
Abstract
Prokineticin 2 (PK2) was reported to be decreased in the hearts of end-state heart failure patients. Our study aimed to explore the effects of PK2 on hypoxia/reoxygenation (H/R) injury and the underlying mechanism. H9c2 cardiomyocytes were treated with 5 nM PK2 in the presence or absence of 5 mM dual phosphatidylinositol 3-kinase (PI3K)/the mammalian target of rapamycin (mTOR) inhibitor (BEZ235) for 24 h and then subjected to H/R treatment. Cell viability and lactate dehydrogenase (LDH) release were evaluated by CCK-8 and LDH release assays, respectively. Apoptosis was determined by flow cytometry analysis. Oxidative stress was assessed by measuring superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) activities and malondialdehyde (MDA) content. Results showed that H/R treatment decreased PK2 expression and inactivated the Akt/mTOR pathway in H9c2 cardiomyocytes. PK2 treatment activated the Akt/mTOR pathway in H/R-exposed H9c2 cardiomyocytes. H/R stimulation suppressed cell viability, increased LDH release, induced apoptosis and oxidative stress in H9c2 cardiomyocytes, while these effects were neutralised by treatment with PK2. However, the inhibitory effects of PK2 on H/R-induced injury in H9c2 cardiomyocytes were abolished by the addition of BEZ235. In conclusion, PK2 relieved H/R-induced injury in H9c2 cardiomyocytes by activation of the Akt/mTOR pathway.
Introduction
Acute myocardial infarction (AMI), the most severe manifestation of coronary artery disease, has long been a leading cause of morbidity and mortality of cardiovascular disease worldwide [Citation1]. It is estimated that there will be approximately 23 million patients with AMI in China by 2030, which seriously threatens people’s health [Citation2]. It is well established that ischaemia/reperfusion (I/R)-induced myocardial injury remains the principal cause of AMI worldwide, which is characterized by the interruption of the myocardial blood supply and subsequent irreversible damage to heart muscle [Citation3]. Several lines of evidence have demonstrated that I/R-induced myocardial injury causes mitochondrial dysfunction with excess production of reactive oxygen species (ROS) and oxidative stress, and if prolonged, leads to the aggravation of cardiomyocyte apoptosis and ultimately cardiac failure [Citation4]. Thus, the exploration of effective therapeutic approaches will open new avenues to protect myocardial cells from I/R injury and improve the clinical outcomes in patients with AMI.
Prokineticin 2 (PK2), also known as Bv8, is a small chemokine-like secreted protein that exerts its biological activities via activation of two cognate G protein-linked receptors, namely prokineticin receptor 1 (PKR1) and 2 (PKR2) [Citation5]. PK2 is well-documented to be highly expressed in activated immune cells and inflamed murine tissues with infiltrating neutrophils, showing proinflammatory activities via PKR1 [Citation6]. Recent literatures report that PK2 is also implicated in regulating diverse essential physiological and/or pathological processes, including circadian rhythms, neuroprotection, angiogenesis and pain perception [Citation7–10]. More importantly, it was reported that the expression of PK2 and its receptors were decreased in the hearts of end-state heart failure patients [Citation11]. PK2 activated the protein kinase B (Akt) pathway to prevent cardiomyocytes from apoptosis against hypoxic insult [Citation11]. Still, little is known about the effects of PK2 on I/R injury in AMI.
In the present study, we aimed to investigate the effects of PK2 on hypoxia/reoxygenation (H/R)-induced injury, apoptosis and oxidative stress in H9c2 cardiomyocytes and further explored the underlying mechanism.
