
Abstract
Smoking is an important factor in the pathogenesis of chronic obstructive pulmonary disease (COPD), which is commonly characterised by cellular senescence and inflammation. Recently, miR-200b has emerged as an important target to cure lung disease; however, the function of miR-200b in reducing cellular senescence and inflammatory responses has not been reported. In this study, we found that miR-200b was downregulated in the lungs of COPD model mice, and its expression is correlated with cellular senescence and inflammatory responses. We hypothesised that miR-200b may be a potential novel therapy for treating COPD. We performed senescence-Associated-β-galactosidase (SA-β-GAL) staining, western blot, qRT-PCR and ELISA; our data suggested that miR-200b is an anti-aging factor in the lungs that is involved in inflammatory responses. We also confirmed that ZEB2 (Zinc finger E-box binding homeobox 2) is a target gene of miR-200b using luciferase reporter assay. In addition, we verified the function of ZEB2 in cellular senescence and inflammatory responses through transfection experiments. Moreover, we found that the protective effects of miR-200b are inhibited when cells overexpress the ZEB2 protein. In conclusion, our results suggest that miR-200b may attenuate cellular senescence and inflammatory responses by targeting ZEB2 in pulmonary emphysema.
Introduction
Recently, smoking has been regarded as the key factor in inducing chronic obstructive pulmonary disease (COPD) and may also induce chronic inflammatory responses to noxious particles or gases in the airways and lungs [Citation1]. Another study has also indicated that cellular senescence plays a critical role in such pathogenesis, and reducing the number of aging cells could be a useful method to attenuate inflammatory responses [Citation2]. In addition, previous studies have shown that cellular senescence exists in COPD, which increases TNF-α and IL-6 levels [Citation3]. Moreover, a snapshot indicates that inhibiting cellular senescence could also reduce inflammatory responses by inhibiting the NF-κB pathway [Citation4].
MicroRNAs (miRNAs) are a subset of small noncoding RNAs that control gene expression by regulating messenger RNA (mRNA) expression or by triggering translational repression or RNA degradation [Citation5]. Recent studies have reported elevated levels of large number of miRNAs in the lungs of COPD patients. However, the functions of miRNAs in the lungs are not known. In the current study, we focussed on miR-200b, which has been described in previous studies; it was reported that miR-200b may attenuate acute lung injury by inhibiting cell apoptosis and fibrosis [Citation6]. However, the interaction between miR-200b and cellular senescence has not been reported.
ZEB2 is a member of the zinc-finger E homeobox-binding protein family, and in mammals, these proteins primarily serve as transcriptional repressors via the SMAD protein [Citation7]. According to previous studies, ZEB2 can also cooperate with miR-200a in inhibiting hepatocyte apoptosis and enhancing hepatocyte proliferation [Citation8]. Here, we hypothesised that ZEB2 could be a gene involved in cellular senescence due to the high correlation between ZEB2 and cellular senescence.
The aim of this study was to illustrate the key functions of miR-200b in cellular senescence and inflammatory responses in smoking-induced lung dysfunction, and to further clarify the molecular relationship between miR-200b and ZEB2. In this study, we detected miR-200b levels by qRT-PCR in elastase-treated mouse lungs. In addition, we transfected plasmids of miR-200b into MLE cells to examine its function in cellular senescence and inflammatory responses. Furthermore, we verified the interaction between miR-200b and ZEB2 by website predating and performing luciferase reporter assays. Our study focussed on ROS-induced cellular senescence and mitochondrial dysfunction-related inflammatory responses, which may suggest a novel therapeutic method for COPD.
