
Abstract
Human hepatocellular carcinoma (HCC) is the most common type of liver cancer, and it has a high mortality rate. Despite surgical treatments, radiotherapy, and chemotherapy, the median survival of patients with advanced HCC is low. Evidence has shown that tanshinone (TA) I exhibits anti-proliferative activity against numerous cancers. However, the role of TA I and its mechanism in HCC remain unknown. Here, we determined the anti-cancer potential of TA I against HCC cell lines HepG2 and Huh7. Cell viability was analyzed using a Cell Counting Kit-8 assay. Flow cytometry was used to analyze cell cycles and apoptosis. Western blotting was used to detect protein expression and phosphorylation levels. TA I was found to inhibit cell proliferation, induce G0/G1 phase arrest, and trigger apoptosis in HepG2 and Huh7 cells. We further explored the molecular mechanism of TA I-mediated apoptosis. Our results showed that TA I induced G0/G1 phase arrest through downregulation of cyclin D1 expression and upregulation of p21 expression. TA I induced cell apoptosis via reactive oxygen species-mediated endoplasmic reticulum stress and by inhibiting p53/damage-regulated autophagy modulator (DRAM)-mediated autophagy in HepG2 and Huh7 cells. Therefore, TA I may be an anti-cancer drug candidate in the treatment of HCC.
Introduction
It is well known that human hepatocellular carcinoma (HCC) is the most common type of liver cancer and the third leading cause of cancer death worldwide [Citation1]. Currently available therapy involves surgical resection, chemotherapy, and radiotherapy which could significantly improve the survival rates of patients with HCC. However, for patients with advanced HCC, surgical resection is not effective, and chemotherapy is often the last choice due to its high toxicity and drug resistance. Hence, it is necessary to develop more effective therapeutic drugs with high efficacy and low toxicity for advanced HCC patients.
Traditional Chinese herbs are considered to have great potential for the treatment of cancer and are increasingly attracting attention. Danshen (Salvia miltiorrhiza Bunge), a traditional Chinese herb, is used for treating numerous diseases including hepatitis, menstrual disorders, stroke, vascular disease, coronary heart disease, and cancer in Asian countries [Citation2]. Tanshinone (TA) I is one of the bioactive components of Danshen and reportedly has various pharmacological and biochemical activities, including antioxidant, anti-inflammatory, and anti-cancer activities [Citation3]. Recently, the majority of studies about TA I have focused on its anti-cancer activity. These studies demonstrated that TA I could induce the apoptosis of cancer cells, including in gastric cancers [Citation4], human breast cancer [Citation5], human lung cancer [Citation6], and human colon cancer [Citation7,Citation8]. Nonetheless, the exact mechanism underlying the anti-cancer activity of TA I in HCC remains unknown.
Accumulated evidence suggests that autophagy plays an important role in cell apoptosis mediated by anti-cancer drugs [Citation9,Citation10]. Furthermore, the p53/damage-regulated autophagy modulator (DRAM) signaling pathway has been reported to be involved in regulating autophagy [Citation11,Citation12]. Many studies have revealed that ROS accumulation in various types of cancers involves cell apoptosis and autophagy induced by anti-tumor drugs [Citation13,Citation14]. Increasing evidence suggests that ROS-mediated ER stress activation is involved in anti-cancer drug-induced apoptosis of cancer cells [Citation15–17].
To understand the anti-cancer activity of TA I in HCC, this study explored the molecular mechanisms by which TA I may inhibit cell proliferation and induce apoptosis in HCC cell lines HepG2 and Huh7. These results are expected to provide evidence that TA I is a potential drug candidate for treating HCC.
Materials and methods
Reagents
The anti-cleaved poly(ADP-ribose) polymerase (PARP, catalog #5625), anti-β-actin (#4967), anti-LC3B (#2775), anti-beclin-1 (#3738), anti-CHOP (#5554), anti-phospho-eIF2α (Ser51) (#9721), and anti-cyclin D1 (#2922) antibodies were purchased from Cell Signal Technology (Danvers, MA, USA). Anti-p21 (#195720) was purchased from R&D Systems (Minneapolis, MN, USA). Annexin V-FITC and a propidium iodide (PI) kit were purchased from BD Biosciences (San Jose, CA, USA). TA I, a cocktail of protease inhibitors, 3-methyladenine (3-MA), and N-acetyl-cysteine (NAC, a reactive oxygen species [ROS] scavenger) were purchased from Sigma-Aldrich (St. Louis, MO, USA). A cell cycle detection kit and ROS assay kit were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). RPMI-1640 medium and fetal bovine serum (FBS) were purchased from Gibco Life Technologies (Grand Island, NY, USA).
