
Abstract
Lung diseases are common health problems in many countries. The dysfunction of bronchial epithelial cells is important for the development of lung diseases. Recent progress reveals that inflammasome is the fundamental mechanism of epithelial activation. Here, we report the protective effect of doxofylline, a theophylline derivative agent, on lipopolysaccharides (LPS)-induced inflammatory response in bronchial epithelial cells. The presence of doxofylline reduces LPS-induced production of NO and PGE2. Doxofylline also inhibits LPS-induced production of mitochondrial ROS. Mechanistically, we show that doxofylline suppresses the expression of NOX4 induced by LPS. Doxofylline inhibits LPS-induced NLRP3-TXNIP inflammasome activation as revealed by its inhibitive effect on NLRP3, caspase 1 (P10 unit), and TXNIP induction as well as weakened induction of IL-1β and IL-18. Furthermore, we show that doxofylline ameliorates LPS-induced Sirtuin 1 (SIRT1) reduction. The silencing of SIRT1 abolishes the inhibitory effect of doxofylline on NLRP3 inflammasome activation. Collectively, our study demonstrates that doxofylline mitigates epithelial inflammation via amelioration of multiple cellular pathways, including NLRP3-TXNIP inflammasome activation.
Introduction
Lung diseases are one type of common health problem worldwide. The most prevalent lung diseases include chronic obstructive pulmonary disease (COPD), asthma, and lung cancers. It has been reported that these common lung diseases accounted for 9.5 million deaths worldwide in 2008 [Citation1]. The respiratory system functions as the primary site for the gas exchange, phonation, and pulmonary Defence. Lung tissue forms as an upside-down pulmonary tree which terminates in alveoli. These highly branched pulmonary units are covered by the epithelium at their surface. In the lung, the bronchial epithelium acts as a defensive barrier to protect its normal function. Bronchial epithelial cells serve as the interface for the exchange between inhaled gas and tissue. These epithelial cells respond directly to pathogens and environmental exposure. Once activated by these external stimuli, bronchial epithelial cells release cytokines and chemokines to recruit other immune cells as the primary defensive mechanism of innate immunity [Citation2]. Activated epithelial cells also produce reactive oxygen species (ROS) which can cause pulmonary inflammation and lung tissue injury. The long-time burden of tissue injury and remodelling can lead to blockage of the small bronchial tract. The activation of bronchial epithelial cells has been linked to many aspects of pulmonary diseases [Citation3].
Lipopolysaccharide (LPS) is the major membrane component of gram-negative bacteria. LPS exhibits strong immune induction when exposed to different immune cells. The structure and modification of LPS from gram-negative bacteria can be extremely virulent to respiratory tract cells. Therefore, LPS is also referred to as an “endotoxin” [Citation4]. LPS-induced inflammatory response in epithelial cells has been well characterised, but ameliorative measures to prevent its activation continue to pose major challenges. Recent progress demonstrates that the inflammasome, an inflammation-associated multiprotein complex, functions as a piece of critical regulatory machinery involved in epithelium-mediated innate immunity [Citation5]. So far, several inflammasomes have been reported, including NLRP1, NLRP2, NLRP3, IPAF and AIM2. The most studied inflammasome NLRP3 features the generation of the NLPR3 protein. The activation of the NLPR3 inflammasome involves the conversion of procaspase-1 to caspase-1, and the secretion of mature IL-1β and IL-18 [Citation6]. In bronchial epithelial cells, LPS has been shown to induce strong NLPR3 inflammasome activation and IL-1β secretion [Citation7]. Recent studies show that the NLPR3 inflammasome is regulated by several upstream signals, including negative regulation of Sirtuin 1 (SIRT1). SIRT1 is an anti-aging factor, and its high expression reduces the activation of the NLPR3 inflammasome [Citation8,Citation9].
