
Abstract
Cytisine is a natural product isolated from plants and is a member of the quinolizidine alkaloid family. This study aims to investigate the effect of cytisine in human lung cancer. Cell viability was determined using the CCK-8 assay, and the results showed that cytisine inhibited the growth of lung cancer cell lines. The apoptotic effects were evaluated using flow cytometry, and the results showed that cytisine induced mitochondrial-dependent apoptosis through loss of the mitochondrial membrane potential; increased expression of BAD, cleaved caspase-3, and cleaved-PARP; and decreased expression levels of Bcl-2, pro-caspase-3, and pro-PARP. In addition, cytisine caused G2/M phase cell cycle arrest that was associated with inhibiting the AKT signalling pathway. During apoptosis, cytisine increased the phosphorylation levels of JNK, p38, and I-κB, and decreased the phosphorylation levels of ERK, STAT3, and NF-κB. Furthermore, cytisine treatment led to the generation of ROS, and the NAC attenuated cytisine-induced apoptosis. In vivo, cytisine administration significantly inhibited the lung cancer cell xenograft tumorigenesis. In conclusion, cytisine plays a critical role in suppressing the carcinogenesis of lung cancer cells through cell cycle arrest and induction of mitochondria-mediated apoptosis, suggesting that it may be a promising candidate for the treatment of human lung cancer.
Introduction
Lung cancer is the most frequent cause of cancer-related death [Citation1]. It is estimated that 1.8 million people died from this disease in 2018, accounting for 18.4% of total estimated cancer deaths [Citation2]. Recently, the morbidity and mortality of lung carcinoma have noticeably increased [Citation3]. Because lung cancer is typically advanced at the time of diagnosis, the feasibility of surgical resection is greatly limited, and systemic drug therapy is focussed on controlling symptoms and prolonging survival [Citation4]. The survival rate for advanced lung cancer patients is still very low despite improvements in drug therapy regimens [Citation5]. Therefore, it is imperative to identify novel and safe anti-tumour agents.
Apoptosis, one of the strategies for cancer treatment, involves activation of a series of molecular events and is induced via various extracellular stresses including reactive oxygen species (ROS). A relatively high level of ROS induces redox imbalance, causing cell apoptosis during the physiological and pathological progression of cancer [Citation6]. Tumour cells with higher ROS levels are more likely to be killed than normal cells with lower ROS levels, as elevated levels of ROS can activate multiple signalling pathways that induce apoptosis such as mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription 3 (STAT3), and nuclear factor-kappa B (NF-κB) signalling pathways [Citation7]. Accordingly, targeting relevant signalling pathways, especially ROS-related pathways, may have potential for lung cancer treatment [Citation8]. Some quinolizidine alkaloids have been reported to exert pro-oxidant actions, which may be an important mechanism for their anticancer and apoptosis-inducing properties [Citation9].
The majority of approved anticancer drugs are unmodified natural products or semi-synthetic derivatives, and natural products are considered a promising source for future drugs [Citation10,Citation11]. Cytisine is a natural quinolizidine alkaloid that is abundant in the roots of Papilionaceae and Caesalpinioideae, medicinal plants that have been used in traditional Chinese medicine [Citation12]. Cytisine is well known for its ability to improve the success of smoking cessation and antitumour activities [Citation13,Citation14].
This study evaluated the therapeutic impact and molecular pathways involved in cytosine-mediated death of human lung cancer cells. In addition, the effects of cytisine on lung cancer cell viability, apoptosis, mitochondrial membrane potential, cell cycle arrest, and ROS generation were studied.
Materials and methods
Materials
Cytisine and 5-FU were purchased from Chengdu Herbpurify Co., Ltd. (Cheng’du, China). Cisplatin was purchased from Solarbio (Beijing, China).
Cell line and cell culture
Lung cancer A549, NCI-H23, and NCI-H460 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). Normal lung fibroblast IMR-90 cell lines, normal liver L-02 cell lines, and normal kidney 293 T cell lines were obtained from Saiqi Biotech Co., Ltd. (Shanghai, China). A549, NCI-H23, NCI-H460, and 293 T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Waltham, MA, USA). The other cells were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640, Gibco). These media were supplemented with 10% heat-inactivated foetal bovine serum (FBS, Gibco), 100 U/mL penicillin (Gibco), and 100 µg/mL streptomycin (Gibco). Cells were cultured in humidified atmosphere of 5% CO2 at 37 °C.
Cell counting kit-8 assay
Cells were seeded into 96-well plates at 1 × 104 cells per well, followed by incubation in a 5% CO2, 95% atmosphere at 37 °C. After exposure of Cytisine (0, 3, 10, 30, 60, and 100 μM) or (0, 6, 12, 24, and 36 h), 10 μL Cell Counting Kit-8 (CCK-8) reagent (Solarbio) was added, and the cells were incubated for 2 h. Then, a microplate reader (BioTek, Instruments Inc., Winooski, VT, USA) was used for detection. Optical density (OD) values were recorded at 450 nm to determine the percentage of viable cells. The percentage of cell viability was calculated as the 50% inhibitory concentration (IC50) and determined using Graphpad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). In each experiment, determinations were performed in triplicate.