Cell culture
Human HCC HepG2, Huh7, and L02 cell lines were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). Cells were cultured and maintained in RPMI-1640 supplemented with 10% FBS and penicillin (100 U/mL)/streptomycin (100 µg/mL) at 37 °C with 5% CO2.
Cell transfection
CHOP siRNA (siCHOP) and control siRNA (siNC) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The HepG2 and Huh7 cells were seeded at 4 × 103 cells/well in 96-well plates for 24 h, and then the cells were transfected with siCHOP and siNC for 48 h according to the Lipofectamine 2000 kit instructions.
Cell Counting Kit-8 assay
The cell viability of HepG2, Huh7, and L02 cells was assessed using a Cell Counting Kit-8 (CCK-8) assay. The HepG2, Huh7, and L02 cells were seeded at 8 × 103 cells/well in 96-well plates for 24 h. Next, HepG2 and Huh7 cells were treated with TA I (0, 2, 4, 6, 8, or 10 μM) for 24 h. L02 cells were treated with TA I (0, 2, 4, 8, 16, or 32 μM) for 24 h. The cells were incubated with CCK8 solution (10 μL CCK8 solution/well) for 4 h. The optical density of each well was detected using an ELISA reader at a wavelength of 450 nm.
Cell cycle analysis
Flow cytometry was used to examine the distribution of the cell cycle phases in HepG2 and Huh7 using a Cell Cycle Detection Kit (KeyGen Biotech, Nanjing, China). Briefly, HepG2 and Huh7 cells were seeded at 2 × 106 cells/well in a 6-well plate for 24 h. After treatment with TA I (0, 2, 4, and 6 μM) for 24 h, the treated cell groups were fixed in cold 70% ethanol overnight. The cell samples were then stained with 50 μL PI (50 μg/mL) for 30 min in the dark at 25 °C. The distribution of cell cycles was detected by a FACSCalibur (BD Biosciences, San Jose, CA, USA), and data were analyzed using Modfit LT version 2.0 software (Verity Software House, Topsham, ME, USA).
Cell apoptosis analysis
Flow cytometry was used to detect the apoptotic rate in HepG2 and Huh7 cells using the Apoptosis Detection Kit (KeyGen Biotech, Nanjing, China). The HepG2 and Huh7 cells were seeded at 2 × 106 cells/well in 6-well plates and treated with TA I (0, 2, 4, and 6 μM) for 24 h. The cells were then resuspended and double incubated with Annexin V-FITC and PI in the dark at room temperature for 15 min. A FACSCalibur flow cytometer was used to assay the apoptotic rate in HepG2 and Huh7 cells. Data were analyzed using FlowJo 7.6 software (FlowJo LLC, Ashland, OR, USA).
Measurement of ROS production
Flow cytometry was used to assay ROS levels in HepG2 and Huh7 using a ROS assay kit (KeyGen Biotech, Nanjing, China). Briefly, the cells were seeded in 6-well plates at a density of 2.0 × 105 cells/well for 24 h. Next, the cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h and then incubated with 10 μM of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) for 15 min at 37 °C in a dark room. A FACSCalibur flow cytometer was used to detect ROS production. FlowJo 7.6 software was used to analyze the data.
Western blot analysis
The HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 24 h and lysed with RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) with a 1% cocktail of protease inhibitors. The protein samples (40 μg/well) were separated using 10–15% SDS-PAGE and transferred onto nitrocellulose membranes (Millipore, USA). Membranes were incubated with primary antibodies against LC3B (1:1000), beclin-1 (1:2000), cyclin D1 (1:1000), cleaved-PARP (1:1000), phospho-eIF2α (Ser51) (1:3000), CHOP (1:1000), β-actin (1:5000), or p21 (1:1000) at 4 °C overnight, followed by incubation with HRP-conjugated secondary antibodies (1:25,000) at 25 °C for 2 h. Protein expression levels were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
All data were presented as mean ± standard deviation. Differences between groups were calculated using Student’s t test and one-way analysis of variance followed by Dunnett’s post hoc tests. Statistical significance was considered at p < .05.