Xanthine derivatives such as theophylline have long been used to treat lung diseases, but many of them have demonstrated limited effect as well as various side effects. Doxofylline is the most recently modified theophylline-based compound and has been approved to treat lung diseases such as COPD and asthma [Citation10]. The chemical name of doxofylline is 7-(1,3 dioxolar-2-ylmethyl)-theophylline. Compared with theophylline, doxofylline has a dioxalane group at position 7. Doxofylline has improved efficacy as compared to theophylline but shows fewer side effects and is viewed as a replacement for theophylline [Citation11]. Doxofylline possesses potent anti-inflammatory and bronchodilatory activities, and it acts as a non-selective phosphodiesterase (PDE) inhibitor. However, whether doxofylline has a protective effect against LPS exposure in bronchial epithelial cells and the underlying mechanisms are still unknown. In this study, we explored its protective mechanism against LPS-induced inflammation in cultured bronchial epithelial cells. Our findings reveal that doxofylline mitigated LPS-induced bronchial epithelial cells inflammation by suppressing the activation of the NLRP3 inflammasome.
Materials and methods
Cell culture and treatment
We purchased human bronchial epithelial cells line 16HBEC from ATCC, USA, and the cells were cultured in DMEM media supplemented with 10% FBS (Gibco, Grand Island, NY) and 1% P/S. All cell cultures were maintained with 5% CO2 and 95% air in a humidified incubator. LPS in this study was from R&D Systems (Minneapolis, MN) and dissolved in PBS. Doxofylline was from Sigma-Aldrich (Saint Louis, MO) and dissolved in PBS. To examine the effects of doxofylline, we co-treated the cells with 5 or 10 μM doxofylline and LPS for 48 h.
Real-time PCR
We extracted RNAs from 16HBEC cells using Qiazol reagent (Qiagen, Germany). Then, 1-2 μg RNA was reverse-transcribed into cDNA using the first-strand cDNA synthesis system (Life Technologies, Waltham, MA). Quantitative real-time PCR was performed using the SYBR Green method. The relative expressions of these genes were assessed via normalization to GAPDH using the 2−ΔΔCT method.
The following primers were used in this study: GAPDH (F, 5′-GGAGCGAGATCCCTCCAAAAT-3′; R, 5′-GGCTGTTGTCATACTTCTCATGG-3′); IL-1β (F, 5′-TTCCTGTTGTCTACACCAATGC-3′;R, 5′-CGGGCTTTAAGTGAGTAGGAGA-3′); TXNIP (F, 5′-ACTCGTGTCAAAGCCGTTAGG-3′; R, 5′-TCCCTGCATCCAAAG CACTT-3′); NOX1 (F, 5′-GCCTGTGCCCGAGCGTCTGC-3′; R, 5′- ACCAATGCCGTGAATCCCTAAGC-3′); NOX2 (F, 5′-GGAGTTTCAAGATGCGTGGAAACTA-3′; F, 5′-GCCAGACTCAGAGTTGGAGATGCT-3′); NOX4 (F, 5′-CTTTTGGAAGTCCATTTGAG-3′; R, 5′-CGGGAGGGTGGGTATCTAA-3′); SIRT1 (F, 5′-CATAGACACGCTGGAACAGG-3′; R, 5′-GCAGATGAGGCAAAGGTT-3′).
Immunoblot
Protein was extracted from 16HBEC cells using RIPA buffer supplemented with protease inhibitor cocktail. After brief sonication and spinning at 15,000 rpm (4 °C, 15 min), the protein concentration of the whole lysate was measured using a BCA assay kit. Protein lysates were then heated to 95 °C for 5 min with 2-mercaptoethanol. The denatured samples were loaded onto 10% PAGE gel and electrophoresed for 2 h to reach optimal separation. The gel was then wet-transferred onto a 0.2 µm PVDF membrane at 100 V in a cold room for 2 h. The blots were blocked with 5% non-fat milk in TBST at room temperature for 1 h, followed by the reaction with primary antibodies overnight at 4 °C and HRP-conjugated secondary antibodies at room temperature for 1 h. Finally, the blots were digitally processed using an iBright Western Blot imaging system (ThermoFisher, Waltham Mass, MA), and the captured images were quantitated by Image J software (NIH). The following antibodies were used in this study: SIRT1 (1:1000, sc-19857; Santa Cruz Biotechnology, Inc., Santa Cruz, CA); TXNIP (1:2000, #14715, Cell Signalling Technology, Danvers, MA); NOX1 (1:1000, #ab55831, Abcam, Cambridge, MA); NOX2 (1:2000, #ab80508, Abcam); NOX4 (1:2000, # ab154244, Abcam); NLRP3 (1:1000, #13158, Cell Signalling Technology); ASC (1:1000, # ab155970, Abcam); cleaved caspase-1 p10 (1:500, # ab2553, Abcam); β-actin (1:10000, # ab8226, Abcam).