Analysis of cell apoptosis
Cell apoptosis was examined with Hoechst-33258/PI (Solarbio) according to the manufacturer’s instructions. Briefly, A549 and NCI-H460 human lung cancer cells were seeded into 12-well plates at 1 × 105 cells/well/100 μL and treated with cytisine at the IC50 concentration for different times (0, 3, 6, 12, and 24 h). Then Hoechst-33258/PI was mixed with the medium in each well and incubated for 15 min at 37 °C. Fluorescence images were captured randomly with an inverted fluorescence microscope (Thermo Fisher Scientific, Waltham, MA, USA) at a 200× magnification. The Annexin V-FITC/PI Apoptosis Detection Kit (Solarbio) was also used to assess cell apoptosis. After treatment with cytisine for different times at the IC50 concentration, cells were centrifuged and resuspended in binding buffer. Then, 5 µL Annexin V-FITC and 5 µL PI were added to each well and incubated for 30 min in the dark. The percentage of cells in each phase was determined using a flow cytometer (Beckman Coulter, Inc., Brea, CA, USA). The apoptosis data were analyzed using CytExpert software 2.0 (Beckman Coulter, Inc., Brea, CA, USA).
Analysis of the mitochondrial membrane potential
The mitochondrial membrane potential (MMP) was examined using the Mitochondrial Membrane Potential Assay Kit (Solarbio) according to the manufacturer’s instructions. Briefly, A549 cells were seeded into 12-well plates at 1 × 105 cells/well/100 μL, treated with cytisine at the IC50 concentration for different times (0, 3, 6, 12, and 24 h), and then incubated with 500 μL JC-1 staining working solution for 20 min at 37 °C. Then, the cells were harvested and washed twice with 500 μL JC-1 staining buffer. The MMP was detected using flow cytometry, and data were analyzed using CytExpert software 2.0.
Analysis of cell cycle arrest
Cell cycle distribution was determined using flow cytometry after treatment with cytisine. Briefly, 1 × 105 cells/well/100 μL were harvested, washed with cold phosphate-buffered saline (PBS), and then fixed in cold 70% ethanol to store at 4 °C for 2 h. Next, fixed cells were pelleted by centrifugation and were cultured with 100 μL RNase A for 30 min at 37 °C and 400 μL PI (Solarbio) for 30 min at 4 °C, followed using flow cytometry. The cell cycle data were analyzed using CytExpert software 2.0.
Assessment of intracellular ROS levels
ROS accumulation was monitored using the fluorescent dye hydroethidine 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Solarbio). After treatment with cytisine for different times, cells were incubated with 10 μmol/L DCFH-DA at 37 °C for 30 min. Then, the cells were resuspended in PBS and immediately subjected to flow cytometry. ROS level was analyzed using CytExpert software 2.0.
Nuclear extract preparation
The nuclear extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagent Kit (Thermo Fisher Scientific). A total of 1 × 106 treated cells were harvested, washed with cold PBS, and gently resuspended with 50 μL Cytoplasmic Extraction Reagent I (CER) for 15 min, followed by incubation with CER II for 2 min. The cell lysates were centrifuged at 13,000 g for 10 min. The precipitate was incubated with Nuclear Extraction Reagent for 1 h, and the collected supernatant contained the nuclear proteins.
Western blot analysis
The expression of specific proteins was detected using western blot analysis. After cytisine treatment at the IC50 concentration for different times (3, 6, 12, and 24 h), we collected the cells and added lysis buffer to lyse on ice for 30 min. Protein extracts were separated on 8–12% SDS-PAGE and then electro-transferred to nitrocellulose membranes (Sigma, St. Louis, MO, USA). The membranes were blocked in 5% (w/v) non-fat milk for 2 h and then overnight at 4 °C with primary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). Horseradish peroxidase-conjugated secondary antibody (ZSGB-bio, Inc., Beijing, China) was used to detect primary antibody binding. Enhanced chemoluminescence (Thermo Fisher Scientific) and the AI600 Imager (GE Healthcare, Fairfield, CT, USA) were used to visualize proteins. The blots were analyzed using ImageJ 1.46r software (National Institutes of Health), and protein levels were normalized to the matching densitometry value of β-actin as the internal control.
In vivo tumour xenografts
All animal experiments and care were performed according to the Animal Care and Treatment Administration of the National Ministry of Health and the requirement of the Heilongjiang Bayi Agricultural University. Six to eight-week-old male BALB/c mice were used for in vivo experiments and were obtained from Beijing Vital River Laboratory Animal Technology Company. 1 × 107 A549 cells were subcutaneously injected into each anterior flank region of nude mice. Treatment was started when the injected cell mass reached a mean volume of 100 mm3. Specifically, the A549 xenograft mice were randomly divided into PBS negative control group, 5-FU positive control group (10 mg/kg), and two cytisine-treated group (1 mg/kg and 10 mg/kg). All groups were used for intraperitoneal administration to the mice every 2 days for 20 days (10 times). Tumour growth and body weight was monitored every 2 days, and tumour volume (mm3) was defined as (l × w2)/2, where l is the length and w is the width (mm) of the tumour. All mice were euthanized and subcutaneous tumours were resected, weighed, photographed, and measured immediately.
