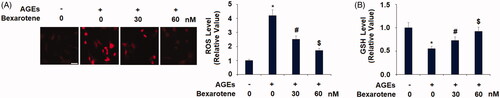
AGEs are able to cause oxidative stress in various cell types, including hepatocytes, retinal cells, and podocytes [Citation30,Citation31,Citation32]. Here, we determined the effects of bexarotene on AGE-induced oxidative stress in SW1353 chondrocytes. As shown in , AGEs stimulation increased the production of ROS to 4.2-fold, which was attenuated by 30 and 60 nM bexarotene to only 2.5- and 1.7-fold, respectively. Interestingly, AGEs reduced the level of GSH to only 55% baseline, which was significantly rescued by the two doses of bexarotene to 73% and 92% baseline ().
Figure 1. Bexarotene inhibits AGE-induced oxidative stress. (A). Levels of ROS in SW1353 cells determined by dihydroethidium (DHE) staining. Scale bar, 100 μm; (B). Levels of reduced glutathione (GSH) (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
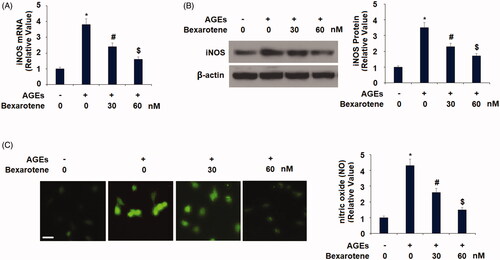
Next, we measured the effects of bexarotene on AGE-induced expression of iNOS and NO. The iNOS pathway has been cited as playing a critical role in the pathogenesis of AGE-induced OA [Citation33]. As demonstrated in , AGEs increased iNOS expression to 3.8-fold at the gene level and 3.5-fold at the protein level, respectively. However, treatment with the two doses of bexarotene resulted in only 2.4- and 1.6-fold expression of iNOS at the mRNA level and only 2.3- and 1.7-fold at the protein level, thereby demonstrating a strong ability of RXR agonism to reduce iNOS pathway activation. The results of DAF-FM DA staining in reveal that bexarotene exerted a similar effect on the AGEs-induced expression of NO. Exposure to AGEs increased NO expression to 4.3-fold baseline, which was reduced by 30 and 60 nM bexarotene to only 2.6- and 1.5-fold, respectively.
Figure 2. Bexarotene inhibits AGE-induced iNOS and nitric oxide (NO). (A). mRNA levels of iNOS; (B). Protein levels of iNOS; (C). Levels of NO. Scale bar, 100 μm (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
Bexarotene reduces AGE-induced expression of proinflammatory cytokines
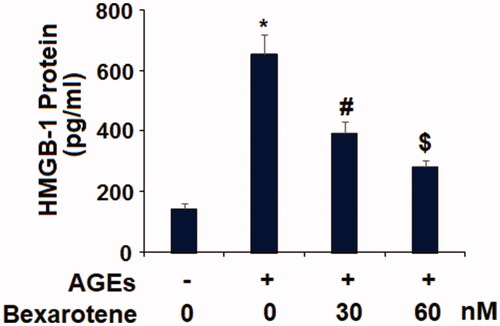
The inflammatory cytokines IL-6, MCP-1, and HMGB1 are considered as important factors driving AGE-induced chronic inflammation in OA cartilage. Real-time PCR and ELISA were performed to measure the effects of bexarotene on AGE-induced expression of IL-6, MCP-1, and HMGB1. As shown in , exposure to 100 µg/mL increased the gene expression of IL-6 and MCP-1 to 5.3- and 6.6-fold, respectively. Meanwhile, the addition of 30 and 60 nM bexarotene remarkably reduced the expression of IL-6 to only 3.1- and 1.8-fold, and of MCP-1 to 2.9- and 1.7-fold. Concordantly, the results of ELISA in show that the two doses of bexarotene reduced AGE-induced protein expression of IL-6 from 4.2-fold to only 2.6- and 1.7-fold, and of MCP-1 from 5.2-fold to only 2.6- and 1.5-fold. As shown in , the results of ELISA reveal that AGEs increased the secretion of HMGB1 to 4.5-fold, which was decreased to only 2.7- and 1.5-fold by the two doses of bexarotene, thereby demonstrating a potentially remarkable dose-dependent anti-inflammatory effect.