Materials and methods
Pulmonary emphysema mouse model preparation
All mice were randomly divided into two groups: the control group and the elastase-injected group (n = at least 6 mice in each group). The mice were modelled according to previous studies. In these protocols, the treatment process required 3 weeks. Mice were suspended at 50–60 °C by securing the upper incisor teeth to a board after they were anaesthetised with pentobarbital. A 100 μl aliquot of saline alone or saline containing 1 U of porcine pancreatic elastase (Sigma, US) was injected into the trachea using a microsyringe with a square tip. Pulmonary emphysema took three weeks to develop after elastase injection. The mice were then sacrificed, and the lungs were stored at −70 °C. All mice were purchase from the Experimental Animal Centre of the Anhui Medical University (Anhui, China). All animal experiments in the current study were performed after obtaining permission from the Ethics Committees of the Anhui Medical University, and all procedures were in accordance with the Guidelines of the Animal Care and Use Committee of the Anhui Medical University.
Haematoxylin and eosin staining
The lung sections were stained by haematoxylin and eosin (H&E) according to a previously reported protocol [Citation9]. First, sections were soaked in an ethanol concentration gradient, which the paraffin can be washed. The sections then were stained with haematoxylin for 5 to 20 min followed by polarisation with the polarisation solution for 30 s. The sections were stained with eosin for 20 s. The sections were then washed through a series of water washing procedures. Finally, the sections were covered with glass after dropping neutral gum.
Immunohistochemistry staining
Immunohistochemistry (IHC) staining was the method used to stain the tissue sections followed by imaging under a microscope. First, all the sections were incubated with primary antibodies (P16, P21, SOD2, Santa Cruz, US). The sections were then washed with PBS and incubated with secondary antibodies. Images were captured under a microscope connected to a computer (Olympus, IX3-CBH).
Bronchoalveolar lavage (BAL)
Mice were injected with pentobarbital and sacrificed by exsanguination. The lungs were subjected to lavage three times with 0.6 ml of saline as described previously. The lavage fluid was centrifuged, and the cell-free supernatant was collected for later analysis.
Enzyme-linked immunosorbent assay (ELISA)
The BAL fluid and MLE cell culture supernatants were collected by centrifugation and stored at −70 °C until further analysis. The concentration of TNF-α was quantified by an indirect enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (JYM0218Mo, JIYINMEI, China).
Senescence-associated beta-galactosidase (SA-β-GAL) activity
SA-β-GAL activity was detected by quantitatively measuring the conversion of 4-methylumbelliferyl-β-D-galactopyranoside (MUG) to the fluorescent hydrolysis product 4-methylumebelliferone (4-MU) at pH 6.0, according to a previous report. First, the lung tissues were homogenised in lysis buffer (Beyotime, China) and kept on ice for 1 h. The lysates were centrifuged for 5 min at 12,000 g, and the supernatant was mixed with 2x reaction buffer (M9659, Sigma, US), after which the sample was placed in a 37 °C water bath for 3 h. Finally, 50 μl of the reaction mix was added to 500 μl of 400 mM sodium carbonate stop solution (pH 11.0), and a 150 μl aliquot of that was added to a 96-well plate and read using a Spectrum ID 3 multifunction reader.
Cigarette smoke extract preparation
For these experiments, Kentucky research 3R4F cigarettes were chosen, and the smoke was bubbled through 10 ml of cell culture medium at a speed of 1 min per cigarette using a negative pressure pump. This aqueous smoke extract was 10% cigarette smoke extract (CSE) with an OD value of 1 at a wavelength of 320 nm after sterile filtering. The CSE was freshly prepared before each experiment to avoid side effects, such as the breakdown of substances in the extract and the evaporation of volatile components.
Cell culture
The mouse lung epithelial cell line MLE-12 was purchased from a global bioresource centre (ATCC) and was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-glutamine. The cells were cultured at standard culture conditions placed in an incubator containing humidified air with 5% CO2 at 37 °C.
Cell transfection
The MLE-12 cells were seeded into 6-well plates and transfected with mouse miR-200b or ZEB2 mimic plasmids at a concentration of 50 nM using Lipofectamine 2000 (Invitrogen) for 24 h, according to the manufacturer’s instructions. After 6 h of transfection, opti-MEM was replaced with DMEM containing 10% FBS, and 0.5% CSE.