ELISA
To measure secreted PGE2, IL-1β, and IL-18 levels, the supernatants from 16HBEC cells were collected by centrifugation at 1000 rpm for 10 min. We purchased commercial ELISA kits for PGE2, IL-1β and IL-18 from R&D Systems and performed the procedures by following the manufacturer’s instructions. The final reaction values were extrapolated from the standard curve using a variable slope. The data are presented as fold change with the non-treated group as a baseline.
SIRT1 knockdown
To silent SIRT1 expression in 16HBEC cells, we purchased siRNA target oligos of human SIRT1 and scrambled siRNA from Santa Cruz Biotechnology Inc., Santa Cruz, CA. For siRNA transfection, 50% confluent 16HBEC cells were transfected by 50 µM oligos using Lipofectamine RNAiMAX reagent (Invitrogen, Waltham, MA). The efficiency of knockdown was evaluated via immunoblot experiment 2 days after transfection.
Mitochondrial ROS
We used MitoSOX™ Red reagent (Thermo Fisher Scientific, Waltham, MA) to target mitochondria in live cells. In brief, 5 µM MitoSOX Red was added to the treated 16HEC cells and allowed to permeate and react for 10 min at 37 °C. Fluorescent signals were visualized with a fluorescent microscope. The fluorescence was excited at 510 nm with emission detection at 580 nm.
Intracellular nitric oxide (NO) measurement
The intracellular NO production was assessed by staining the living 16HBEC cells with the cell-permeable fluorescent probe DAF-FM DA (Thermo Fisher Scientific). Briefly, 10,000 cells were seeded onto a 96-well plate and allowed to reach full confluence, and then 5 µM probe was added to the cells and incubated for 10 min. Fluorescent signals were visualized with a fluorescence microscope. The fluorescence was excited at 495 nm with emission detection at 515 nm.
Statistical analysis
All experimental data in this study are presented as means ± SEM For experiments with more than two groups, ANOVA was performed with the Bonferroni multiple-comparisons method. p < .05 was considered statistically significant. All plots in this study were generated using GraphPad Prism 9 (GraphPad Software).
Results
Doxofylline inhibits LPS-induced generation of nitro oxide (NO) and prostaglandin E2 (PGE2)
To test the pharmacological effect of doxofylline, we assessed the LPS–induced expression of inflammatory mediators with or without doxofylline. We measured the released levels of NO and PGE2 in cultured human bronchial epithelial cell line 16HBE when LPS and doxofylline were added. Our experiment showed that 48 h of LPS treatment induced more than 3-fold NO production when compared to non-treated controls. However, the addition of 5 and 10 μM doxofylline weakened NO induction with a dose-dependent effect (). Meanwhile, the same dose and duration of LPS treatment induced roughly 8-fold induction on PGE2, but the presence of the two doses doxofylline showed robust dose-dependent inhibition of PGE induction ().
Figure 1. Doxofylline reduces lipopolysaccharides (LPS)-induced NO and PGE2 in human 16HBE cells. Cells were stimulated with 1 μg/ml LPS with or without doxofylline (5 and 10 μM) for 48 h. (A) Production of NO as measured by DAF-FM DA staining; (B) production of prostaglandin E2 (PGE2) (****p < .0001 vs. vehicle control; ###p < .001; ####p < .0001 vs. LPS treatment group).
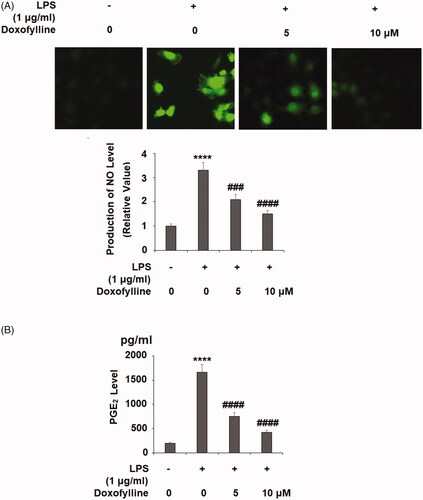
Doxofylline inhibits LPS-induced production of mitochondrial ROS
Epithelial mitochondrial dysfunction has been linked to lung diseases. LPS-mediated epithelial mitochondrial impairment generates high production of ROS. We measured mitochondrial ROS status upon the addition of doxofylline was added to bronchial 16HBE cells. When compared to non-treated cells, doxofylline induced roughly 4-fold ROS production, but the addition of two doses of doxofylline exerted dose-dependent inhibition on LPS-induced mitochondrial ROS production ().