Figure 3. Bexarotene inhibits AGE-induced expressions and secretions of IL-6 and MCP-1. (A). mRNA levels of IL-6 and MCP-1; (B). Protein levels of IL-6 and MCP-1 (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
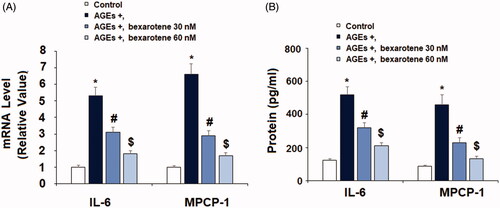
Figure 4. Bexarotene inhibits AGE-induced secretions of HMGB-1. Secretions of HMGB-1 were determined by ELISA (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 6).
Bexarotene ameliorates AGE-induced type II collagen degradation
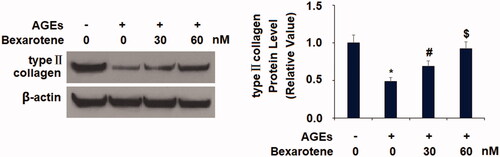
AGEs treatment significantly promoted the expression of two key degradative enzymes, increasing the expression of MMP-3 and MMP-13 to 6.2- and 6.7-fold at the mRNA level and to 5.6- and 5.8-fold at the protein level, respectively (). However, treatment with the two doses of bexarotene markedly mitigated AGE-induced expression of these enzymes, reducing MMP-3 expression to only 3.5- and 2.1-fold and MMP-13 expression to 3.3- and 1.8-fold at the mRNA level, respectively. Concordantly, bexarotene reduced the protein expression of MMP-3 to only 3.2- and 1.9-fold, and of MMP-13 to only 2.9- and 1.8-fold. Next, we confirmed that the inhibitory effect of bexarotene against AGE-induced expression of MMPs resulted in reduced loss of type II collagen. As shown in , we employed western blot analysis to measure type II collagen degradation. Our findings show that exposure to AGEs triggered a 51% reduction in type II collagen, which was rescued by treatment with the two doses of bexarotene to 69% and 92%, thereby revealing a potent protective capacity of RXR agonism against AGE-induced cartilage degradation.
Figure 5. Bexarotene inhibits AGE-induced expressions of MMP-3, and MMP-13. (A). mRNA of MMP-3 and MMP-13; (B). Protein of MMP-3 and MMP-13 (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
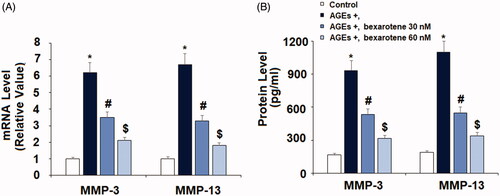
Figure 6. Bexarotene inhibits AGE-induced loss of type II collagen. Protein levels of type II collagen were measured (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
The effects of bexarotene are mediated through the IκBα/NF-κB pathway
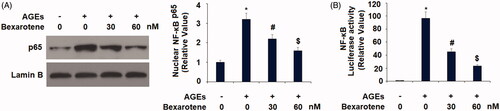
Finally, we assessed the involvement of the NF-κB signalling pathway in mediating the effects of bexarotene against AGE-induced oxidative stress, inflammation, and degradation of articular ECM. Phosphorylation of IκBα facilitates nuclear translocation of p65 protein and activation of its downstream targets. As shown in , we first determined the levels of phosphorylated IκBα in Sw1353 chondrocytes exposed to AGEs with or without 30 and 60 nM bexarotene and found that AGEs increased the level of p-IκBα to 3.7-fold, which was reduced to only 2.2- and 1.6-fold by the two doses of bexarotene. Next, we confirmed that the inhibition of IκBα phosphorylation by bexarotene resulted in decreased NF-κB activation. AGEs increased nuclear translocation of NF-κB to 3.2-fold, which was reduced to only 2.2- and 1.6-fold by the two doses of bexarotene (). The results in indicate that the luciferase activity of NF-κB was increased roughly 96.8-fold by exposure AGEs alone, while the two doses of bexarotene reduced NF-κB luciferase activity to only 45.2- and 23.3-fold, thereby demonstrating a powerful inhibitory effect against NF-κB signalling via downregulation of phosphorylation of IκBα.