RNA extraction and real-time PCR
The total RNA was extracted from lungs and MLE cells using TRIzol reagent (Invitrogen, US), according to the manufacturer’s instructions. Total RNA was quantified using a Nanodrop 2000 spectrophotometer (Thermo Scientific, US). The RNeasy Mini Kit (Qiagen, Germany) was used to purify the miRNAs from the total RNAs, and the miRNA abundance was assessed by quantitative real-time PCR (qRT-PCR) using One-Step miR qRT-PCR reagent kits and validated primers (Biomics, China). In addition, the fold changes in the RNA expression were analysed by the 2−ΔΔCt method, and the level of the endogenous U6 RNA served as the internal control. The CFX-96 PCR system was used to perform the PCR (Bio-Rad, US).
Western blot
The Lung and MLE-12 cells were lysed using the RIPA buffer containing 1% PMSF. The protein concentration of the extracts was quantified by using a Nanodrop 2000 spectrophotometer (Thermo Scientific, US). Equal amount of protein was loaded onto each lane of a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel, and the protein bands were then transferred onto the PVDF membranes (Millipore Corp, Billerica, MA, USA). After approximately 2 h of blocking in non-fat milk, the membranes were then incubated with primary antibodies diluted in primary antibody dilution buffer (Beyotime, China). The membranes were then washed three times with PBS and incubated with HRP-conjugated secondary antibodies (1:8000) for 1 h. Finally, the membranes were treated with an ECL chemiluminescent kit (ECL-plus, Thermo Scientific), and the membranes were then imaged with a Bio-Rad chemiluminescence capture system [Citation10].
Ethics statement
This study was carried out in accordance with the recommendations of Animal Care and Use Committee of the Anhui Medical University. All the protocols were approved by the Ethics Committee of the Anhui Medical University.
Statistical analysis
All the data were analysed using PRISM software version 7.0, and all the results are presented as the mean ± standard deviation (SD). The statistical significances were determined by one-way ANOVA or Student’s t-test.
Results
miR-200b was down-regulated in the lungs of mice with elastase-induced pulmonary disease
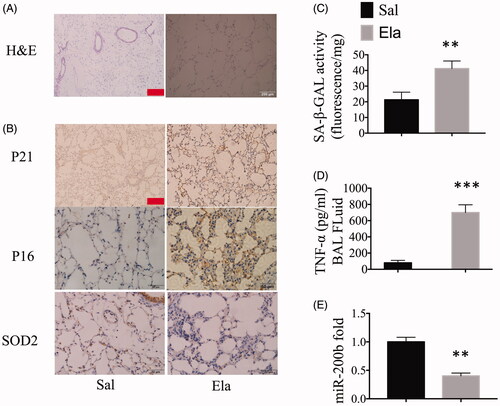
The mice were randomly divided into two groups: a control group and an elastase-treated group. A significant increase in the mean linear intercept (Lm) was observed in mice treated with elastase. In contrast, no derangement of the cell cord or dilation of the intercellular spaces was observed in the control group (). As depicted by the IHC staining, the expression levels of P16 and P21 increased significantly in elastase-treated mice as compared to the control mice. In contrast, we observed extremely low expression of SOD2 in elastase-treated mice as compared to the control mice (). In addition, the result of GOLD rule in cellular senescence (SA-β-GAL enzyme detection) showed high levels of aging in elastase-treated mice as compared to the control mice (). Moreover, the ELISA data showed that the levels of TNF-α in elastase-treated mice were higher than in the control group (). We next measured the fold change in the expression levels of miR-200b in both the groups by qRT-PCR, and found that the expression of miR-200b was lower in the elastase-treated mice as compared to that in the control mice (). Consequently, miR-200b was down-regulated in our pulmonary model, and its expression had a negative correlation with cellular senescence and inflammatory responses.
Figure 1. miR-200b was down-regulated in COPD mice and had a negative correlation with cellular senescence and inflammation. (A) Representative haematoxylin and eosin (H&E) staining of lung tissues. Scale bar = 200 μm. (B) IHC staining of lung tissues, which indicate the P21, P16 and SOD2 protein expression levels. Scale bar = 50 μm. (C) SA-β-GAL activity in mouse lungs. (D) Cytokine (TNF-α) levels in the BAL fluid. (E) qRT-PCR results of miR-200b levels in the lungs. **p < .01, ***p < .001 versus saline group.