Materials and methods
Cell culture and treatment
The H9c2 cardiomyocytes derived from rat embryonic heart were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and routinely maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose, 10% heat-inactivated foetal bovine serum (FBS; Hyclone, Logan, UT, USA) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) at 37 °C in a humidified incubator containing 95% air and 5% CO2. To establish H/R injury model, H9c2 cardiomyocytes were transferred to FBS- and glucose-free DMEM and incubated under hypoxic conditions in an incubator supplied with a gas mixture containing 1% O2, 94% N2 and 5% CO2 for 12 h at 37 °C, followed by reoxygenation (95% air and 5% CO2) in complete medium in a normoxic chamber for another 12 h at 37 °C. H9c2 cardiomyocytes in the normal control were cultured under normoxic conditions. PK2 and BEZ235 (a dual phosphatidylinositol 3-kinase (PI3K)/the mammalian target of rapamycin (mTOR) inhibitor) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in dimethyl sulphoxide (DMSO) to a final concentration of 5 nM and 5 mM, respectively. In some experiments, H9c2 cardiomyocytes were incubated in FBS- and glucose-free DMEM supplemented with 5 nM PK2 in the presence or absence of 5 mM BEZ235 and then subjected to H/R treatment.
Cell viability assay
Cell viability was assessed using the cell counting kit-8 (CCK-8) assay. In brief, H9c2 cardiomyocytes were inoculated in 96-well plates at an optimal density of 5 × 104 cells/well. Subsequent to the aforementioned treatments, cells were incubated with 10 μl CCK-8 reagent (Beyotime, Shanghai, China) at 37 °C for another 2 h. The optical density at a wavelength of 450 nm was detected using a microplate reader (MQX 200, BioTek Instruments, Winooski, VT, USA).
Lactate dehydrogenase (LDH) release assay
Cell necrosis induced by the H/R injury was evaluated by measuring the release of LDH, a biochemical indicator of cellular damage. Following experimental treatments, H9c2 cardiomyocytes were harvested and lysed, and the supernatants were then centrifuged for 15 min. LDH release in the supernatant was measured using a commercial LDH kit (Takara, Dalian, China) according to the manufacturer’s protocols.
Analysis of oxidative stress
The activities of superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) in the supernatant were measured using a SOD Activity Assay Kit (BioVision, Milpitas, CA, USA), CAT and GSH-Px assay kits (Nanjing Jiancheng Bioengineer Company, Nanjing, China), respectively. The content of malondialdehyde (MDA), an index of lipid peroxidation, was determined by a Lipid Peroxidation MDA Assay Kit (Beyotime) according to the manufacturer’s instructions.
Flow cytometry analysis of apoptosis
Apoptosis of H9c2 cardiomyocytes was analysed using an Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Jose, CA, USA). Briefly, H9c2 cardiomyocytes were harvested by trypsinization following treatments and washed twice with PBS. After being resuspended in 100 μL of 1× binding buffer, H9c2 cardiomyocytes were incubated with 5 μL annexin V-FITC and 5 μL PI in the dark for 15 min. The apoptotic cells were detected by a FACScan flow cytometry (BD Biosciences, San Jose, CA, USA) equipped with CellQuest™ software (BD Biosciences).
Caspase-3 and caspase-9 activity assay
The activities of caspase-3 and caspase-9 in the supernatant were detected using the colorimetric caspase-3 and caspase-9 activity assay kits (Beyotime), respectively. Absorbance at 405 nm was measured with a microplate reader (MQX 200, BioTek Instruments).
Western blot analysis
Proteins were extracted from treated H9c2 cardiomyocytes with ice-cold RIPA buffer Protein Extraction Kit (Pierce, Rockford, IL, USA) and protein concentration was determined by the Bradford method (Beyotime). Subsequently, 20 μg of protein samples were subjected to 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred onto nitrocellulose membranes (Millipore, Boston, MA, USA). After being blocked with 5% non-fat milk for 1 h at 37 °C, the membranes were probed with primary antibodies overnight against PK2 (Abcam, Cambridge, MA, USA), Akt (Abcam), phosphorylated Akt (p-Akt) (Ser473) (Abcam), mTOR (Abcam), phosphorylated mTOR (p-mTOR) (Ser2448) (Abcam), Bax (Cell Signalling Technology, Danvers, MA, USA), Bcl-2 (Cell Signalling Technology) and β-actin (Cell Signalling Technology). Then, the membrane was incubated with corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signalling Technology) for 2 h at room temperature. The protein bands were visualised using an enhanced chemiluminescence (Amersham Pharmacia, Piscataway, NJ, USA) and quantified with an image analyser Quantity One System (Bio-Rad Laboratories, Hercules, CA, US).