Figure 2. Doxofylline mitigates lipopolysaccharides (LPS)-induced production of mitochondrial ROS in human 16HBE cells. Cells were stimulated with 1 μg/ml LPS with or without doxofylline (5 and 10 μM) for 48 h. Mitochondrial ROS was examined by MitoSOX Red staining (****p < .0001 vs. vehicle control; ##p < .01; ###p < .001 vs. LPS treatment group).
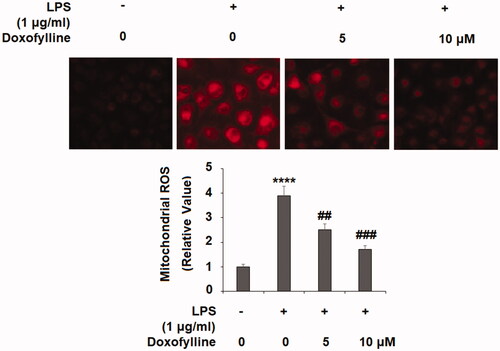
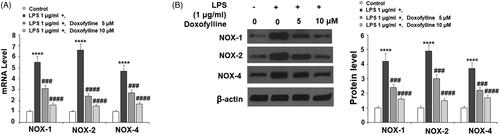
Doxofylline suppresses LPS-induced NADPH oxidase subunits expression
Next, we examined the producer of cellular ROS NADPH oxidase. NOX1, NOX2 and NOX4 are critical catalytic units of NADPH oxidase; we assessed their mRNA and protein expression in our experiment. When compared to non-treated cells at the mRNA level, LPS induced a significant increase of NOX1, NOX2 and NOX4, but the addition of the two doses of doxofylline exhibited dose-dependent suppression of the induction of these molecules ().
Figure 3. Doxofylline suppresses lipopolysaccharides (LPS)-induced expressions of the NADPH oxidases NOX1, NOX2 and NOX4 in human 16HBE cells. Cells were treated with 1 μg/ml LPS in the presence or absence of Doxofylline (5 and 10 μM) for 48 h. (A) mRNA of NOX1, NOX2 and NOX4; (B) protein of NOX1, NOX2 and NOX4 (****p < .0001 vs. vehicle control; ###p < .001; ####p < .0001 vs. LPS treatment group).
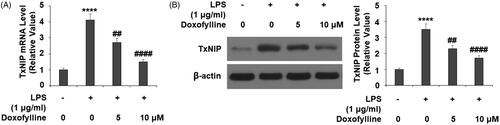
Doxofylline suppresses LPS-induced TXNIP expression
Next, we tested the effects of doxofylline on the inflammation and oxidative stress mediator thioredoxin-interacting protein (TXNIP) and found that doxofylline suppressed TXNIP expression. When comparing its mRNA expression to non-treated 16HBE cells, LPS caused more than 4-fold induction of TXNIP, but the addition of doxofylline exhibited dose-dependent suppression of this increase (). We confirmed the suppressive effect of doxofylline on TXNIP at the protein level ().
Figure 4. Doxofylline reduces lipopolysaccharides (LPS)-induced expressions of TXNIP in human 16HBE cells. Cells were stimulated with 1 μg/ml LPS with or without doxofylline (5 and 10 μM) for 48 h. (A) mRNA of TXNIP; (B) protein of TXNIP (****p < .0001 vs. vehicle control; ##p < .01; ####p < .0001 vs. LPS treatment group).
Doxofylline inhibits LPS-induced NLRP3 inflammasome activation
The pathogen-activated inflammasome is an important mechanism of lung epithelial activation. We assessed the widely studied NLRP3 inflammasome complex by examining the activation of the NLPR3 receptor and cleaved caspase 1 (P10). When compared to non-treated 16HBE cells, LPS induced a several-fold increase in NLPR3 and P10, but the presence of doxofylline showed a dose-dependent suppression of this increase, indicating that doxofylline suppressed epithelial NLPR3 inflammasome activation ().