Overexpression of miR-200b attenuates CSE-induced cellular senescence and inflammatory responses in vitro
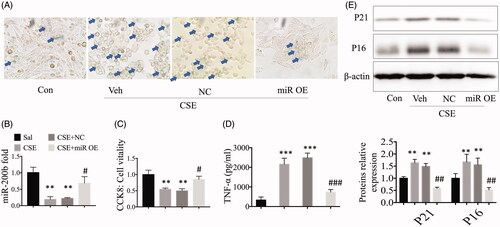
We treated MLE cells with CSE (0.5%), and then the MLE-12 cells were transfected with miR-200b mimic plasmids and control plasmids. We found that the overexpression of miR-200b attenuated the CSE-induced enhanced levels of SA-β-GAL in the cell staining experiments, as shown earlier. We also found that the cell morphology in miR-200b overexpressing cells changed to the normal fusiform type, rather than remaining as point-like cells (). The qRT-PCR results showed that the miR-200b expression was enhanced in the CSE-treated cells while miR-200b expression levels were significantly higher after transfection with the mimic plasmids (), as compared to the control cells. In addition, according to the CCK-8 kit results, we found that the overexpression of miR-200b could significantly revive cell vitality as compared to the CSE-treated cells (). To explore the function of miR-200b in inflammatory responses, we evaluated the inflammatory cytokine levels in the MLE-12 cells. The data showed that TNF-α was significantly increased after CSE treatment, while the overexpression of miR-200b deterred this this increase in the TNF-α levels (). Moreover, protein expression levels were also significantly altered. The expression levels of the P16 and P21 proteins were high in CSE-treated cells, but were significantly reduced the overexpression of miR-200b, as determined by western blot, thus indicating a potential role for miR-200b in deterring cellular senescence (). These findings indicate that the overexpression of miR-200b is beneficial for intervening in the development of COPD by reducing cellular senescence and inflammatory responses.
Figure 2. Overexpression of miR-200b reduced cellular senescence and inflammatory responses in vitro. (A) SA-β-GAL staining of cells. (B) qRT-PCR results of miR-200b levels in the cells. (C) CCK-8 results. (D) Cytokine (TNF-α) levels in the cell culture supernatants. (E) Western blot analysis of P16, P21, and ZEB2 protein expression in the cells. ** p < .01, ***p < .001 versus control group. #p < .05, ##p < .01, ###p < .001 versus CSE-treated group.
ZEB2 is the downstream target gene of miR-200b
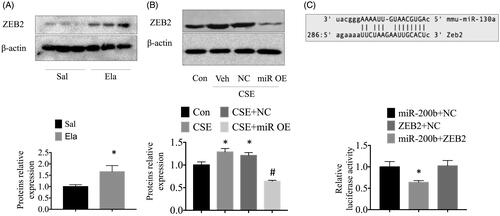
First, we measured ZEB2 protein levels in control and elastase-treated mice by western blot. We found that the expression of the ZEB2 protein was significantly higher in the elastase-treated mice than in the control mice (). In addition, we found that the overexpression of miR-200b may diminish the high ZEB2 protein expression that was induced by CSE in vitro (). Next, we considered whether there was any correlation between ZEB2 and miR-200b. To ascertain whether miR-200b binds to ZEB2 in MLE cells, we assessed the miRNA expression profile using bioinformatics software. We found that ZEB2, which is implicated in COPD, could be a target gene of miR-200b in MLE cells. Interestingly, we found that ZEB2 had putative binding sites for the 3’-UTR of miR-200b. To further test whether ZEB2 is a miR-200b target gene, we used a luciferase reporter assay to define the correlation between miR-200b and ZEB2. We found that the fold change in the expressions of ZEB2 mRNA declined by treatment with the miR-200b mimic, while no change was observed in the NC-treated group, thus, these data indicated that miR-200b may regulate ZEB2 gene expression ().