Statistical analysis
The data are presented as the mean ± standard deviation (SD) of at least three independent experiments. Statistical differences between controls and treated groups were determined by Tukey’s post hoc test using SPSS 20.0 software. Analysis of variance was used to compare differences between groups, and *p < .05, **p < .01, ***p < .001 were considered statistically significant.
Results
Cytisine inhibits cell proliferation in human lung cancer cells
The growth and survival inhibitory properties of cytisine were investigated in lung cancer cell lines including A549, NCI-H23, and NCI-H460 cells using the CCK-8 assay. show that cytisine caused a steep decline in viable lung cancer cell number in a concentration- and time-dependent manner, with IC50 values of 26.83 μM (A549), 42.79 μM (NCI-H23), and 32.45 μM (NCI-H460). At the same dose, cytisine had no obvious cytotoxic effects on three normal cell lines (). The CCK-8 assay results showed that the cytotoxic effects of cytisine were less than 5-FU and cisplatin on three normal cell lines ().
Figure 1. Cytotoxic effects of 5-FU, cisplatin, and cytisine in lung cancer cells and normal cells determined using the CCK-8 assay. (A) Three human lung cancer cell lines (A549, NCI-H23, and NCI-H460) were treated with different concentrations of 5-FU, cisplatin, and cytisine for 24 h, followed by assessment of cell viability with the CCK-8 assay. (B) Three human normal cell lines (IMR-90, L-02, and 293 T) were treated with different concentrations of 5-FU, cisplatin, and cytisine for 24 h, followed by assessment of cell viability with the CCK-8 assay. (C) Three human lung cancer cell lines (A549, NCI-H23, and NCI-H460) were treated with the IC50 of 5-FU, cisplatin, and cytisine for different times, followed by assessment of cell viability using the CCK-8 assay. (D) Three human normal cell lines (IMR-90, L-02, and 293 T) were treated with the IC50 of 5-FU, cisplatin, and cytisine for different times, followed by assessment of cell viability with the CCK-8 assay. The data are expressed as the means ± SDs of the results from three independent experiments (*p < .05, **p < .01, ***p < .001).
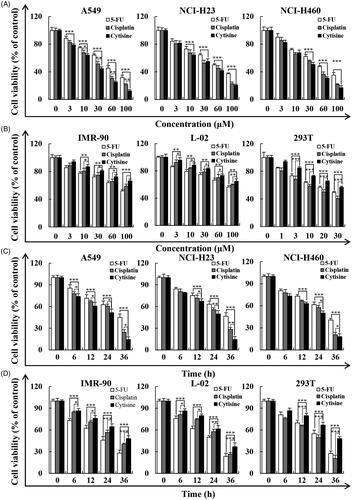
Cytisine induces apoptosis in A549 cells via a mitochondria-dependent pathway
To examine the apoptosis caused by cytisine, Hochest/PI double staining was performed and showed that after treatment with cytisine for different times, the fluorescence intensities gradually increased in A549 and NCI-H460 cells (). The flow cytometry results showed that the proportion of early and terminal phase apoptotic cells increased after cytisine treatment compared to untreated cells (). A marked shift from the upper-right to lower-right quadrant indicated dissociation of JC-1 from the dimer to monomer form and a reduction of MMP (). To explore the molecular mechanism underlying cytisine-induced apoptosis, western blot analysis was used to assess apoptosis-related proteins. Cytisine caused increased expression levels of Bcl-2-associated death promoter (BAD), cytochrome c (Cyt-c), cleaved caspase-3, and poly(ADP-ribose) polymerase (PARP), and a decrease in B-cell lymphoma 2 (Bcl-2) protein expression levels ().
Figure 2. Apoptotic effects of cytisine in A549 cells. (A) Cells were treated with 26 μM cytisine for different times (3, 6, 12, and 24 h), and stained with Hoechst 33342/PI. Data shown represent fluorescence microscopic images (original magnifications, 200×). (B) Quantification of fluorescent intensity. (C) Cells were incubated with Annexin V-FITC/PI and analyzed by flow cytometry. (D) Cells were incubated with JC-1 mitochondrial membrane potential dye and analyzed using flow cytometry. (E) The protein expression levels of Cyt-c, BAD, Bcl-2, pro-caspase-3, cleaved caspase-3, pro-PARP, and cleaved PARP were measured using western blotting, and β-actin was used as the internal control. The intensities of the bands were quantified using Image J (*p < .05, **p < .01, ***p < .001).
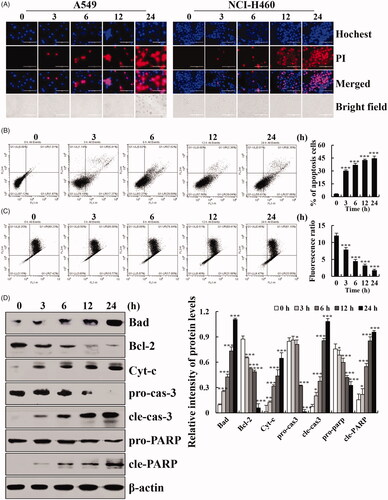
Cytisine causes G2/M cell cycle arrest in A549 cells
To examine the distribution of cell cycle progression, the effects of cytisine on various cell cycle phases were confirmed using flow cytometry. The treatment of A549 cells with cytisine (26 μM) for different times showed that compared with untreated cells, G2/M phase arrest was significantly induced in cytisine-treated cells. The percentage of cells in the G2/M phase at 24 h increased from 21.06% to 50.04% (). G2/M-related protein expression levels were also investigated using western blotting. The expression levels of p21 and p27 were increased, whereas the expression levels of cyclin B1, cyclin-dependent kinase 1/2 (CDK1/2), and p-AKT were decreased in A549 cells treated with cytisine ().