Results
TA I inhibits the growth of HepG2 and Huh7 cells
To explore the anti-tumor activity of TA I on HepG2 and Huh7 cells, the viability of HepG2 and Huh7 cells were measured using a CCK-8 assay. The HepG2 and Huh7 cells were treated with TA I (0, 2, 4, 6, 8, or 10 μM) for 24 h. As shown in ), TA I significantly inhibited the growth of HepG2 and Huh7 cells in a concentration-dependent manner. The viability of HepG2 cells was reduced to 89.34 ± 3.15%, 77.25 ± 1.86%, 54.51 ± 2.56%, 38.23 ± 2.41%, and 30.06 ± 1.98% at doses of 2, 4, 6, 8, and 10 µM TA I, respectively, after treatment for 24 h. Furthermore, the viability of Huh7 cells was reduced to 88.07 ± 1.50%, 74.91 ± 1.57%, 52.34 ± 1.73%, 34.25 ± 1.53%, and 23.13 ± 1.41% at doses of 2, 4, 6, 8, and 10 µM TA I, respectively, after treatment for 24 h. Additionally, the viability of L02 cells was reduced to 91.97 ± 2.46%, 68.11 ± 1.84,% and 43.20 ± 0.95% at doses of 8, 16 and 32 µM TA I, respectively, after treatment for 24 h (). These data indicated that TA I exerted lower cytotoxic effects on L02 cells but markedly cytotoxic effects on HepG2 and Huh7 cells by suppressing cell growth.
Figure 1. TA I inhibits the proliferation of HepG2, Huh7, and L02 cells. The growth inhibitory effect of TA I was measured using the CCK-8 assay. (A) Cell viability was observed after treatment with TA I (0, 2, 4, 6, 8, and 10 μM) for 24 h in HepG2 cells. (B) Cell viability was observed after treatment with TA I (0, 2, 4, 6, 8, and 10 μM) for 24 h in Huh7 cells. (C) Cell viability was observed after treatment with TA I (0, 2, 4, 8, 16, and 32 μM) for 24 h in L02 cells. Data are presented as means ± standard deviations of three separate experiments. *p < .05, ***p < .001, ****p < .0001. TA: tanshinone; CCK-8: cell counting Kit-8.
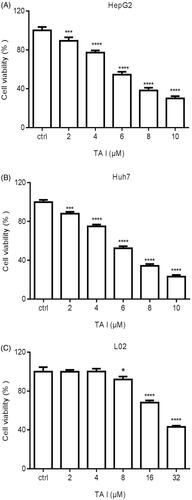
TA I induces G0/G1 phase arrest in HepG2 and Huh7 cells
To explore the mechanism of TA I anti-tumor activity on HepG2 and Huh7 cells, cell cycle distribution was analyzed using flow cytometry in the cells treated with TA I (0, 2, 4, and 6 µM) for 24 h. Our results illustrated that TA I significantly induced cell cycle arrest at the G0/G1 phase in a concentration-dependent manner in HepG2 and Huh7 cells (). The percentage of HepG2 cells in the G0/G1 phase increased to 53.89 ± 2.07%, 58.91 ± 1.70,% and 63.28 ± 2.07% at doses of 2, 4, and 6 µM TA I after treatment for 24 h (). Furthermore, the percentage of Huh7 cells in the G0/G1 phase increased to 51.94 ± 0.36%, 57.64 ± 0.78%, and 67.36 ±1.48% at doses of 2, 4, and 6 µM TA I after treatment for 24 h (). Moreover, our results showed that TA I markedly downregulated the expression of cyclin D1 protein and upregulated the expression of p21 protein (). These results revealed that TA I exerted effective anti-tumor activity by inducing G0/G1 phase arrest in HepG2 and Huh7 cells.
Figure 2. Effects of TA I on cell cycle distribution in HepG2 and Huh7 cells. (A) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 24 h, and cell cycle distribution was analyzed by flow cytometry. (B) Bar graphs show the quantification of cell cycle distribution in HepG2 and Huh7 cells. (C) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h, and the protein expression levels of cyclin D1 and p21 were assayed by western blot. β-actin served as a loading control. **p < .01, ***p < .001, ****p < .0001. TA: tanshinone.
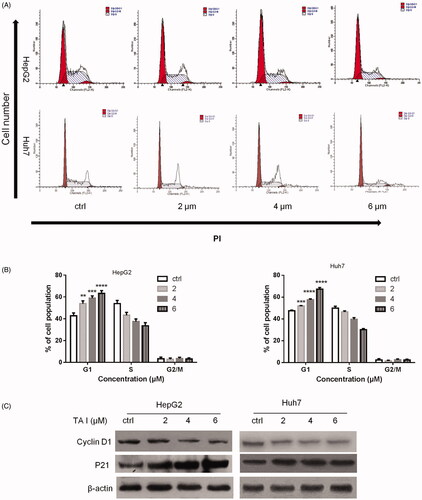
TA I triggers apoptosis in HepG2 and Huh7 cells
To further explore the mechanism of TA I anti-tumor activity in HepG2 and Huh7 cells, cell apoptosis was analyzed using flow cytometry in the cells treated with TA I (0, 2, 4, and 6 µM) for 24 h. As shown in , TA I significantly induced apoptosis in HepG2 and Huh7 cells in a concentration-dependent manner. The apoptotic rate in HepG2 cells increased to 9.96 ± 1.37%, 22.31 ± 1.46,% and 36.14 ± 2.23% at 2, 4, and 6 μM TA I, respectively (). Furthermore, the apoptotic rate in Huh7 cells increased to 11.94 ± 1.76%, 19.77 ± 2.73%, and 42.47 ± 2.09% at 2, 4, and 6 μM TA I (). We also examined the expression of apoptosis-related protein cleaved-PARP using western blot in HepG2 and Huh7 cells. As shown in , TA I upregulated the expression of cleaved PARP protein. These results suggested that TA I exerted effective anti-tumor activity by inducing cell apoptosis in HepG2 and Huh7 cells.