Figure 3. miR-200b directly regulated the expression of ZEB2. (A) Western bolt results showing the high expression of ZEB2 in the lungs. (B) Western blot analysis of ZEB2 protein expression in the cells. (C) Bioinformatics analyses show that the miR-200b seed sequence binds to ZEB2 mRNA. A luciferase reporter assay was performed with MLE cells to detect the relative luciferase levels. *p < .05 versus saline/control group. #p < .05 versus CSE-treated group.
Overexpression of ZEB2 increases cellular senescence and inflammatory responses
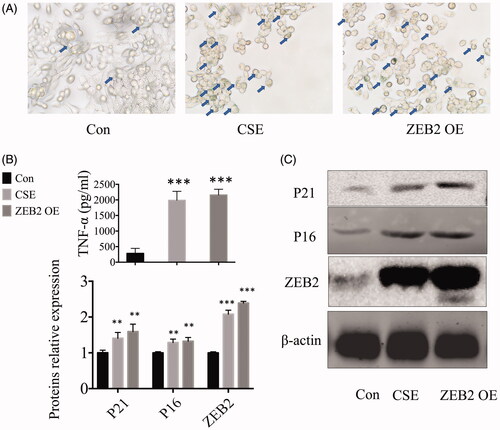
We transfected MLE cells with ZEB2 overexpression plasmids. First, the SA-β-GAL staining results indicated that the ZEB2 overexpression can increase cellular senescence. In addition, the cellular morphology was greatly influenced, with normal fusiform type cells changing into point-like cells similar to the CSE treated cells (). Next, we collected the cell culture supernatants and measured TNF-α levels by ELISA. We found that ZEB2 overexpression increased the TNF-α levels (). We then examined the protein levels by western blotting, and found that the expression levels of senescence-associated proteins, such as P16 and P21, were increased with the increase in ZEB2 levels. Moreover, these were no obvious differences between the CSE-treated cells and the ZEB2 overexpressing cells ().
Figure 4. ZEB2 caused cellular senescence and inflammatory responses. (A) SA-β-GAL staining of cells. (B) Cytokine (TNF-α) levels in cell culture supernatants. (C) Western blot analysis of P16, P21, and ZEB2 protein expression in the cells. **p < .01 ***p < .001 versus control group.
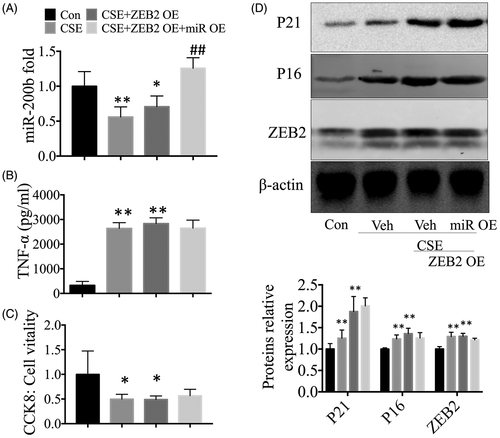
The effect of overexpressed-miR-200b on cellular senescence and inflammatory responses is inhibited in ZEB2 overexpressing cells
We co-transfected cells with miR-200b and ZEB2 plasmids. First, the qRT-PCR results showed that miR-200b expression levels increased significantly after transfection with the mimic plasmids, while the ZEB2 overexpresing plasmids had no influence on miR-200b expression levels, as compared to the control cells (). These results confirmed that ZEB2 is the downstream target of miR-200b. Interestingly, we did not see the protective effect of miR-200b knockdown in the cells with high expression of ZEB2. For instance, our ELISA results showed that the overexpression of miR-200b could not reduce the high levels of TNF-α in the culture medium in ZEB2 overexpressing cells (). We observed the same trend in our CCK-8 results, in which no protective effect of miR-200b was observed in the cells with highly expressed ZEB2, as compared to the controls (). The senescence associated proteins P16 and P21 were increased in the CSE-treated cells and ZEB2 overexpressing cells, while miR-200b mimics did not affect the levels of these proteins ().