Statistical analysis
All experimental results were shown as mean ± standard deviation (SD). All statistical analysis was evaluated through Student’s t-test or one-way analysis of variance using GraphPad Prism software version 6.0 (GraphPad Software Inc., San Diego, CA, USA). p values less than .05 were considered to be statistically significant.
Results
H/R treatment decreased PK2 expression and inactivated the Akt/mTOR pathway in H9c2 cardiomyocytes
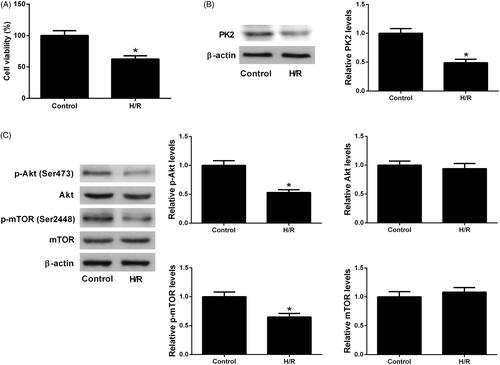
As compared with the control group, H/R treatment resulted in a significant suppression of cell viability in H9c2 cardiomyocytes (). In addition, western blot analysis showed that PK2 protein level was reduced in response to H/R exposure with respect to control group (). It has been reported that the Akt/mTOR pathway is involved in cardiovascular diseases including AMI [Citation12]. The effect of H/R treatment on the Akt/mTOR pathway in H9c2 cardiomyocytes was explored. As displayed in , we found that the protein levels of p-Akt (Ser473) and p-mTOR (Ser2448) were decreased, whereas the expression of total Akt and mTOR were not changed in the H/R treatment group relative to control group, suggesting that H/R treatment inhibited the Akt/mTOR pathway in H9c2 cardiomyocytes.
Figure 1. Effects of H/R treatment on PK2 expression and the Akt/mTOR pathway in H9c2 cardiomyocytes. (A) CCK-8 assay was conducted to assess the viability of H9c2 cardiomyocytes following H/R treatment. (B) PK2 protein level in H9c2 cardiomyocytes after H/R treatment was determined by western blot. (C) The protein levels of p-Akt, Akt, p-mTOR and mTOR in H9c2 cardiomyocytes exposed to H/R were estimated by western blot. *p < .05.
PK2 activated the Akt/mTOR pathway in H/R-treated H9c2 cardiomyocytes
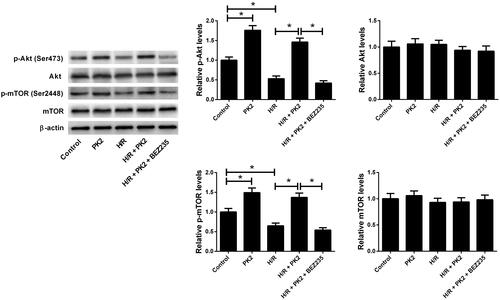
Next, we examined the effect of PK2 on the Akt/mTOR pathway in H/R-treated H9c2 cardiomyocytes. Western blot analysis indicated that administration with PK2 increased the phosphorylation of Akt and mTOR in H9c2 cardiomyocytes versus control group. Notably, treatment with PK2 evidently reversed the reduction of p-Akt and p-mTOR expression induced by H/R in H9c2 cardiomyocytes, while these effects were abrogated following the addition of BEZ235, a dual PI3K/mTOR inhibitor (). These results suggested that PK2 activated the Akt/mTOR pathway in H/R-treated H9c2 cardiomyocytes.
Figure 2. Effect of PK2 on the Akt/mTOR pathway in H/R-treated H9c2 cardiomyocytes. H9c2 cardiomyocytes were treated with 5 nM PK2 in the presence or absence of 5 mM BEZ235, followed by H/R treatment for 12/12 h, or H9c2 cardiomyocytes were only treated with 5 nM PK2 for 24 h. Western blot was performed to measure the protein levels of p-Akt, Akt, p-mTOR, and mTOR. *p < .05.