Figure 7. Bexarotene inhibits the phosphorylation and degradation of IκBα. Phosphorylated IκBα was determined (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
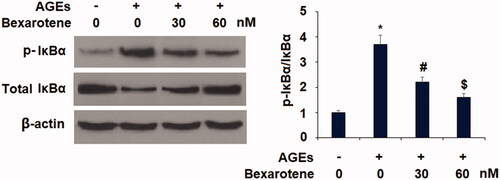
Figure 8. Bexarotene inhibits the activation of NF-κB. (A). Nuclear levels of NF-κB; (B). Luciferase activity of NF-κB reporter gene (*, p < .01 vs. vehicle group; #, p < .01 vs. AGE group; $, p < .01 vs. AGE + 30 nM bexarotene, n = 5–6).
Discussion
Age-related disease will become increasingly more common and treatments to prevent or ameliorate such diseases more desirable. The accumulation of AGEs in joint tissue triggers the development of OA by causing excessive impairment of ECM via oxidative stress and proinflammatory cytokines, chemokines, and degradative enzymes. Here, we examined the effects of the specific RXR agonist bexarotene on these markers of AGE-induced OA using the SW1353 human chondrosarcoma cell line. To our knowledge, this study is the first to examine the impact of bexarotene on cells exposed to AGEs. Firstly, our findings provide evidence of an antioxidant role of bexarotene in chondrocytes exposed to AGEs. AGE-induced production of ROS, iNOS, and NO were significantly downregulated by bexarotene. Additionally, bexarotene treatment rescued the reduction in the expression of GSH, a key antioxidant, induced by AGEs. Recent investigations have shown the importance of ameliorating oxidative stress caused by overproduction of ROS, iNOS, and NO and suppression of GSH in the treatment and prevention of OA [Citation34,Citation35]. However, there has been some conflicting evidence regarding the role of bexarotene in oxidative stress. Previous research has suggested an anti-oxidative stress role of RXR agonism [Citation36,Citation37], but the findings of a recent study indicate that bexarotene treatment caused oxidative stress in erythrocytes [Citation20]. However, the present study shows that in chondrocytes, bexarotene significantly inhibited oxidative stress.
Reducing increased expression of inflammatory mediators is considered a promising avenue for the treatment and prevention of OA. IL-6 is one of the most widely studied cytokines in the context of OA, as IL-6 triggers increased the production of ROS, MMPs, and subsequent degradation of the articular ECM [Citation38]. Concordant with our findings, AGEs treatment upregulates the expression of IL-6, which is dependent on NF-κB activation [Citation39]. A contemporary study displayed the ability of bexarotene to reduce IL-6 expression in a rat model of subarachnoid haemorrhage [Citation40]. Similarly, our findings indicate that bexarotene significantly inhibited AGE-induced expression of IL-6 in chondrocytes. MCP-1 has been shown to contribute to OA by increasing MMP-3 and MMP-13, thereby inducing excessive cartilage degradation, and injection of MCP-1 into mouse knees can induce cartilage degradation [Citation41]. Here, it was shown that agonism of RXR with bexarotene significantly reduced MCP-1 induced by AGEs. We also investigated the effect of bexarotene on AGE-induced expression of HMGB1 and found similar results. HMGB1 plays a key role in OA by mediating activation of the master inflammatory pathway NF-κB, and neutralisation of HMGB1 has been cited as a potential treatment for OA [Citation42,Citation43]. Finally, we investigated the role of RXR agonism by bexarotene in preventing AGE-induced cartilage degradation. Consistent with our finding that bexarotene reduced expression of MCP-1, which in turn reduces MMP expression, we found that bexarotene treatment significantly reduced MMP-3 and MMP-13 and exerted a corresponding protective effect against AGE-induced reduction of type II collagen. However, the exact mechanisms behind these promising findings remain to be elucidated. In a recent RXR-α knockdown experiment, RXR agonism was shown to confer a protective effect on articular ECM homeostasis in immature murine articular chondrocytes by modulating lipid metabolism [Citation44]. Our findings support this by showing that bexarotene significantly reduced AGE-induced oxidative stress, expression of inflammatory cytokines, and MMP-mediated degradation of type II collagen. Additionally, we demonstrate the involvement of the IκBα/NF-κB cellular signalling pathway in mediating the protective effects of bexarotene. A previous study showed that RXR mediates the transactivation potential of NF-κB without affecting NF-κB translocation and binding [Citation45]. Our findings show that agonism of RXR can prevent phosphorylation of IκBα, thereby greatly inhibiting activation of NF-κB. Together, our results introduce a new therapeutic potential of bexarotene in ameliorating the pathogenesis of OA induced by AGEs. Further research is required to fully understand the mechanisms by which bexarotene exerts these effects in OA chondrocytes.