Figure 5. The protective effects of miR-200b overexpression were inhibited by high expression of ZEB2. (A) qRT-PCR results of miR-200b levels in the lungs. (B) Cytokine (TNF-α) levels in the cell culture supernatants. (C) CCK-8 results. (D) Western blot analysis of P16, P21, and ZEB2 protein expression in the cells. *p < .05 **p < .01 versus control group. ##p < .01 versus CSE-treated group.
Discussion
Recent studies have shown that several cellular conditions, such as ROS imbalance, mitochondrial dysfunction, cellular senescence, and inflammatory responses may be involved in the development of COPD [Citation2]. These studies have highlighted the importance of clarifying the role that cellular senescence plays in COPD. Cellular senescence is a state in which cells no longer divide and grow. The senescent cells are not metabolically quiet because of the mitochondrial dysfunction. On the contrary, these cells tend to produce inflammatory cytokines and matrix metalloproteinases, such as TNF-α, IL-1β, and collagen I, which further induce and reinforce senescence and destroy the adjacent cells. In addition, recent studies have indicated that senescence and inflammatory factors can cooperate and support each other. For instance, senescent cells can secrete inflammatory factors, and the inflammatory factors can induce cellular senescence [Citation11].
Cellular senescence is an important factor in the pathogenesis of chronic lung diseases, including COPD and pulmonary fibrosis. For this reason, we first treated mice with elastase via the lung injections to develop a COPD model, and these mice fulfilled the basic criteria of COPD indicators [Citation3,Citation11]. The smoke exposure models are widely accepted because they can mimic the microenvironment and cause cells to age. However, with regards to emphysema, mice treated with smoke for 4 months did not produce any better results as compared to elastase injection [Citation1]. In addition, our H&E and IHC staining results indicate that elastase-treated mice can develop senescent cells because of the high expression of P16 and P21, combined with the low expression of SOD2. Moreover, in vitro experiments, we treated MLE cells, a normal mouse lung airway cell line, with CSE. Such in vitro methods may lack cell transdifferentiation as compared to the straightforward stimulation with TGF-β (the common cytokine in cell experiments), but they can mimic the microenvironment of mice lungs [Citation12]. We chose MLE cells because of their typical type II cellular functions that allow them to retain their cell proliferation ability, while they can still transform into type I cells if the immune system is activated.
Previous studies have indicated that miR-200 family members, including miR-200a, miR-200b, and miR-200c, are effective in reducing epithelial to mesenchymal transition (EMT) in lung disease, which may be beneficial in repair [Citation6,Citation13,Citation14]. Such functions can also be seen in hepatocyte apoptosis related studies [Citation8]. However, other mechanisms have not been described well, and only oxidative stress has been reported in studies on skeletal muscle and liver cells. In our study, a dysfunctional mitochondria could have led to oxidative stress because impaired mitochondria cannot manage the increased ROS, resulting in serious damage to the membrane.
The stressors leading to senescence include telomere shortening due to replicative exhaustion, DNA or chromatin structure and mitochondrial dysfunction [Citation15–19]. Recent reports indicated that mitochondrial dysfunction plays an important role in initiating inflammatory responses and cellular senescence and has significance in the pathogenesis of lung disease [Citation20]. ROS generation occurs via tissue damage-activated RAC1 and dysfunctional mitochondrial. Other metabolic perturbations can also induce a TP53-dependent arrest via AMPK activation, and in previous studies, AMPK activation could attenuate inflammatory responses in COPD [Citation13]. Another study indicated that cis-regulatory regions of SASP genes contain NF-κB and C/EBPß binding sites, and their increased expression promotes a positive feedback loop that reinforces the senescent state through intra-, auto-, and paracrine signalling. In response to DNA damage, a DDR-activated PARP1-NF-κB axis induces the expression of a CCL-2-dominated inflammatory SASP response that confers metastatic properties in vivo [Citation4,Citation21]. In other words, inhibiting oxidative stress also reduces cellular senescence, which can inhibit the NF-κB pathway [Citation4]. Considering these functions of miR-200 family proteins in inhibiting oxidative stress, we hypothesised that miR-200b could play a critical role in reducing cellular senescence and inflammatory responses and could be a novel therapeutic option for COPD.