Figure 3. Effects of cytisine on the cell cycle distribution and cell cycle checkpoint related proteins in A549 cells. (A) Cells were treated with 26 μM cytisine for different time points (3, 6, 12, or 24 h), and stained with PI. DNA content was analyzed for cell cycle phase distribution using flow cytometry. (B) Protein expression levels of p21, p27, cyclin B1, and CDK1/2 were measured using western blotting, and β-actin was used as the internal control. The intensities of the bands were quantified using Image J (*p < .05, **p < .01, ***p < .001).
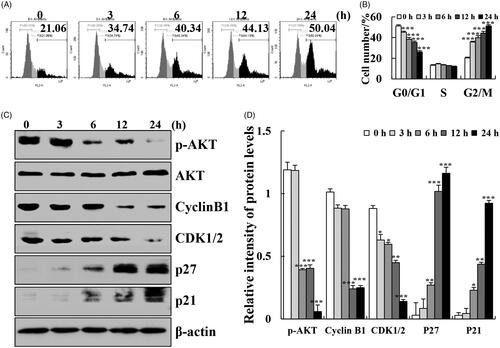
Cytisine induces apoptosis and cell cycle arrest via the MAPK/STAT3/NF-κB signalling pathways in A549 cells
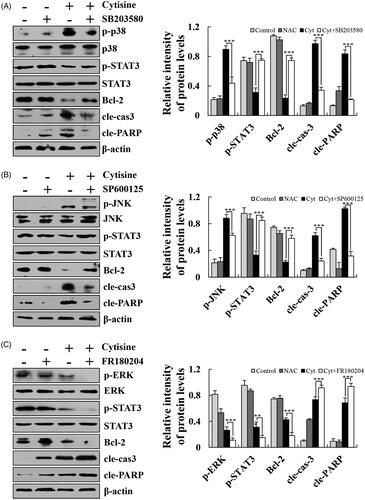
To elucidate the exact mechanism underlying cytisine-induced apoptosis in human lung cancer cells, the expression levels of MAPK/STAT3/NF-κB signalling pathway-related proteins were determined using western blot analysis. After treatment with cytisine, the expression levels of phosphorylated extracellular signal-related kinase (p-ERK), p-STAT3, and NF-κB (p65) were significantly decreased, whereas those of p-p38, phosphorylated c-Jun N-terminal kinase (p-JNK), and ikappaB (I-κB) were increased (. Then, we pre-treated A549 cells with FR180204 (ERK inhibitor), SP600125 (JNK inhibitor), or SB203580 (p38 inhibitor) for 30 min before add cytisine. The expression levels of p-STAT3 was increased in the presence of JNK inhibitor or p38 inhibitor, whereas expression levels were inhibited upon treatment with the ERK inhibitor, and further found that the expression levels of Bcl-2, cleaved caspase-3, and cleaved PARP were reversed ().
Figure 4. Effects of cytisine on MAPK, STAT3, and NF-κB signalling pathways in A549 cells. (A) Cells were treated with 26 μM cytisine for different time points (3, 6, 12, and 24 h), and the expression levels of protein were measured using western blotting; and β-actin was used as the internal control. (B) The expression levels of nuclear proteins were measured using western blotting; Lamin B1 was used as the nuclear loading control. The data are expressed as the means ± SDs of the results from three independent experiments (*p < .05, **p < .01, ***p < .001).
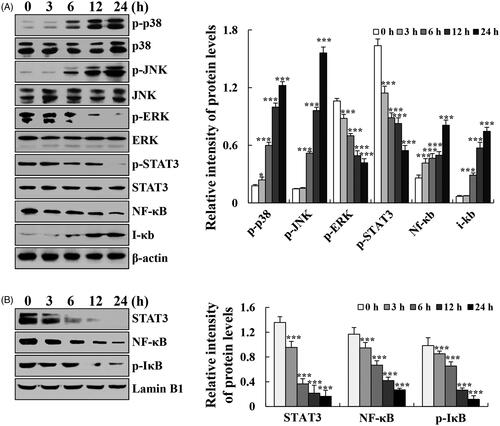
Figure 5. Effects of MAPK on the STAT3 signalling pathway in A549 cells. (A) Cells were treated with a p38 inhibitor, and the expression levels of p-p38, p-STAT3, Bcl-2, cleaved caspase-3, and cleaved PARP were measured using western blotting. (B) Cells were treated with a JNK inhibitor, and the expression levels of p-JNK, p-STAT3, Bcl-2, cleaved caspase-3, and cleaved PARP were measured using western blotting. (C) Cells were treated with an ERK inhibitor, and the expression levels of p-ERK, p-STAT3, Bcl-2, cleaved caspase-3, and cleaved PARP were measured using western blotting, and β-actin was used as the internal control. The data are expressed as the means ± SDs of the results from three independent experiments (*p < .05, **p < .01, ***p < .001).