Disclosure statement
There is no conflict interest need to be disclosed
References
- Osteoarthritis. Centers for Disease Control and Prevention Centers for Disease Control and Prevention. 2018.
- Arden N, Nevitt MC. Osteoarthritis: epidemiology. Best Pract Res Clin Rheumatol. 2006;20(1):3–25.
- Zhang Y, Jordan JM. Epidemiology of osteoarthritis. Clin Geriatr Med. 2010;26(3):355–369.
- Johnson VL, Hunter DJ. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. 2014;28(1):5–15.
- Aigner T, Haag J, Martin J, et al. Osteoarthritis: aging of matrix and cells-going for a remedy. Curr Drug Targets. 2007;8(2):325–331.
- Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(7):412.
- Singh R, Barden A, Mori T, et al. Advanced glycation end-products: a review. Diabetologia. 2001;44(2):129–146.
- Schett G, Kleyer A, Perricone C, et al. Diabetes is an independent predictor for severe osteoarthritis: results from a longitudinal cohort study. Diabetes Care. 2013;36(2):403–409.
- Nonaka K, Kajiura Y, Bando M, et al. Advanced glycation end-products increase IL-6 and ICAM-1 expression via RAGE, MAPK and NF-κB pathways in human gingival fibroblasts. J Periodont Res. 2018;53(3):334–344.
- Nowotny K, Jung T, Höhn A, et al. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5(1):194–222.
- van Zoelen MA, Yang H, Florquin S, et al. Role of Toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1–induced inflammation in vivo. Shock. 2009;31(3):280.
- Chen YJ, Chan DC, Chiang CK, et al. Advanced glycation end‐products induced VEGF production and inflammatory responses in human synoviocytes via RAGE-NF-κB pathway activation. J Orthop Res. 2016;34(5):791–800.
- Lin SS, Yuan LJ, Niu CC, et al. Hyperbaric oxygen inhibits the HMGB1/RAGE signaling pathway by upregulating Mir-107 expression in human osteoarthritic chondrocytes. Osteoarthr Cartilage. 2019;27(9):1372.
- Nah SS, Choi IY, Yoo B, et al. Advanced glycation end products increases matrix metalloproteinase-1,-3, and-13, and TNF-α in human osteoarthritic chondrocytes. FEBS Lett. 2007;581(9):1928–1932.
- Akkiraju H, Nohe A. Role of chondrocytes in cartilage formation, progression of osteoarthritis and cartilage regeneration. J Dev Biol . 2015;3(4):177–192.
- Verzijl N, DeGroot J, Thorpe SR, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275(50):39027–39031.
- Farol LT, Hymes KB. Bexarotene: a clinical review. Expert Rev Anticancer Ther. 2004;4(2):180–188.
- Hurst RE. Bexarotene ligand pharmaceuticals. Curr Opin Investig Drugs. 2000;1(4):514–523.
- Abbott RA, Whittaker SJ, Morris SL, et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160(6):1299–1307.
- Bhuyan AA, Bissinger R, Cao H, et al. Triggering of suicidal erythrocyte death by bexarotene. Cell Physiol Biochem. 2016;40(5):1239–1251.
- Medarametla L, Casali BT, Landreth GE. Effects of bexarotene on neuroinflammation in an ad mouse model. Ohio J Sci. 2017;117(1):A18.
- O'hare E, Jeggo R, Kim EM, et al. Lack of support for bexarotene as a treatment for Alzheimer's disease. Neuropharmacology. 2016;100:124–130.
- Faria RX, Gonzaga DTG, Pacheco PAF, et al. Searching for new drugs for Chagas diseases: triazole analogs display high in vitro activity against Trypanosoma cruzi and low toxicity toward mammalian cells. J Bioenerg Biomembr. 2018;50(2):81–91.