It is evident that miR-200b expression is significantly higher in the elastase-treated group as compared to the control group, as determined by qRT-PCR. To identify the functions of miR-200b in CSE-induced pulmonary disease, miR-200b mimic plasmids were transfected into MLE cells. Interestingly, compared with the group, SA-β-gal activity was significantly decreased in the overexpression group. In addition, the expression of P16 and P21, which separately indicate the senescence level, was significantly inhibited in the overexpression group as compared to the control group. Moreover, we found that TNF-α, a key factor of NF-κB pathway, was greatly inhibited in the knockdown group as compared to the control group. Here, we confirmed that reducing miR-122 expression could inhibit inflammatory responses of the NF-κB pathway by reducing cellular senescence. We could only detect TNF-α in the cell culture medium, but no change in the NF-κB pathway proteins, such as p-P65 and p-IκB protein levels. We believed it was enough to indicate the microenvironment because previous studies have indicated that p-P65 or p-IκB may not be activated even if the NF-κB pathway is activated, and P65 and IκB knockout mice survive well when they suffer from inflammation [Citation22]. Consequently, to simplify the question of how reducing cellular senescence could inhibit inflammatory responses, we decided to measure the key inflammatory factor, TNF-α.
In the current study, we measured the P16 and P21 protein levels by western blot, and we obtained the same results as we have previously. Although P16 and P21 have been widely used in aging research [Citation23], we should note other functions in which they are involved. For instance, P16 is a good marker for acute injury, such as acute kidney injury, in which p16 plays a critical role not only in cellular senescence but also in inflammation, apoptosis, and the excessive accumulation of reactive oxygen species (ROS). A recent study indicated that cinobufagin could indirectly increase the expression of p21 to increase G2/M arrest [Citation24], inducing cell apoptosis, which indicates that p21 functions in apoptosis as well as in cellular senescence. Consequently, here, we chose SA-β-GAL staining to define cellular senescence, which is widely accepted in senescent research as the gold standard. In addition, we stained MLE cells immediately and obtained accurate results according to the instructions, which were defined by professional technicians.
We next considered whether there is any correlations between miR-200b and the ZEB2 protein. Here, we first used a pharmacological method to identify the relationship between miR-200b and ZEB2 and found that they have a highly negative correlation. Moreover, the treatment of COPD is a complex process that involves reducing fibrosis, inflammatory responses and cellular senescence. Similar to previous studies, we found that the ZEB1/2 proteins can deter fibrosis by attenuating EMT progression in COPD and in liver fibrosis [Citation7,Citation25,Citation26]; however, the remaining functions of ZEB2 are not involved in cellular senescence. In our previous study, we confirmed that miR-200b works closely with cellular senescence and inflammatory responses, and there was a negative correlation between miR-200b and ZEB2. Although there was no direct evidence, it was easy for us to hypothesise that the ZEB2 protein may function in cellular senescence and inflammatory responses.
In summary, we found that miR-200b was negatively correlated with the ZEB2 protein. High miR-200b expression was negatively associated with the pathological stage, in which cellular senescence was induced. In addition, aging cells could transform into secretory cells and cause inflammatory responses. We further demonstrated that ZEB2 is also highly correlated with cellular senescence, which may induce inflammatory responses. Consequently, elucidating the regulatory network that is involved with the miR-200b/ZEB2 axis may provide a better understanding of smoking-induced cellular senescence and inflammatory responses in chronic obstructive pulmonary disease. In conclusion, we demonstrated that miR-200b may attenuate cellular senescence and inflammatory responses by targeting ZEB2 in pulmonary emphysema, which possibly suggest a novel therapeutic target for COPD.
Disclosure statement
The authors declare that there is no conflict of interest with the contents of the manuscript.
Additional information
Funding
References
- Yao H, Chung S, Hwang JW, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122(6):2032–2045.