Cytisine-induced apoptosis through ROS-mediated MAPK/STAT3/NF-κB signalling pathways in A549 cells
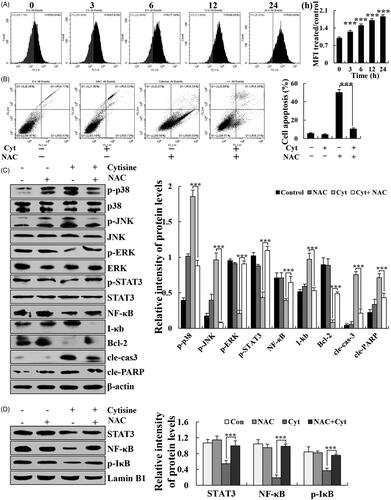
The generation of ROS is a well-known marker of mitochondrial damage. Cytisine-treated A549 cells were stained with DCFH-DA to assess ROS generation with flow cytometry. Cytisine treatment increased intracellular ROS levels as early as 3 h after treatment initiation and increased in a time-dependent manner (. To further explore the role of ROS in cytisine-induced apoptosis, the A549 cells were preincubated with N-acetyl cysteine (NAC) (a ROS scavenger), which significantly attenuated cytisine-induced apoptosis (. In addition, apoptosis-associated proteins were further tested to validate the role of ROS in cytisine-induced apoptosis. NAC greatly upregulated the expression levels of p-ERK, p-STAT3, NF-κB, p-STAT3, and Bcl-2, and downregulated the expression levels of p-JNK, p-p38, I-κB, and cleaved caspase-3 in the cytoplasm (. Subsequently, the expression of nuclear transcription proteins was detected. NAC markedly upregulated the expression levels of STAT3, NF-κB (p65), and p-IκB in the nucleus ().
Figure 6. Cytisine-induced apoptosis through ROS-mediated MAPK/STAT3/NF-κB signalling pathways. (A) Cells were treated with 26 μM cytisine for different time points (3, 6, 12, or 24 h) and stained with DCFH-DA (10 μM). Intracellular ROS levels were determined using flow cytometry. (B) A549 cells were treated with NAC for 30 min and then incubated with cytisine for 24 h. Cell apoptosis was determined using flow cytometry. (C) The expression levels of p-p38, p-JNK, p-ERK, p-STAT3, NF-κB, I-κB, Bcl-2, cleaved caspase-3, and cleaved PARP were measured using western blotting. (D) Quantitative analysis of C. (E) Expression levels of nuclear STAT3, NF-κB (p65), and p-IκB were detected using western blotting; Lamin B1 was used as the nuclear loading control. (F) Quantitative analysis of E. The data are expressed as the means ± SDs of the results from three independent experiments. (*p < .05, **p < .01, ***p < .001).
Cytisine-induced cell cycle arrest through the ROS-mediated AKT signalling pathway
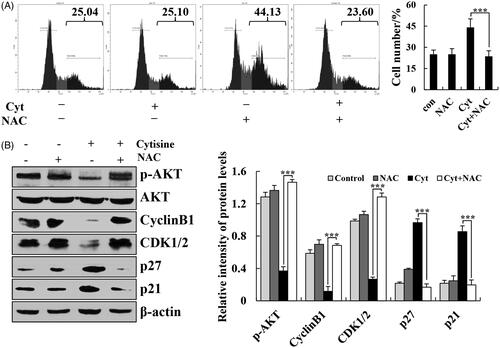
To further explore the role of ROS in cytisine-induced cell cycle arrest, the A549 cells were preincubated with NAC, which significantly attenuated the cytisine-induced cell cycle arrest (. In addition, G2/M phase-associated proteins were further tested to validate the role of ROS in cytisine-induced cell arrest. NAC markedly upregulated the expression levels of p-AKT, cyclin B1, and CDK1/2 and downregulated the expression levels of p27 and p21 (. These results suggested that the observed cytisine-induced mitochondrial apoptosis and G2/M phase cell cycle arrest may, at least in part, be caused by the generation of ROS in A549 cells.
Figure 7. Cytisine-induced cell cycle arrest through ROS-mediated AKT signalling pathways. (A) A549 cells were treated with NAC for 30 min and then incubated with cytisine for 24 h. Cell cycle arrest was determined using flow cytometry. (B) Expression levels of p-AKT, cyclin B1, CDK1/2, p27, and p21 were measured using western blotting, and β-actin was used as the internal control. The data are expressed as the means ± SDs of the results from three independent experiments (*p < .05, **p < .01, ***p < .001).
Cytisine suppressed tumour growth in mouse xenograft models
Tumorigenicity experiment was performed in nude mice using A549 cell xenograft model to confirm the function of cytisine in vivo. Cytisine-treated and control mice did not show any evidence of significant side effects, and their body weights did not differ from each other (. By compared with negative control group, treat with cytisine showed an obvious inhibition of lung tumour growth in vivo. Apparently reduction in tumour volume was observed at 4th administration in cytisine treated group and the inhibition of tumour growth induced by cytisine was better than that induced by 5-FU (). Compared with the control group and 5-FU group, the cytisine group showed significantly decreased tumour weight, and the inhibitory effect of 10 mg/kg group was better than 10 mg/kg group ().