Figure 3. Effects of TA I on cell apoptosis in HepG2 and Huh7 cells. (A) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 24 h, and cell apoptosis was analyzed by flow cytometry. (B) Bar graphs show the quantification of apoptosis in HepG2 and Huh7 cells. (C) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h, and the protein expression level of cleaved PARP was assayed by western blot. β-actin served as a loading control. *p < .05, ***p < .001, ****p < .0001. TA: tanshinone; PARP: poly(ADP-ribose) polymerase.
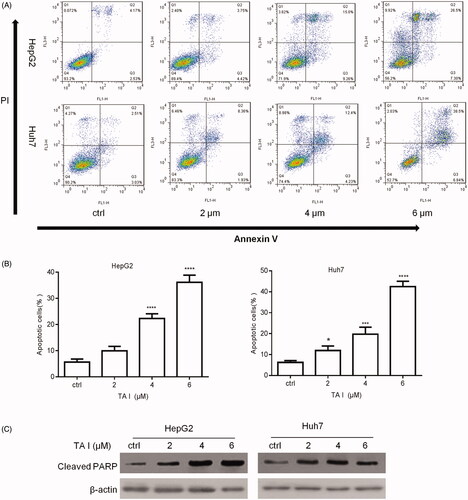
TA I triggers cell apoptosis by suppressing p53/DRAM-mediated autophagy in HepG2 and Huh7 cells
We examined TA I-activated autophagy in HepG2 and Huh7 cells. Western blot analysis was used to examine the expression level of autophagy-associated proteins, including LC3B and beclin-1. Our results showed that the expression levels of LC3B-II and beclin-1 were significantly decreased by TA I in HepG2 and Huh7 cells ().
Figure 4. TA I promotes apoptosis in HepG2 and Huh7 cells by inhibiting autophagy through suppressing the p53/DRAM signaling pathway. (A) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h, and the protein expression levels of LC3B and beclin-1 were detected by western blot. (B) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h, and the protein expression levels of p53 and DRAM were detected by western blot. (C) HepG2 and Huh7 cells were pre-treated with 3-MA (2 mM) for 1 h, and then incubated with TA I (4 µM) for 24 h. Cell viability was examined by CCK-8 assay. **p < .01, ***p < .001. TA: tanshinone; DRAM: damage-regulated autophagy modulator; 3-MA: 3-methyladenine; CCK-8: cell counting Kit-8.
The expression levels of p53 and DRAM were also examined using western blot analysis. Our results showed that TA I markedly downregulated p53 and DRAM expression in HepG2 and Huh7 cells (). In addition, we pretreated HepG2 and Huh7 cells with or without 3-MA (an autophagy inhibitor) for 1 h, followed by treatment with TA I for 24 h, and examined cell viability using a CCK-8 assay. As shown in , 3-MA pretreatment significantly increased TA I-mediated inhibition of cell proliferation in HepG2 and Huh7 cells. These data indicated that TA I mediated apoptosis in HepG2 and Huh7 cells by inhibiting p53/DRAM-activated autophagy.
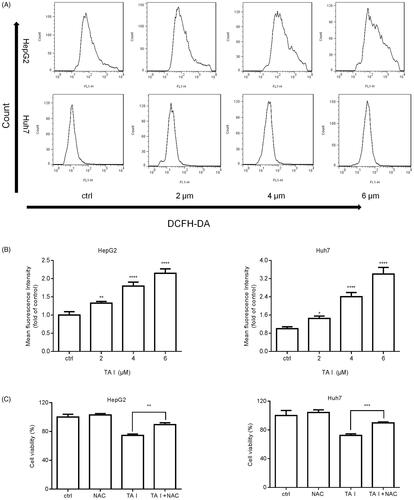
TA I induces cell apoptosis via ROS accumulation in HepG2 and Huh7 cells
We investigated intracellular ROS accumulation using flow cytometry following TA I treatment (0, 2, 4, or 6 μM) for 12 h. We found that TA I significantly increased intracellular ROS accumulation in a concentration-dependent manner in HepG2 and Huh7 cells (). To further test whether increased ROS accumulation plays an important role in TA I-induced cell death, HepG2 and Huh7 cells were pretreated with NAC for 1 h before TA I treatment, and then the viability was examined using a CCK-8 assay. Our results revealed that pretreatment with NAC effectively prevented TA I-induced growth inhibition of HepG2 and Huh7 cells (). Collectively, our results demonstrated that TA I-induced cell apoptosis was associated with ROS accumulation in HepG2 and Huh7 cells.