PK2 suppressed H/R-induced injury by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes
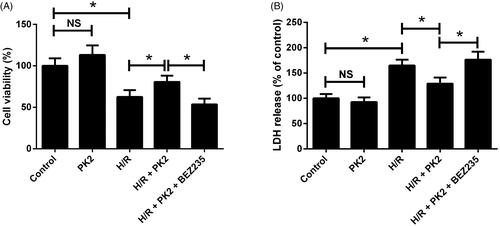
To explore the effects of PK2 on H/R-induced injury in H9c2 cardiomyocytes, cell viability and LDH release were evaluated by CCK-8 assay and LDH release assay, respectively. The results demonstrated that PK2 treatment alone showed no effect on cell viability and LDH release in H9c2 cardiomyocytes (). Besides, H/R-induced decrease of cell viability and increase of LDH release were restored by treatment with PK2, which were offset following exposure to a combination of PK2 and BEZ235 (). Together, these data indicated that PK2 impeded H/R-induced injury by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes.
Figure 3. Effects of PK2 on H/R-induced injury in H9c2 cardiomyocytes. H9c2 cardiomyocytes were treated with 5 nM PK2 in the presence or absence of 5 mM BEZ235, followed by H/R treatment for 12/12 h, or H9c2 cardiomyocytes were only treated with 5 nM PK2 for 24 h. (A) cell viability was evaluated by CCK-8 assay. (B) LDH release was estimated by LDH release assay. *p < .05. NS: not significant.
PK2 inhibited H/R-induced apoptosis by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes
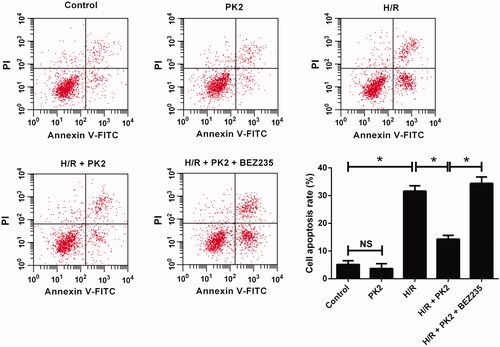
To clarify the effect of PK2 on apoptosis in H/R-treated H9c2 cardiomyocytes, flow cytometry analysis was applied to detect cell apoptosis. As presented in , no significant difference in apoptotic rate was observed between PK2-treated group and control group. H/R exposure led to a higher ratio of apoptosis in H9c2 cardiomyocytes, while this effect was significantly attenuated by PK2 treatment. However, inhibition of the Akt/mTOR pathway by BEZ235 successfully abolished the suppressive effect of PK2 on H/R-induced apoptosis in H9c2 cardiomyocytes. Thus, we concluded that PK2 inhibited H/R-induced apoptosis by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes.
Figure 4. Effect of PK2 on H/R-induced apoptosis in H9c2 cardiomyocytes. H9c2 cardiomyocytes were treated with 5 nM PK2 in the presence or absence of 5 mM BEZ235, followed by H/R treatment for 12/12 h, or H9c2 cardiomyocytes were only treated with 5 nM PK2 for 24 h. Cell apoptosis was evaluated by flow cytometry. *p < .05. NS, not significant.