Figure 8. Cytisine exert antitumor effect and low toxicity in vivo. (A) The mice body weights after injection. (B) The tumour size was recorded once every two days using Vernier calliper measurement and calculated as [(length × width2)/2]. (C) Tumour images after injection. (D) The tumour weights after injection. The data are expressed as the means ± SDs of the results from three independent experiments (*p < .05, **p < .01, ***p < .001).
![Figure 8. Cytisine exert antitumor effect and low toxicity in vivo. (A) The mice body weights after injection. (B) The tumour size was recorded once every two days using Vernier calliper measurement and calculated as [(length × width2)/2]. (C) Tumour images after injection. (D) The tumour weights after injection. The data are expressed as the means ± SDs of the results from three independent experiments (*p < .05, **p < .01, ***p < .001).](/cms/asset/bba8e77e-4476-4d7f-9ec8-9afafb0450d1/ianb_a_1699813_f0008_c.jpg)
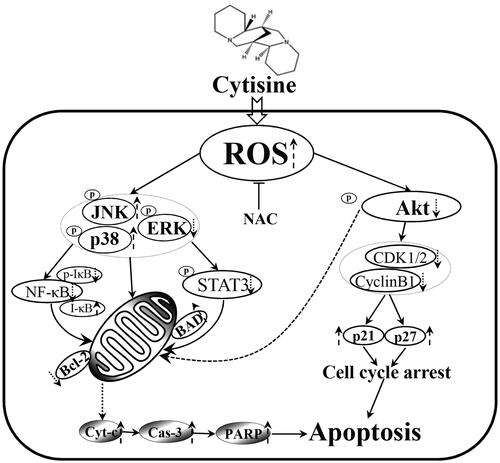
Figure 9. Schematic presentation of the signalling pathways in A549 human lung cancer cells affected by cytisine.
Discussion
Cytisine is a natural product isolated from plants and is a member of the quinolizidine alkaloid family; its five-ring skeleton structure makes cytisine highly efficient in biological activity [Citation15]. Cytisine has good anticancer activity in various types of cancer including hepatocellular and breast cancers [Citation6]. However, to the best of our knowledge, no previous studies have evaluated the effects of cytisine on lung carcinoma. The results of CCK-8 assay showed that cytisine inhibited the proliferation of human lung cancer cells in a dose-dependent manner (). We also detected the effects of cytisine on apoptosis induction, cell cycle arrest, and ROS generation in lung cancer cells.
Mitochondria are key factors of apoptotic signalling and act as integrating sensors for various death stimuli including ROS and DNA-damaging reagents [Citation16]. Many chemotherapeutic agents appear to eliminate tumour cells by apoptosis induction through the activation of mitochondria-mediated apoptotic signalling pathway [Citation17]. In this study, both Hochest/PI double staining and flow cytometry confirmed the induction of apoptosis by cytisine in A549 cells (). The results of the JC-1 dye assay demonstrated that cytisine markedly reduced the levels of MMP (. The decrease of MMP is the earliest event in the process of apoptosis, which can initiate the apoptosis cascade. Cyt-c can activate the caspase cascade after mitochondrial release, inducing the expression of the effector protein caspase-3, and activating cleavage of PARP protein to induce mitochondria-dependent apoptosis. The dynamic balance of Bcl-2/Bax also affects the occurrence of apoptosis [Citation18]. In our study, cytisine caused an increase in expression levels of Cyt-c, BAD, cleaved caspase-3, and PARP protein and a decrease in Bcl-2 protein levels (. These data suggested that cytisine induced apoptosis through the intrinsic apoptotic pathway involving mitochondrial dependence.
An increasing number of studies have focussed on cell cycle regulation-mediated apoptosis as an effective way to inhibit cancer cell growth [Citation19]. During the cell cycle, the G2/M checkpoint is a possible target for cancer therapy, and CDK/cyclin are critical and complex regulators of the G2/M phase [Citation20]. Recent studies have demonstrated that various anticancer drugs induce G2/M arrest accompanied by downregulating the expression levels of AKT [Citation21]. In our study, the results showed that cytisine treatment resulted in G2/M arrest and decreased expression of p-AKT, CDK1/2, and cyclin B1, and increased the expression of p27 and p21, suggesting that the AKT signalling pathway may be the molecular mechanism through which cytisine induced G2/M phase arrest (). Furthermore, the AKT pathway also participates in the regulation of Bcl-2 family proteins, which are key regulators of the apoptotic pathway.