Figure 5. TA I promotes apoptosis in HepG2 and Huh7 cells via ROS production. (A) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h, and ROS levels were detected by flow cytometry. (B) Bar graphs show the quantification of ROS levels in HepG2 and Huh7 cells. (C) HepG2 and Huh7 cells were pre-treated with NAC (4 mM) for 1 h, and then incubated with TA I (4 µM) for 24 h. Cell viability was examined by CCK-8 assay. *p < .05, **p < .01, ***p < .001, ****p < .0001. TA: tanshinone; ROS: reactive oxygen species; NAC: N-acetyl-cysteine; CCK-8: cell counting Kit-8.
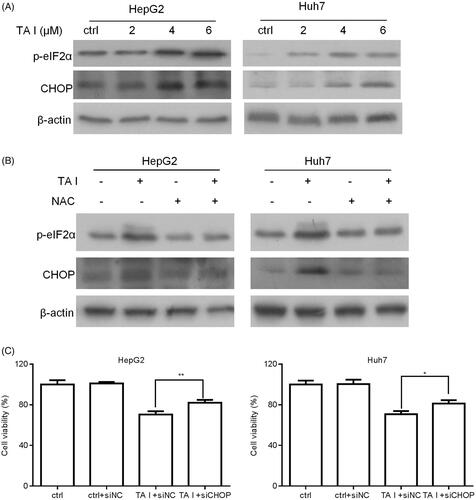
TA I induces cell apoptosis via the ROS-mediated endoplasmic reticulum (ER) stress pathway in HepG2 and Huh7 cells
We firstly examined the effect of TA I on the ER stress-associated proteins CHOP and phospho-eIF2α in HepG2 and Huh7 cells. As shown in , TA I markedly upregulated the expression of CHOP and phospho-eIF2α proteins in HepG2 and Huh7 cells. Furthermore, we investigated whether ROS contributed to TA I-mediated ER stress activation in HepG2 and Huh7 cells. Both cells were pretreated with NAC for 1 h and then treated with TA I for 12 h. The levels of CHOP and phospho-eIF2α proteins were detected using western blot analysis. Our results demonstrated that NAC pretreatment markedly decreased the expression of CHOP and phospho-eIF2α proteins induced by TA I (). In addition, downregulation of CHOP protein expression markedly attenuated TA I-mediated cell growth inhibition in HepG2 and Huh7 cells (). These results demonstrated that TA I induced cell apoptosis via ROS-induced activation of ER stress in HepG2 and Huh7 cells.
Figure 6. TA I promotes apoptosis in HepG2 and Huh7 cells by activating ER stress through ROS generation. (A) HepG2 and Huh7 cells were treated with TA I (0, 2, 4, and 6 μM) for 12 h, and the expression levels of phospho-eIF2α and CHOP proteins were assayed by western blot. (B) HepG2 and Huh7 cells were pre-treated with NAC (4 mM) for 1 h, and then incubated with TA I (4 µM) for 12 h; the expression levels of phospho-eIF2α and CHOP proteins were assayed by western blot. β-actin served as a loading control. (C) HepG2 and Huh7 cells were transfected with or without siNC or siCHOP for 48 h and then incubated with or without TA I (4 µM) for 24 h. The cell viability was measured using the CCK-8 assay. *p < .05, **p < .01. TA: tanshinone; ER: endoplasmic reticulum; ROS: reactive oxygen species; NAC: N-acetyl-cysteine; siRNA: control siRNA; CCK-8: cell counting Kit-8.