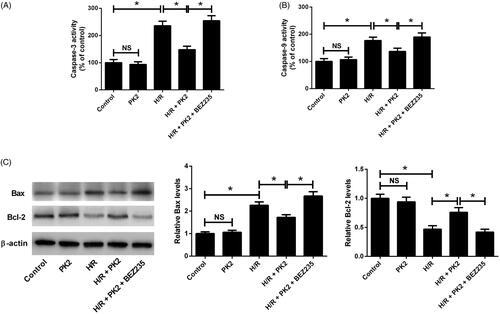
PK2 regulated caspase-3/9 activities and apoptosis-related protein expression by activation of the Akt/mTOR pathway
To further confirm whether PK2 could inhibit H/R-induced apoptosis in H9c2 cardiomyocytes, caspase-3 and caspase-9 activities were measured by caspase-3 and caspase-9 activity assays. The results revealed that PK2 alone did not affect caspase-3 and caspase-9 activities in H9c2 cardiomyocytes (). H/R stimulation enhanced the activities of caspase-3 and caspase-9 in H9c2 cardiomyocytes, while PK2 treatment apparently undermined H/R-induced increase of caspase-3 and caspase-9 activities (). However, the presence of BEZ235 nullified the inhibitory effects of PK2 on H/R-induced increase of caspase-3 and caspase-9 activities in H9c2 cardiomyocytes (). Meanwhile, western blot analysis demonstrated that the protein levels of Bax and Bcl-2 were unchanged after PK2 treatment in H9c2 cardiomyocytes (). H/R exposure resulted in a significant increase of Bax and an evident decrease of Bcl-2 in H9c2 cardiomyocytes (). PK2 treatment effectively reduced Bax level and elevated Bcl-2 level in H9c2 cardiomyocytes in response to H/R, which was overturned following treatment with BEZ235 (). Collectively, these results proved that PK2 regulated caspase-3/9 activities and apoptosis-related protein expression by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes.
Figure 5. Effects of PK2 on the apoptosis-related protein activities and expression in H9c2 cardiomyocytes. H9c2 cardiomyocytes were treated with 5 nM PK2 in the presence or absence of 5 mM BEZ235, followed by H/R treatment for 12/12 h, or H9c2 cardiomyocytes were only treated with 5 nM PK2 for 24 h. (A and B) The activities of caspase-3 and caspase-9 in treated H9c2 cardiomyocytes were examined by caspase-3 and caspase-9 activity assays. (C) The protein levels of Bax and Bcl-2 in treated H9c2 cardiomyocytes were determined by western blot. *p < .05. NS, not significant.
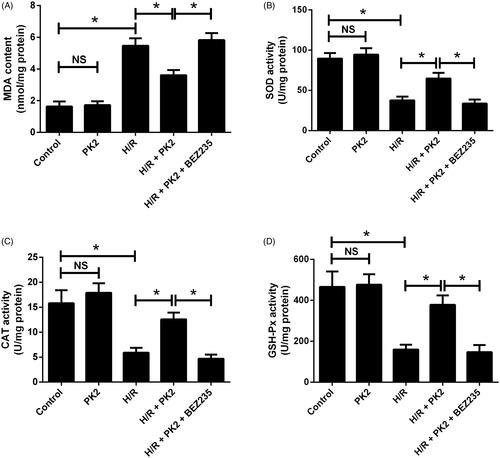
PK2 suppressed H/R-induced oxidative stress by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes
To investigate the effect of PK2 on oxidative stress in H/R-exposed H9c2 cardiomyocytes, the level of oxidative stress indicator MDA and the activities of anti-oxidant enzymes including SOD, CAT, and GSH-Px were measured. As shown in ), PK2 treatment alone showed no significant effect on MDA content and SOD, CAT and GSH-Px activities, while H/R stimulation notably augmented MDA content and blocked the activities of SOD, CAT and GSH-Px in H9c2 cardiomyocytes. Interestingly, treatment with PK2 recuperated H/R-induced enhancement of MDA content and inhibition of SOD, CAT and GSH-Px activities in H9c2 cardiomyocytes, while these effects were abolished by the addition of BEZ235. Therefore, we concluded that PK2 retarded H/R-induced oxidative stress by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes.
Figure 6. Effects of PK2 on H/R-induced oxidative stress in H9c2 cardiomyocytes. H9c2 cardiomyocytes were treated with 5 nM PK2 in the presence or absence of 5 mM BEZ235, followed by H/R treatment for 12/12 h, or H9c2 cardiomyocytes were only treated with 5 nM PK2 for 24 h. MDA content (A), and the activities of SOD (B), CAT (C), and GSH-Px (D) were measured in H9c2 cardiomyocytes. *p < .05. NS, not significant.