In subsequent experiments, we paid further attention to the apoptosis and underlying cell cycle arrest mechanisms. MAPKs are a large family of seine/threonine kinases that mediate cellular signals [Citation22]. ERK is usually associated with cell proliferation and growth. In contrast, JNK and p38 are induced by cellular stress and are closely associated with cell death [Citation23,Citation24]. NF-κB and STAT3 signalling pathways play pivotal roles in the cellular transcription process in a number of malignancies. Upon activation of the NF-κB signalling pathway, NF-κB enters the nucleus and binds to target genes with an NF-κB binding site to initiate transcriptional processes. At the same time, expression of the I-κB gene is activated to inhibit the expression of p-IκB and cause DNA damage [Citation25]. Our results showed that cytisine induced activation of MAPK, STAT3, and NF-κB signalling pathways in human lung cancer A549 cells (. The expression levels of STAT3, NF-κB, and p-IκB in the nucleus were decreased, suggested that cytisine regulates the transcription of DNA in cells via the NF-κB signalling pathway (. In addition, SB203580 (p38 inhibitor), SP600125 (JNK inhibitor), or SB203580 (p38 inhibitor) completely blocked the expression of apoptotic proteins such as Bcl-2, cleaved caspase-3, and cleaved PARP, suggesting that activation of MAPKs is required and involved in cytisine-induced apoptosis in A549 cells. It is interesting that SB203580, SP600125, and SB203580 reversed activation of the STAT3 signalling pathway, which indicates that MAPK not only contributes to cytisine-induced apoptosis but also regulates STAT3 throughout the apoptotic signalling pathway ().
There is increasing evidence indicating that ROS mediates intracellular signal cascades and excessive ROS production leads to intracellular stress, mitochondria dysfunction, and disturbance of the balance of redox reactions, ultimately causing cell apoptosis or necrosis [Citation26,Citation27]. Association between lung cancer risk and oxidative stress has been well recognized. Many quinolizidine alkaloids cause elevated levels of ROS in cells. Sophoridine significantly induces apoptosis in BRL-3A rat liver cells with associated accumulation of ROS [Citation28]. Subditine, a quinolizidine alkaloid from the bark of nauclea subdita, induces apoptosis in human prostate cancer cells by promoting ROS production [Citation29]. In this study, we found that cytisine induced ROS generation in A549 cells in a dose-dependent manner (. Moreover, the antioxidant NAC significantly abolished apoptosis and cell cycle arrest ( and . These results suggest that ROS markedly mediates mitochondria-related apoptosis in cytisine-treated A549 cells. Similarly, NAC not only significantly inhibited the expression of MAPK, STAT3, AKT, and NF-κB proteins in the cytoplasm, but also reversed the decreased expression of STAT3, NF-κB, and p-IκB in the nucleus ( and ), suggesting that ROS generation activates these signalling pathways, which are supposedly upstream of the cytisine-induced apoptosis pathway in A549 cells.
Along with the in vitro studies, the in vivo studies were also carried out. We confirmed the enhanced antitumor effects of cytisine using A549 tumour xenografts in mice. Our in vivo study indicated that cytisine markedly inhibited tumour growth in a dose-dependent manner during the 20-day treatment. The tumour tissues developed in the cytisine-treated mice were much smaller compared with the control group (. Among cytisine-treated mice, the group treated with 10 mg/kg dose exhibited the smallest tumour size, followed by 1 mg/kg dose. At the same drug weight doses (10 mg/kg), cytisine showed stronger in vivo cancer therapeutic effect than 5-FU (. These in vivo results are similar to our in vitro study, which further suggests that cytisine may be an applicable approach to enhance the antitumor activity against lung cancer.
In conclusion, we found that cytisine induced mitochondria-dependent apoptosis and increased the levels of ROS, subsequently regulating the expression of MAPK/STAT3/NF-κB/AKT pathway-related proteins (). These data suggest that cytisine exerts anticancer activity by inducing oxidative stress; thus, it may be an attractive bioactive phytochemical for lung cancer chemoprevention and/or treatment.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389 (10066):299–311.
- Feng C, Xia Y, Zou P, et al. Curcumin analog L48H37 induces apoptosis through ROS-mediated endoplasmic reticulum stress and STAT3 pathways in human lung cancer cells. Mol Carcinog. 2017;56(7):1765–1777.
- Zhou M, Shen S, Zhao X, et al. Luteoloside induces G0/G1 arrest and pro-death autophagy through the ROS-mediated AKT/mTOR/p70S6K signalling pathway in human non-small cell lung cancer cell lines. Biochem Biophys Res Commun. 2017;494(1–2):263–269.
- Yao J, Ma C, Gao W, et al. Fentanyl induces autophagy via activation of the ROS/MAPK pathway and reduces the sensitivity of cisplatin in lung cancer cells. Oncol Rep. 2016;36(6):3363–3370.
- Song Y, Kong L, Sun B, et al. Induction of autophagy by an oleanolic acid derivative, SZC017, promotes ROS-dependent apoptosis through AKT and JAK2/STAT3 signaling pathway in human lung cancer cells. Cell Biol Int. 2017;41(12):1367–1378.
- Liu J, Chang F, Li F, et al. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun. 2015;463(3):262–267.
- Duan F, Yu Y, Guan R, et al. Vitamin K2 induces mitochondria-related apoptosis in human bladder cancer cells via ROS and JNK/p38 MAPK signal pathways. PLoS One. 2016;11(8):e0161886.
- Yang L, Wu L, Du S, et al. 1,25(OH)2D3 inhibits high glucose-induced apoptosis and ROS production in human peritoneal mesothelial cells via the MAPK/P38 pathway. Mol Med Rep. 2016;14(1):839–844.