Figure 5. Doxofylline inhibits lipopolysaccharides (LPS)-induced NLRP3 inflammasome activation in human 16HBE cells. Cells were stimulated with 1 μg/ml LPS with or without doxofylline (5 and 10 μM) for 48 h. Protein of NLRP3 and cleaved caspase 1 (P10) were measured (****p < .0001 vs. vehicle control; ###p < .001; ####p < .0001 vs. LPS treatment group).
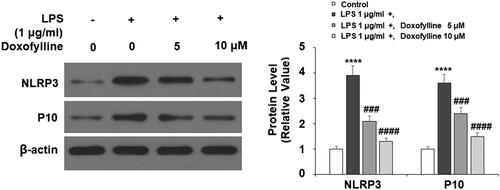
Doxofylline inhibits LPS-induced secretion of IL-1β and IL-18
The activation of NLRP3 inflammasome features the release of pro-inflammatory cytokines IL-1β and IL-18. Next, we measured these two cytokines using ELISA. When compared to non-treated 16HBE cells, LPS treatment induced more than 25-fold release of IL-1β, but doxofylline exhibited strong dose-dependent inhibition on its production (). In the same way, LPS induced about 30-fold IL-18 release, but doxofylline showed similar dose-dependent inhibition on its release ().
Figure 6. Doxofylline reduces lipopolysaccharides (LPS)-induced secretion of IL-1β and IL-18 in human 16HBE cells. Cells were treated with 1 μg/ml LPS in the presence or absence of doxofylline (5 and 10 μM) for 48 h. (A) Secretion of IL-1β; (B) secretion of IL-18 (****p < .0001 vs. vehicle control; ####p < .0001 vs. LPS treatment group).
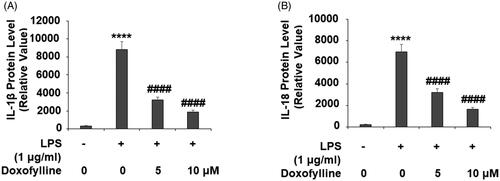
Doxofylline mitigates LPS-mediated SIRT1 reduction
The anti-aging factor SIRT1 has been shown to be an important regulator of NLRP3 inflammasome activation. We investigated the effect of doxofylline on SIRT1 expression. When comparing SIRT1 mRNA expression to non-treated 16HBE cells, LPS reduced SIRT1 transcription by about 60%, but the addition of doxofylline dose-dependently mitigated this reduction (). Meanwhile, the addition of doxofylline showed consistent mitigation of LPS-mediated reduced SIRT1 protein expression ().
Figure 7. Doxofylline reduces lipopolysaccharides (LPS)-induced reduction of SIRT1 in human 16HBE cells. Cells were treated with 1 μg/ml LPS in the presence or absence of doxofylline (5 and 10 μM) for 48 h. (A) mRNA of SIRT1; (B) protein of SIRT1 (***p < .0001 vs. vehicle control; ##p < .01; ####p < .0001 vs. LPS treatment group).
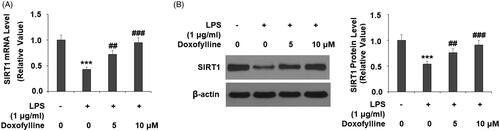
Silencing of SIRT1 abolishes doxofylline-mediated NLRP3 inflammasome activation
Finally, we examined the association of SIRT1 expression with the action of doxofylline. We silenced SIRT1 and assessed the effects of its loss on LPS-induced activation of the NLRP3 inflammasome. We were able to knockdown SIRT1 by roughly 60% as determined by its protein expression (). As shown before, 10 μM doxofylline dramatically inhibited LPS-induced NLRP3 expression in SIRT1-expressing cells but failed to exert the same inhibitory effect in SIRT1-silent cells (). At the same time, ELISA experiments revealed that doxofylline had a suppressive effect on IL-18 induction in SIRT1-expressing cells, but lost this effect in SIRT1-silent cells (). Due to the essential function of TXNIP in the activation of the NLRP3 inflammasome, we then investigated whether the expression of TXNIP is regulated by SIRT1. As expected, the inhibitory effects of doxofylline against LPS-induced TXNIP expression were abolished by knockdown of SIRT1 (). Consistently, the silencing of SIRT1 blocked the inhibitory effects of doxofylline on mitochondrial ROS production (). These findings suggest that the effects of doxofylline on NLRP3 inflammasome and oxidative stress in 16HBEC cells are dependent on SIRT1.