Discussion
AMI is a complex event responsible for cardiac fibrosis, malignant arrhythmia and heart failure [Citation13], and searching for novel therapeutic targets to treat AMI has become a research focus in the cardiovascular field. Increasing experimental data have shown that H/R-induced myocardial injury in AMI is closely implicated in ROS-induced oxidative stress, calpain activation and cardiomyocyte apoptosis [Citation14]. Wide range of evidence proves that cellular hypoxia disrupts microenvironment homeostasis, resulting in cardiomyocyte necrosis and apoptosis [Citation15]. Cardiomyocyte apoptosis is an important pathological process of H/R-induced myocardial injury and strongly associated with heart dysfunction [Citation16]. In addition, it is extensively believed that oxidative stress is a major cause of cardiomyocyte apoptosis in the development of H/R-induced myocardial injury [Citation17]. Therefore, inhibition of cardiomyocyte apoptosis and oxidative stress might be an effective therapeutic strategy for preventing H/R-induced myocardial injury in AMI. Herein, we demonstrated that H/R stimulation suppressed cell viability, increased LDH release, induced apoptosis and oxidative stress in H9c2 cardiomyocytes, indicating that H/R induced myocardial injury.
Prokineticins (PKs), a newly identified peptide family, has been shown to participate in cardiac repair possibly by inducing angiogenesis or regenerating cardiomyocytes [Citation18]. Dysregulation of PKs and their receptors has been reportedly associated with heart failure [Citation19]. PK2, a member of PK family that located at chromosome 3p21.1, is a powerful angiogenic factor. Recent studies have documented that PK2/PKR1 signalling acts as a new linker between development of obesity, diabetes and cardiovascular diseases [Citation20]. Previously, it was reported that PK2 or overexpressing PKR1 protected cardiomyocytes against hypoxia-mediated apoptosis by activating Akt pathway [Citation11]. In addition to its role in cardiomyocyte protection against hypoxia-induced injury, PK2 was also reported to inhibit serum starvation-induced apoptosis in adrenal cortical capillary endothelial cells and luteal endothelial cells [Citation21,Citation22]. A previous study indicated that the expression profile of PKR1 and PKR2 might manage the effects of PK2 on coronary endothelial cells, favouring endothelial cell barrier dysfunction or compensatory angiogenesis during myocardial infarction [Citation23]. However, whether PK2 has a protective effect on H/R-induced injury in endothelial cells is unknown and deserves further research. It is worth noting that PK2 has been demonstrated to be activated by hypoxia-ischemia in primary cortical cultures, and blocking PK2 decreases infarct volume, suppresses central inflammation, and improves behavioural functional outcome in an in vivo stroke model [Citation24]. In the present study, we demonstrated that PK2 expression was decreased in response to H/R stimulation. PK2 treatment activated the Akt/mTOR pathway in H/R-exposed H9c2 cardiomyocytes. We further manifested that PK2 treatment suppressed H/R-induced reduction of viability, increase of LDH release, apoptosis and oxidative stress in H9c2 cardiomyocytes, which were abolished following the addition of BEZ235, a dual PI3K/mTOR inhibitor. These results suggested that PK2 protected cardiomyocytes from H/R-induced injury by activation of the Akt/mTOR pathway.