- Zhang C, Jia X, Bao J, et al. Polyphyllin VII induces apoptosis in HepG2 cells through ROS-mediated mitochondrial dysfunction and MAPK pathways. BMC Complement Altern Med. 2015;16(1):58.
- Yu L, Wang X, Chen ZF, et al. Cytisine induces apoptosis of HepG2 cells. Mol Med Rep. 2017;16(3):3363–3370.
- Peng TT, Sun XR, Liu RH, et al. Cytisine-pterocarpan-derived compounds: biomimetic synthesis and apoptosis-inducing activity in human breast cancer cells. Molecules. 2018;23(12):E3059.
- Yu L, Jiang B, Chen Z, et al. Cytisine induces endoplasmic reticulum stress caused by calcium overload in HepG2 cells. Oncol Rep. 2018;39(3):1475–1484.
- Li YJ, Yang Q, Zhang K, et al. Cytisine confers neuronal protection against excitotoxic injury by down-regulating GluN2B-containing NMDA receptors. Neurotoxicology. 2013;34:219–225.
- Tutka P, Kondrat-Wróbel MW, Zaluska K, et al. Cytisine inhibits the protective activity of various classical and novel antiepileptic drugs against 6 Hz-induced psychomotor seizures in mice. Psychopharmacology (Berl). 2017;234(2):281–291.
- Paduszyńska A, Banach M, Rysz J, et al. Cytisine-from the past to the future. CPD. 2019;24(37):4413–4423.
- Xu C, Wang X, Zhu Y, et al. Rapamycin ameliorates cadmium-induced activation of MAPK pathway and neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A. Neuropharmacology. 2016;105:270–284.
- Poornima P, Weng CF, Padma VV. Neferine, an alkaloid from lotus seed embryo, inhibits human lung cancer cell growth by MAPK activation and cell cycle arrest. Biofactors. 2014;40(1):121–131.
- Lim HS, Kang YJ, Sung B, et al. Novel dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative, MHY-449, induces cell cycle arrest and apoptosis via the downregulation of AKT in human lung cancer cells. Oncol Rep. 2015;34(5):2431–2438.
- Tu Y, Kim E, Gao Y, et al. Theaflavin-3, 3'-digallate induces apoptosis and G2 cell cycle arrest through the AKT/MDM2/p53 pathway in cisplatin-resistant ovarian cancer A2780/CP70 cells. Int J Oncol. 2016;48(6):2657–2665.
- Yang B, Zhao Y, Lou C, et al. Eupalinolide O, a novel sesquiterpene lactone from Eupatorium lindleyanum DC., induces cell cycle arrest and apoptosis in human MDA-MB-468 breast cancer cells. Oncol Rep. 2016;36(5):2807–2813.
- Sampson VB, Vetter NS, Kamara DF, et al. Vorinostat enhances cytotoxicity of SN-38 and temozolomide in ewing sarcoma cells and activates STAT3/AKT/MAPK pathways. PLoS One. 2015;10(11):e0142704.
- Wang Y, Luo YH, Piao XJ, et al. Novel 1,4-naphthoquinone derivatives induce reactive oxygen species-mediated apoptosis in liver cancer cells. Mol Med Rep. 2019;19(3):1654–1664.
- Zuo D, Zhou Z, Wang H, et al. Alternol, a natural compound, exerts an anti-tumour effect on osteosarcoma by modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol Med. 2017;21(2):208–221.
- Zhang ZR, Gao MX, Yang K. Cucurbitacin B inhibits cell proliferation and induces apoptosis in human osteosarcoma cells via modulation of the JAK2/STAT3 and MAPK pathways. Exp Ther Med. 2017;14(1):805–812.
- Richardson JSM, Aminudin N, Abd Malek SN, et al. Chalepin: Compound from Ruta angustifolia L. Pers exhibits cell cycle arrest at S phase, suppresses nuclear factor-kappa B (NF-κB) pathway, signal transducer and activation of transcription 3 (STAT3) phosphorylation and extrinsic apoptotic pathway in non-small cell lung cancer carcinoma (A549). Phcog Mag. 2017;13(3):S489–S498.
- Wang JR, Luo YH, Piao XJ, et al. Mechanisms underlying isoliquiritigenin-induced apoptosis and cell cycle arrest via ROS-mediated MAPK/STAT3/NF-κB pathways in human hepatocellular carcinoma cells. Drug Dev Res. 2019;80(4):461.
- Meng LQ, Wang Y, Luo YH, et al. Quinalizarin induces apoptosis through reactive oxygen species (ROS)-mediated mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription 3 (STAT3) signaling pathways in colorectal cancer cells. Med Sci Monit. 2018;24:3710–3719.
- Qiu M, Shi F, Dai F, et al. A reactive oxygen species activation mechanism contributes to sophoridine-induced apoptosis in rat liver BRL-3A cells. J Ethnopharmacol. 2018;213:376–383.
- Liew SY, Looi CY, Paydar M, et al. Subditine, a new monoterpenoid indole alkaloid from bark of Nauclea subdita (Korth.) Steud. induces apoptosis in human prostate cancer cells. PLoS One. 2014;9(2):e87286.