Figure 8. Silencing of SIRT1 abolished the inhibitory effects of doxofylline on NLRP3 inflammasome activation in human 16HBE cells. Cells were transfected with siSIRT1, followed by treatment with 1 μg/ml LPS in the presence or absence of doxofylline (10 μM) for 48 h. (A) Western blot revealed successful knockdown of SIRT1; (B) NLRP3 expression; (C) secretion of IL-18; (D) TXNIP expression; (E) mitochondrial ROS (***p < .001 vs. previous group).
Discussion
Clinical studies have concluded that doxofylline has a better clinical outcome than the original drug theophylline. Compared to theophylline, administration of doxofylline does not increase the frequency of cardiac events, and patients show improved tolerability and a favourable risk-to-benefit ratio. Thus, doxofylline has better efficacy and fewer side effects, making it a promising bronchodilator [Citation11]. Preclinical research has shown that doxofylline inhibits LPS-induced lung inflammation in mice [Citation12]. In a rat model, doxofylline administration significantly improved cigarette smoke-induced COPD [Citation13]. Several studies have shown that doxofylline administration has a significant anti-inflammatory effect in different lung diseases including bronchitis [Citation14–16]. This evidence indicates that doxofylline is a favourable anti-inflammatory agent for the treatment of lung diseases.
In our study, we explored the molecular mechanism of anti-inflammation by doxofylline. We tested the cellular effect of doxofylline in a well-established in vitro epithelial cell model using the 16HBEC cell line. By treating 16HBEC with LPS, we mimicked gram-negative bacterial infection to provoke epithelial inflammation. As expected, doxofylline showed potent protection against LPS-induced epithelial inflammation, as reflected by reduced PGE2, NO release, and decreased mitochondrial ROS generation. In epithelial cells, the release of PGE2 and NO suppresses bronchial contractions and causes hyper-responsiveness of the airway [Citation17]. Epithelial NOX1, NOX2 and NOX4 are vital regulators of ROS and contribute to ROS-induced cell death and pulmonary fibrosis [Citation18]. The inhibitive effect of doxofylline on these responsive mediators demonstrates its broad anti-inflammatory effects. Oxidative stress-induced epithelial activation is the key event in the pathogenesis of lung diseases [Citation19]. Based on the influence of doxofylline on mitochondrial ROS and NOXs, we conclude that doxofylline mitigates both mitochondrial and NADPH oxidase-derived ROS. Furthermore, our study reveals that doxofylline is a robust modulator of the epithelial inflammasome as revealed by the following experiments. Firstly, doxofylline showed strong inhibition on NLRP3 and caspase 1 (P10 subunit) activation. Secondly, doxofylline suppressed IL-1β and IL-18, two final products of inflammasome activation. Activation of the NLRP3-caspase 1 complex promotes the cleavage of pro-IL-1β and pro-IL-18 to release these cytokines. Thus, doxofylline-mediated NLRP3 inflammasome suppression results in the low yield of IL-1 and IL-18 observed in our experiment. Thirdly, we show that doxofylline suppressed the expression of TXNIP, a binding protein involved in NLPR3 inflammasome activation. It has been shown that LPS primes the NLRP3 inflammasome by binding to TXNIP [Citation20]. The suppressive role of doxofylline on TXNIP indicates that this agent suppressed epithelial inflammation through an NLPR3-TXNIP-ROS pathway. Finally, we showed that doxofylline protected against LPS-induced reduced SIRT1 but the silencing of SIRT1 abolished the protective role of doxofylline on NLPR3 inflammasome activation. This evidence indicates that SIRT1 is an upstream negative regulator of NLPR3 inflammasome activation in epithelial cells. SIRT1 is an important anti-aging factor and has been shown to regulate NLRP3 in endothelial cells [Citation21,Citation22], and it is conceivable that epithelial SIRT1 has a similar regulatory effect on the NLRP3 inflammasome. Thus, our data demonstrate that SIRT1 could be a critical molecular target of doxofylline.