The Akt/mTOR signalling pathway is a well-recognized signal transduction pathway that participates in the regulation of cell proliferation, adhesion, apoptosis and survival under pathological conditions [Citation25]. Accumulating evidence shows that the Akt/mTOR pathway is involved in cardiomyocyte protection against hypoxia-induced injury [Citation26]. It was documented that ginsenoside Rg1 protected cardiomyocytes from hypoxia-induced cell injury through activation of the PI3K/Akt/mTOR pathway [Citation27]. Moreover, a previous study reported that miR-21 inhibited H/R-induced autophagy and apoptosis in H9c2 cells by the activation of the Akt/mTOR pathway [Citation28]. In addition, it was demonstrated that dragon’s blood extracts exerted cardioprotective efficacy against myocardial injury in AMI mouse model through activating the PI3K/Akt/mTOR pathway [Citation12]. In our study, we showed that H/R exposure suppressed the activation of Akt/mTOR pathway in H9c2 cardiomyocytes, consistent with the previous study [Citation29]. PK2 activated the Akt/mTOR pathway in H/R-exposed H9c2 cardiomyocytes, which was in accordance with the previous report [Citation11]. We also found that PK2 treatment recuperated H/R-induced enhancement of MDA content and inhibition of SOD, CAT and GSH-Px activities by activation of the Akt/mTOR pathway in H9c2 cardiomyocytes. It has been demonstrated that NF-E2-related factor (Nrf2) is an important factor associated with the expression of cytoprotective genes in response to oxidative stress [Citation30]. Nrf2 knockdown inhibited the expression of Nrf2 downstream target genes, including SOD, CAT, and GSH-Px, at the mRNA and protein levels in H9c2 cells [Citation31]. Nrf2 expression was mediated by the Akt/mTOR pathway [Citation31,Citation32]. Therefore, we inferred that PK2 could increase protein levels of SOD, CAT, and GSH-Px in H/R-exposed H9c2 cardiomyocytes.
Conclusion
In summary, our study provided the evidence that PK2 relieved H/R-induced injury in H9c2 cardiomyocytes by attenuating apoptosis and oxidative stress through activation of Akt/mTOR pathway, heightening our understanding of the roles and molecular mechanisms of PK2 in H/R-induced injury in AMI. Therefore, our study suggested that PK2 might be a potential therapeutic target to protect against H/R-induced injury.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Hartley A, Marshall DC, Salciccioli JD, et al. Trends in mortality from ischemic heart disease and cerebrovascular disease in Europe: 1980 to 2009. Circulation. 2016;133(20):1916–1926.
- Xue-Juan W, Hao W, Cai-Ying G, et al. Impact of an intelligent chronic disease management system on patients with type 2 diabetes mellitus in a Beijing community. BMC Health Serv Res. 2018;18(1):821.
- Araszkiewicz A, Grygier M, Lesiak M, et al. The impact of ischemia-reperfusion injury on the effectiveness of primary angioplasty in ST-segment elevation myocardial infarction. PWKI. 2013;9:275–281.
- Ansari J, Kaur G, Gavins F. Therapeutic potential of annexin A1 in ischemia reperfusion injury. IJMS. 2018;19(4):1211.
- Lin DC, Bullock CM, Ehlert FJ, et al. Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J Biol Chem. 2002;277(22):19276–19280.
- Giannini E, Lattanzi R, Nicotra A, et al. The chemokine Bv8/prokineticin 2 is up-regulated in inflammatory granulocytes and modulates inflammatory pain. Proc Natl Acad Sci USA. 2009;106(34):14646–14651.
- Monnier J, Samson M. Prokineticins in angiogenesis and cancer. Cancer Lett. 2010;296(2):144–149.
- Negri L, Lattanzi R, Giannini E, et al. Bv8, the amphibian homologue of the mammalian prokineticins, modulates ingestive behaviour in rats. Br J Pharmacol. 2004;142(1):181–191.
- Landucci E, Lattanzi R, Gerace E, et al. Prokineticins are neuroprotective in models of cerebral ischemia and ischemic tolerance in vitro. Neuropharmacology. 2016;108:39–48.
- Hu WP, Zhang C, Li JD, et al. Impaired pain sensation in mice lacking prokineticin 2. Mol Pain. 2006;2:35.
- Urayama K, Guilini C, Messaddeq N, et al. The prokineticin receptor-1 (GPR73) promotes cardiomyocyte survival and angiogenesis. FASEB J. 2007;21(11):2980–2993.
- Li C, Zhang Y, Wang Q, et al. Dragon's blood exerts cardio-protection against myocardial injury through PI3K-Akt-mTOR signaling pathway in acute myocardial infarction mice model. J Ethnopharmacol. 2018;227:279–289.