Discussion
Here, we presented the first investigation of the anti-cancer effects of TA I on HCC cell lines HepG2 and Huh7. Our results demonstrated that TA I inhibited cell proliferation, induced G0/G1 arrest, and triggered apoptosis in HepG2 and Huh7 cells. Furthermore, TA I could induce cell apoptosis through ROS-induced activation of ER stress and the inhibition of p53/DRAM-mediated autophagy in HepG2 and Huh7 cells. Abnormal cell cycle progression often mediates uncontrolled growth in cancer cells [Citation18,Citation19]. Therefore, anti-cancer drug-mediated cell cycle arrest is considered an attractive strategy for cancer therapy. It is well known that cell cycle progression is dependent on the activity of cyclin-dependent kinase complexes (cyclin-CDKs). Downregulation of cyclin D1 may arrest the cell cycle at the G0/G1 phase [Citation20,Citation21]. p21, a broad-spectrum CDK inhibitor, has been demonstrated to promote cell cycle arrest by inhibiting the activity of various cyclin-CDK complexes [Citation22,Citation23]. Moreover, some studies have suggested that upregulating p21 expression plays an essential role in G0/G1 arrest [Citation24,Citation25]. Wang et al. revealed that TA I inhibited cell cycle progression by decreasing cyclin B and Cdk2 proteins [Citation5]. Su et al. also revealed that TA I induced G0/G1 arrest by upregulating p21 expression in human colon cancer Colo 205 cells [Citation26]. Our results demonstrated that TA I increased p21 expression and decreased cyclin D1 expression. Therefore, TA I may induce G0/G1 arrest through upregulating p21 and down-regulating cyclin D1.
Apoptosis is a necessary physiological process of cell death, and the induction of apoptosis in cancer cells is a good strategy for cancer therapy. Various reports suggested that TA I induces apoptosis in a variety of human cancer cells. TA I-induced apoptotic death in human breast cancer cells is mediated by the activation of caspase 3, the downregulation of Bcl-2, and the upregulation of Bax [Citation27]. In human colon cancer Colo 205 cells, TA I promotes apoptosis by increasing the expression of bax and caspase-3 proteins [Citation26]. In this study, we determined that treatment with TA I significantly induced apoptosis by upregulating the expression of cleaved PARP in HepG2 and Huh7 cells. It is known that a large number of anti-cancer drugs induce cell apoptosis and autophagy in cancer cells via a ROS-dependent pathway [Citation28,Citation29]. Moreover, total tanshinones reportedly induce cell apoptosis by increasing intracellular ROS production in lung cancer 95D cells [Citation30]. In human osteosarcoma MG-63 cells, ROS induces caspase-dependent apoptosis [Citation31]. Lee et al. also reported that TA I induces ROS generation and caspase-dependent apoptosis in HepG2 cells [Citation32]. Our results demonstrated that TA I induced ROS generation in a concentration-dependent manner in HepG2 and Huh7 cells. Furthermore, pre-treatment with NAC reversed the TA I-mediated cell growth inhibition in HepG2 and Huh7 cells. In addition, ER stress has been reported to be closely associated with anti-cancer drug-induced cancer cell apoptosis, which is an important downstream target of ROS. Zhu et al. reported that apoptosis is induced in non-small cell lung cancer cells via the ROS-mediated ER stress pathway [Citation17]. Moreover, Liu et al. demonstrated that shikonin triggers cell apoptosis via the ROS-induced activation of the ER stress pathway [Citation33]. In this study, TA I markedly increased the expression of CHOP and phospho-eIF2α proteins in HepG2 and Huh7 cells. Western blot analysis also showed that pretreatment with NAC significantly decreased the expression levels of CHOP and phospho-eIF2α proteins induced by TA I in HepG2 and Huh7 cells. Additionally, TA I-mediated cell growth inhibition was restored by the downregulation of CHOP protein expression in HepG2 and Huh7 cells. These results suggest that TA I induced cell apoptosis via ROS-induced activation of an ER stress pathway in HepG2 and Huh7 cells.
Emerging evidence has shown that autophagy may serve as a potential therapeutic target for cancer therapy, and autophagy exerts a contradictory effect in different cancer cell types. Recently, it has been reported that TA IIA induces autophagic cell death in KBM-5 and SCC-9 cells [Citation34,Citation35]. In A375 cells, TA IIA inhibits the proliferation of A375 cells by activating the autophagy pathway [Citation36]. However, Lee et al. revealed that TA I induces protective autophagy in malignant pleural mesothelioma cells [Citation37]. Jing et al. also revealed that TA I induces pro-survival autophagy in gastric cancers [Citation4]. Our results showed that TA I significantly decreased the expression of autophagy-related proteins LC3B-II and beclin-1 in HepG2 and Huh7 cells. Furthermore, 3-MA and TA I significantly increased cell growth inhibition compared with TA I treatment alone in HepG2 and Huh7 cells. Additionally, Cui et al. reported that p53/DRAM-mediated autophagy contributes to radiation-induced death in MCF-7 breast cancer cells [Citation38]. Hsieh et al. reported that sedanolide promotes autophagy via the p53/DRAM signaling pathway in human liver cancer J5 cells [Citation39]. Wei et al. also demonstrated that XingNaoJing inhibits autophagy by suppressing the p53/DRAM signaling pathway in PC12 cells [Citation12]. Interestingly, our results demonstrated that TA I decreased p53 and DRAM expression in HepG2 and Huh7 cells. These data indicated that TA I induced cell apoptosis by repressing p53/DRAM-mediated autophagy in HepG2 and Huh7 cells.