In summary, we provide ample evidence that doxofylline is a potent anti-inflammatory and anti-ROS agent in bronchial epithelial cells. Doxofylline exhibits a regulatory effect on multiple molecular pathways, including its mitigation on cellular inflammatory mediators including PGE2 and NO as well as its reduction of cellular ROS. Mechanistically, doxofylline suppresses the endotoxin-activated NLPR3 inflammasome via the protection of SIRT1. It should be noted that bacterial infection and LPS exposure-induced lung injury are complex pathological processes. A single treatment of bronchial epithelial cells with LPS may not completely imitate the actual pathological mechanisms. Animal models are an important tool in pulmonary disease studies. Interestingly, our results demonstrate that the administration of doxofylline could alleviate LPS-induced acute lung injury in rodent models (Data not showed). Further clinical trials will help to clarify the underlying mechanism. Our study concludes that doxofylline is a robust inhibitor of inflammation in bronchial epithelial cells.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- The burden of lung disease [internet]. Sheffield (UK): ers white book; https://www.erswhitebook.org/chapters/the-burden-of-lung-disease/
- Kato A, Schleimer RP. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol. 2007;19(6):711–720.
- Gao W, Li L, Wang Y, et al. Bronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology. 2015;20(5):722–729.
- Munford RS. Sensing gram-negative bacterial lipopolysaccharides: a human disease determinant? Infect Immun. 2008;76(2):454–465.
- Rongvaux A. Innate immunity and tolerance toward mitochondria. Mitochondrion. 2018;41:14–20.
- Shao BZ, Xu ZQ, Han BZ, et al. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262.
- Tran HB, Lewis MD, Tan LW, et al. Immunolocalization of NLRP3 inflammasome in normal murine airway epithelium and changes following induction of ovalbumin-induced airway inflammation. J Allergy. 2012;2012:819176.
- Li Y, Wang P, Yang X, et al. SIRT1 inhibits inflammatory response partly through regulation of NLRP3 inflammasome in vascular endothelial cells. Mol Immunol. 2016;77:148–156.
- Hardeland R. Aging, melatonin, and the pro- and anti-inflammatory networks. Int J Mol Sci. 2019;20(5):1223.
- Page CP. Doxofylline: a “novofylline”. Pulm Pharmacol Ther. 2010;23(4):231–234.
- Matera M, Page C, Cazzola M. Doxofylline is not just another theophylline! Int J Chronic Obstr Pulmonary Dis. 2017;12:3487–3493.
- Riffo-Vasquez Y, Man F, Page CP. Doxofylline, a novofylline inhibits lung inflammation induced by lipopolysacharide in the mouse. Pulm Pharmacol Ther. 2014;27(2):170–178.
- Cogo R, Castronuovo A. Effects of oral doxofylline on inflammatory changes and altered cell proliferation in chronic obstructive bronchitis. Eur Rev Med Pharmacol Sci. 2000;4(1–2):15–20.
- Xu GH, Shen J, Sun P, et al. Anti-inflammatory effects of potato extract on a rat model of cigarette smoke-induced chronic obstructive pulmonary disease. Food Nutr Res. 2015;59(1):28879.
- Riffo-Vasquez Y, Venkatasamy R, Page CP. Steroid sparing effects of doxofylline. Pulm Pharmacol Ther. 2018;48:1–4.
- Cazzola M, Calzetta L, Rogliani P, et al. Impact of doxofylline in COPD: a pairwise meta-analysis. Pulm Pharmacol Ther. 2018;51:1–9.
- Folkerts G, Nijkamp FP. Airway epithelium: more than just a barrier! Trends Pharmacol Sci. 1998;19(8):334–341.
- Carnesecchi S, Deffert C, Donati Y, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15(3):607–619.
- Kleniewska P, Pawliczak R. The participation of oxidative stress in the pathogenesis of bronchial asthma. Biomed Pharmacother. 2017;94:100–108.
- Feng H, Gu J, Gou F, et al. High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J Diabetes Res. 2016;2016:1–11.
- Cordero MD, Williams MR, Ryffel B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol Metab. 2018;29(1):8–17.
- Li Y, Yang X, He Y, et al. Negative regulation of NLRP3 inflammasome by SIRT1 in vascular endothelial cells. Immunobiology. 2017;222(3):552–561.