- Al Rajoub B, Noureddine S, El Chami S, et al. The prognostic value of a new left bundle branch block in patients with acute myocardial infarction: A systematic review and meta-analysis. Heart Lung. 2017;46(2):85–91.
- Yu P, Zhang J, Yu S, et al. Protective effect of sevoflurane postconditioning against cardiac ischemia/reperfusion injury via ameliorating mitochondrial impairment, oxidative stress and rescuing autophagic clearance. PLoS One. 2015;10(8):e0134666.
- Zhang B, Zhou M, Li C, et al. MicroRNA-92a inhibition attenuates hypoxia/reoxygenation-induced myocardiocyte apoptosis by targeting Smad7. PLOS One. 2014;9(6):e100298.
- Baines CP. How and when do myocytes die during ischemia and reperfusion: the late phase. J Cardiovasc Pharmacol Ther. 2011;16(3-4):239–243.
- de Vries DK, Kortekaas KA, Tsikas D, et al. Oxidative damage in clinical ischemia/reperfusion injury: A reappraisal. Antioxid Redox Signal. 2013;19(6):535–545.
- Khurana R, Simons M, Martin JF, et al. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation. 2005;112(12):1813–1824.
- Urban JD, Clarke WP, von Zastrow M, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320(1):1–13.
- Nebigil CG. Prokineticin is a new linker between obesity and cardiovascular diseases. Front Cardiovasc Med. 2017;4:20.
- LeCouter J, Lin R, Tejada M, et al. The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: localization of Bv8 receptors to endothelial cells. Proc Natl Acad Sci USA. 2003;100(5):2685–2690.
- Kisliouk T, Podlovni H, Spanel-Borowski K, et al. Prokineticins (endocrine gland-derived vascular endothelial growth factor and BV8) in the bovine ovary: expression and role as mitogens and survival factors for corpus luteum-derived endothelial cells. Endocrinology. 2005;146(9):3950–3958.
- Guilini C, Urayama K, Turkeri G, et al. Divergent roles of prokineticin receptors in the endothelial cells: angiogenesis and fenestration. Am J Physiol Heart Circ Physiol. 2010;298(3):H844–852.
- Cheng MY, Lee AG, Culbertson C, et al. Prokineticin 2 is an endangering mediator of cerebral ischemic injury. Proc Natl Acad Sci USA. 2012;109(14):5475–5480.
- Karar J, Maity A. PI3K/Akt/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51.
- Li C, Tian J, Li G, et al. Asperosaponin VI protects cardiac myocytes from hypoxia-induced apoptosis via activation of the PI3K/Akt and CREB pathways. Eur J Pharmacol. 2010;649(1-3):100–107.
- Qin L, Fan S, Jia R, et al. Ginsenoside Rg1 protects cardiomyocytes from hypoxia-induced injury through the PI3K/Akt/mTOR pathway. Pharmazie. 2018;73(6):349–355.
- Huang Z, Wu S, Kong F, et al. MicroRNA-21 protects against cardiac hypoxia/reoxygenation injury by inhibiting excessive autophagy in H9c2 cells via the Akt/mTOR pathway. J Cell Mol Med. 2017;21(3):467–474.
- Xu Y, Gu Q, Tang J, et al. Substance P attenuates hypoxia/reoxygenation-induced apoptosis via the Akt signalling pathway and the NK1-receptor in H9C2cells. Heart Lung Circ. 2018;27(12):1498–1506.
- Chen M, Zhang M, Zhang X, et al. Limb ischemic preconditioning protects endothelium from oxidative stress by enhancing Nrf2 translocation and upregulating expression of antioxidases. PLoS One. 2015;10(6):e0128455.
- Duan J, Guan Y, Mu F, et al. Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: involvement of the AMPK/GSK-3β/Nrf2 signaling pathway. Sci Rep. 2017;7(1):41491.
- Yao H, Han X, Han X. The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. Am J Cardiovasc Drugs. 2014;14(6):433–442.