In summary, our results demonstrated that TA I inhibited the proliferation of HCC HepG2 and Huh7 cells via the induction of apoptosis and G0/G1 cell cycle arrest. TA I mediated G0/G1 cell cycle arrest in HepG2 and Huh7 cells by upregulating p21 and downregulating cyclin D1. TA I induced apoptosis via the ROS-mediated ER stress pathway and by suppressing p53/DRAM-mediated autophagy in HepG2 and Huh7 cells. Therefore, TA I might be an anti-cancer drug candidate in the treatment of HCC.
Authors’ contributions
XL conducted the experiments and analyzed the data. XL and JKL made substantial contributions to the design of the present study and prepared the manuscript. XL performed the western blotting and analyzed the data. All authors read and approved the final manuscript.
Data avaialability
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
- Wu CY, Cherng JY, Yang YH, et al. Danshen improves survival of patients with advanced lung cancer and targeting the relationship between macrophages and lung cancer cells. Oncotarget. 2017;8(53):90925–90947.
- Tian XH, Wu JH. Tanshinone derivatives: a patent review (January 2006–September 2012). Exp Opin Ther Pat. 2013;23(1):19–29.
- Jing X, Xu Y, Cheng W, et al. Tanshinone I induces apoptosis and pro-survival autophagy in gastric cancers. Cancer Chemother Pharmacol. 2016;77(6):1171–1181.
- Wang L, Wu J, Lu J, et al. Regulation of the cell cycle and PI3K/Akt/mTOR signaling pathway by tanshinone I in human breast cancer cell lines. Mol Med Rep. 2015;11(2):931–939.
- Li Y, Gong Y, Li L, et al. Bioactive tanshinone I inhibits the growth of lung cancer in part via downregulation of Aurora A function. Mol Carcinog. 2013;52(7):535–543.
- Lu M, Wang C, Wang J. Tanshinone I induces human colorectal cancer cell apoptosis: the potential roles of Aurora A-p53 and survivin-mediated signaling pathways. Int J Oncol. 2016;49(2):603–610.
- Kim MK, Park GH, Eo HJ, et al. Tanshinone I induces cyclin D1 proteasomal degradation in an ERK1/2 dependent way in human colorectal cancer cells. Fitoterapia. 2015;101:162–168.
- Hsiao YT, Kuo CL, Lin JJ, et al. Curcuminoids combined with gefitinib mediated apoptosis and autophagy of human oral cancer SAS cells in vitro and reduced tumor of SAS cell xenograft mice in vivo. Environ Toxicol. 2018;33:821–832.
- Cho SW, Na W, Choi M, et al. Autophagy inhibits cell death induced by the anti-cancer drug morusin. Am J Cancer Res. 2017;7(3):518–530.
- Lorin S, Pierron G, Ryan KM, et al. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy. 2010;6(1):153–154.
- Wei G, Huang Y, Li F, et al. XingNaoJing, prescription of traditional Chinese medicine, prevents autophagy in experimental stroke by repressing p53-DRAM pathway. BMC Complement Altern Med. 2015;15:377.
- Zhou GZ, Li AF, Sun YH, et al. A novel synthetic curcumin derivative MHMM-41 induces ROS-mediated apoptosis and migration blocking of human lung cancer cells A549. Biomed Pharmacother. 2018;103:391–398.
- Lin CL, Lee CH, Chen CM, et al. Protodioscin induces apoptosis through ROS-mediated endoplasmic reticulum stress via the JNK/p38 activation pathways in human cervical cancer cells. Cell Physiol Biochem. 2018;46(1):322–334.
- Huang H, Xie H, Pan Y, et al. Plumbagin triggers ER stress-mediated apoptosis in prostate cancer cells via induction of ROS. Cell Physiol Biochem. 2018;45(1):267–280.
- Xu Y, Tong Y, Ying J, et al. Chrysin induces cell growth arrest, apoptosis, and ER stress and inhibits the activation of STAT3 through the generation of ROS in bladder cancer cells. Oncol Lett. 2018;15(6):9117–9125.
- Zhu M, Jiang Y, Wu H, et al. Gambogic acid shows anti-proliferative effects on Non-Small Cell Lung Cancer (NSCLC) cells by activating Reactive Oxygen Species (ROS)-induced Endoplasmic Reticulum (ER) stress-mediated apoptosis. Med Sci Monit. 2019;25:3983–3988.
- Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17(2):93–115.
- Visconti R, Della Monica R, Grieco D. Cell cycle checkpoint in cancer: a therapeutically targetable double-edged sword. J Exp Clin Cancer Res: CR. 2016;35(1):153.
- Liu Q, Cao Y, Zhou P, et al. Panduratin A inhibits cell proliferation by inducing G0/G1 phase cell cycle arrest and induces apoptosis in breast cancer cells. Biomol Ther. 2018;26(3):328–334.
- Xia R, Sheng X, Xu X, et al. Hesperidin induces apoptosis and G0/G1 arrest in human non-small cell lung cancer A549 cells. Int J Mol Med. 2018;41(1):464–472.
- Wang ST, Ho HJ, Lin JT, et al. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8(2):e2626.
- El-Deiry WS. p21(WAF1) mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 2016;76(18):5189–5191.
- Jiang L, Wang Y, Liu G, et al. C-Phycocyanin exerts anti-cancer effects via the MAPK signaling pathway in MDA-MB-231 cells. Cancer Cell Int. 2018;18:12.
- Wu MH, Lin CL, Chiou HL, et al. Praeruptorin A inhibits human cervical cancer cell growth and invasion by suppressing MMP-2 expression and ERK1/2 signaling. Int J Mol Sci. 2017;19(1):10.
- Su CC, Chen GW, Lin JG. Growth inhibition and apoptosis induction by tanshinone I in human colon cancer Colo 205 cells. Int J Mol Med. 2008;22(5):613–618.
- Nizamutdinova IT, Lee GW, Son KH, et al. Tanshinone I effectively induces apoptosis in estrogen receptor-positive (MCF-7) and estrogen receptor-negative (MDA-MB-231) breast cancer cells. Int J Oncol. 2008;33(3):485–491.
- Hseu YC, Tsai TJ, Korivi M, et al. Antitumor properties of Coenzyme Q0 against human ovarian carcinoma cells via induction of ROS-mediated apoptosis and cytoprotective autophagy. Sci Rep. 2017;7(1):8062.
- Wang B, Zhou TY, Nie CH, et al. Bigelovin, a sesquiterpene lactone, suppresses tumor growth through inducing apoptosis and autophagy via the inhibition of mTOR pathway regulated by ROS generation in liver cancer. Biochem Biophy Res Commun. 2018;499(2):156–163.
- Gao H, Sun W, Zhao W, et al. Total tanshinones-induced apoptosis and autophagy via reactive oxygen species in lung cancer 95D cells. Am J Chin Med. 2015;43(6):1265–1279.
- Ma K, Zhang C, Huang MY, et al. Crosstalk between Beclin-1-dependent autophagy and caspasedependent apoptosis induced by tanshinone IIA in human osteosarcoma MG-63 cells. Oncology reports. 2016;36(4):1807–1808.
- Lee WY, Liu KW, Yeung JH. Reactive oxygen species-mediated kinase activation by dihydrotanshinone in tanshinones-induced apoptosis in HepG2 cells. Cancer Lett. 2009;285(1):46–57.
- Liu Y, Kang X, Niu G, et al. Shikonin induces apoptosis and prosurvival autophagy in human melanoma A375 cells via ROS-mediated ER stress and p38 pathways. Artif Cells Nanomed Biotechnol. 2019;47(1):626–635.
- Yun SM, Jung JH, Jeong SJ, et al. Tanshinone IIA induces autophagic cell death via activation of AMPK and ERK and inhibition of mTOR and p70 S6K in KBM-5 leukemia cells. Phytother Res. 2014;28(3):458–464.
- Qiu Y, Li C, Wang Q, et al. Tanshinone IIA induces cell death via Beclin-1-dependent autophagy in oral squamous cell carcinoma SCC-9 cell line. Cancer Med. 2018;7(2):397–407.
- Li X, Li Z, Li X, et al. Mechanisms of Tanshinone II a inhibits malignant melanoma development through blocking autophagy signal transduction in A375 cell. BMC Cancer. 2017;17(1):357.
- Lee J, Sohn EJ, Yoon S, et al. Activation of JNK and IRE1 is critically involved in tanshinone I-induced p62 dependent autophagy in malignant pleural mesothelioma cells: implication of p62 UBA domain. Oncotarget. 2017;8(15):25032–25045.
- Cui L, Song Z, Liang B, et al. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol Rep. 2016;35(6):3639–3647.
- Hsieh SL, Chen CT, Wang JJ, et al. Sedanolide induces autophagy through the PI3K, p53 and NF-kappaB signaling pathways in human liver cancer cells. Int J Oncol. 2015;47(6):2240–2246.