- Ahmad T, Sundar IK, Tormos AM, et al. Shelterin telomere protection protein 1 reduction causes telomere attrition and cellular senescence via sirtuin 1 deacetylase in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2017;56(1):38–49.
- Cheng XY, Li YY, Huang C, et al. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget. 2017;8(14):22513–22523.
- Martinez-Zamudio RI, Robinson L, Roux PF, et al. SnapShot: cellular senescence pathways. Cell. 2017;170(4):816–816.
- Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–222.
- Magenta A, Cencioni C, Fasanaro P, et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18(10):1628–1639.
- Scott CL, Omilusik KD. ZEBs: novel players in immune cell development and function. Trends Immunol. 2019;40(5):431–446.
- Zhao YX, Sun YY, Huang AL, et al. MicroRNA-200a induces apoptosis by targeting ZEB2 in alcoholic liver disease. Cell Cycle. 2018;17(2):250–262.
- Lu X, Liu Y, Xuan W, et al. Circ_1639 induces cells inflammation responses by sponging miR-122 and regulating TNFRSF13C expression in alcoholic liver disease. Toxicol Lett. 2019;314:89–97.
- Bu FT, Chen Y, Yu HX, et al. SENP2 alleviates CCl4-induced liver fibrosis by promoting activated hepatic stellate cell apoptosis and reversion. Toxicol Lett. 2018;289:86–98.
- Al-Salama ZT, Frampton JE. Glycopyrronium/formoterol: a review in COPD. Drugs. 2019;79(13):1455–1466.
- Wang X, Song S, Li H, et al. Protective effect of quercetin in LPS-induced murine acute lung injury mediated by cAMP-Epac pathway. Inflammation. 2018;41(3):1093–1103.
- Yongtao Xiao WY, Lu L, Wang Y, et al. Wei Cai: p38 p53 miR 200a 3p feedback loop promotes oxidative stress-mediated liver cell death. Cell Cycle. 2015;10:10.
- Snyder-Talkington BN, Dong C, Sargent LM, et al. mRNAs and miRNAs in whole blood associated with lung hyperplasia, fibrosis, and bronchiolo-alveolar adenoma and adenocarcinoma after multi-walled carbon nanotube inhalation exposure in mice. J Appl Toxicol. 2016;36(1):161–174.
- He Y, Feng D, Li M, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology. 2017;66(1):220–234.
- Hara H, Kuwano K, Araya J. Mitochondrial quality control in COPD and IPF. Cells. 2018;7(8):86.
- Ng Kee Kwong F, Nicholson AG, Harrison CL, et al. Is mitochondrial dysfunction a driving mechanism linking COPD to nonsmall cell lung carcinoma? Eur Respir Rev. 2017;26(146):170040.
- Yue L, Yao H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: pathogenesis and pharmacological targets for chronic lung diseases. Br J Pharmacol. 2016;173(15):2305–2318.
- Kwangwon Lee AH, Osme A, Kim C, et al. Hepatic mitochondrial defects in a mouse model of NAFLD are associated with increased degradation of oxidative phosphorylation subunits. Mol Cell Proteomics. 2018;17(12):2371–2386.
- Li Y, Xue Y, Xu X, et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019;38(3):e98786.
- Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene. 2016;35(39):5079–5092.
- Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734.
- Wan Y, McDaniel K, Wu N, et al. Regulation of cellular senescence by miR-34a in alcoholic liver injury. Am J Pathol. 2017;187(12):2788–2798.
- Deng X, Sheng J, Liu H, Wang N, et al. Cinobufagin promotes cell cycle arrest and apoptosis to block human esophageal squamous cell carcinoma cells growth via the p73 signalling pathway. Biol Pharm Bull. 2019; 42:10.
- Bagadia P, Huang X, Liu TT, et al. An Nfil3-Zeb2-Id2 pathway imposes Irf8 enhancer switching during cDC1 development. Nat Immunol. 2019;20(9):1174–1185.
- Izumchenko E, Chang X, Michailidi C, et al. The TGF -miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res. 2014;74(14):3995–4005.