- Eftekhari A, Ahmadian E, Panahi-Azar V, et al. Hepatoprotective and free radical scavenging actions of quercetin nanoparticles on aflatoxin B1-induced liver damage: in vitro/in vivo studies. Artif Cells Nanomed Biotechnol. 2018;46(2):411–420.
- Dechandt CRP, Zuccolotto-Dos-Reis FH, Teodoro BG, et al. Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. J Bioenerg Biomembr. 2017;49(5):399–411.
- Shajari N, Davudian S, Kazemi T, et al. Silencing of BACH1 inhibits invasion and migration of prostate cancer cells by altering metastasis-related gene expression. Artif Cells Nanomed Biotechnol. 2018;46(7):1495–1504.
- Amini Y, Moradi B, Fasihi-Ramandi M. Aluminum hydroxide nanoparticles show strong activity to stimulate Th-1 immune response against tuberculosis. Artif Cells Nanomed Biotechnol. 2017;45(7):1331–1335.
- Zhang P, Ma J, Gao J, et al. Downregulation of monocarboxylate transporter 1 inhibits the invasion and migration through suppression of the PI3K/Akt signaling pathway in human nasopharyngeal carcinoma cells. J Bioenerg Biomembr. 2018;50(4):271–281.
- Jolly AJ, Wild CP, Hardie LJ. Sodium deoxycholate causes nitric oxide mediated DNA damage in oesophageal cells. Free Radic Res. 2009;43(3):234–240.
- Guimarães EL, Empsen C, Geerts A, et al. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52(3):389–397.
- Yamagishi SI, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev. 2010;3(2):101–108.
- Yamagishi SI, Ueda S, Matsui T, et al. Role of advanced glycation end products (AGEs) and oxidative stress in diabetic retinopathy. Curr Pharm Des. 2008;14(10):962–968.
- Huang CY, Hung LF, Liang CC, et al. COX-2 and iNOS are critical in advanced glycation end product-activated chondrocytes in vitro. Eur J Clin Invest. 2009;39(5):417–428.
- Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. 2016;1862(4):576–591.
- Qin R, Sun J, Wu J, et al. Pyrroloquinoline quinone prevents knee osteoarthritis by inhibiting oxidative stress and chondrocyte senescence. Am J Transl Res. 2019;11(3):1460–1472.
- Shan P, Pu J, Yuan A, et al. RXR agonists inhibit oxidative stress-induced apoptosis in H9c2 rat ventricular cells. Biochem Bioph Res Co. 2008;375(4):628–633.
- Chai D, Wang B, Shen L, et al. RXR agonists inhibit high-glucose-induced oxidative stress by repressing PKC activity in human endothelial cells. Free Radical Bio Med. 2008;44(7):1334–1347.
- Laavola M, Leppänen T, Hämäläinen M, et al. IL-6 in osteoarthritis: effects of pine stilbenoids. Molecules. 2018;24(1):109.
- Rasheed Z, Akhtar N, Haqqi TM. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-κB in human osteoarthritis chondrocytes. Rheumatology. 2011;50(5):838–851.
- Zuo Y, Huang L, Enkhjargal B, et al. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARγ/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J Neuroinflammation. 2019;16(1):47.
- Xu YK, Ke Y, Wang B, et al. The role of MCP-1-CCR2 ligand-receptor axis in chondrocyte degradation and disease progress in knee osteoarthritis. Biol Res. 2015;48(1):64.
- Wang X, Guo Y, Wang C, et al. MicroRNA-142-3p inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting HMGB1. Inflammation. 2016;39(5):1718–1728.
- Aulin C, Palmblad K, Klareskog L, et al. Cartilage-saving effects of local HMGB1-neutralizing therapy in experimental osteoarthritis. Osteoarthr Cartilage. 2017;25:S435.
- Sun MG, Ratneswaran A, Dupuis H, et al. Investigating the role of Retinoid X Receptor in cartilage development and homeostasis. Osteoarthr Cartilage. 2018;26:S98.
- Cras A, Politis B, Balitrand N, et al. Bexarotene via CBP/p300 induces suppression of NF-κB–dependent cell growth and invasion in thyroid cancer. Clin Cancer Res. 2012;18(2):442–453.