
Abstract
Taccalonolide A has been reported to have anti-tumour efficiency. However, the underlying mechanism for taccalonolides A therapy of hepatocellular carcinoma (HCC) is still obscure. Cell viability was evaluated by cell counting kit-8 (CCK-8) assay. Apoptosis was determined by flow cytometry. Protein expression of B cell lymphoma (Bcl-2), Bcl-2 associated X (Bax), sonic hedgehog (Shh), Smoothened (Smo) and Gli family zinc finger 1 (Gli1) was analyzed by western blot. The expression of Shh, Smo and Gli1 mRNA was determined using quantitative real-time polymerase chain reaction (qRT-PCR). Results showed that taccalonolide A inhibited cell proliferation, induced apoptosis and cell cycle arrest at the G0/G1 phase, and improved the cytotoxicity of sorafenib in HCC cells. The expressions of Shh, Smo, Gli1 mRNA and protein were decreased after taccalonolide A treatment. More importantly, activation of the Shh pathway attenuated taccalonolide A-induced inhibition on cell viability and promotion on apoptosis and cell cycle arrest in HCC. Also, activation of the Shh pathway neutralized the effect of taccalonolide A on sorafenib cytotoxicity in HCC. We clarified that taccalonolide A suppressed cell viability facilitated apoptosis, and improved the cytotoxicity of sorafenib in HCC by inhibition of the activation of the Shh pathway, providing alternative treatments for HCC.
Introduction
Hepatocellular carcinoma (HCC) is a type of refractory primary liver cancer with high incidence and mortality [Citation1]. It is characterized by heterogeneity since there are a variety of aetiological factors, such as alcohol consumption, liver cirrhosis, diabetes-induced liver injury and hepatitis C virus infection [Citation2,Citation3]. Despite multiple advanced strategies that have improved the therapeutic outcomes, the 5-year survival rate is still undesirable [Citation4–6]. Therefore, it is imperative to develop novel anti-tumour drugs for HCC.
Taccalonolides are microtubule stabilizers with considerable biological activities isolated from the tubers of Tacca leontopetaloides [Citation7]. Taccalonolide A and E are unique structurally since they lack the acetoxy group at C11, thus leading to interphase microtubules bundling and mitotic arrest and stimulate apoptosis of cancer cells [Citation8–10]. Previous studies clarified that taccalonolide A could overcome drug resistance because of the inhibition of drug efflux transporter P-glycoprotein (P-gp) [Citation11,Citation12]. However, whether taccalonolide A has anti-hepatoma activity is largely obscure.
Hedgehog (Hh) is a critical signalling pathway that mediates intercellular communication, embryonic development, oxidative stress regulation and organ differentiation [Citation13]. Hh is classified as sonic hedgehog (Shh), Indian hedgehog (Ihh) and desert hedgehog (Dhh) [Citation14]. Alteration of the Shh signalling pathway abnormally has been identified as the pathogenesis of multiple diseases and the Gli family zinc finger 1 (Gli1) transcription factor is a significant mediator during the activation of Shh signalling pathway [Citation15,Citation16]. For example, early inactivation and late reactivation of Shh pathway facilitated mesenchymal stromal cell differentiation to insulin-producing cells, representing an alternative therapeutic approach for diabetes [Citation17]. Suppression of the Shh signalling pathway weakened tumour aggressiveness in cervical carcinoma by inhibiting epithelial to mesenchymal transition process [Citation18].
We hypothesized that taccalonolide A regulated HCC cell progression by affecting the Shh pathway. We found that taccalonolide A suppressed the viability and enhanced sorafenib cytotoxicity in HCC cells by blockage of the Shh pathway.
Materials and methods
Cell culture and treatment
HCC cell lines HepG2 and Huh7 were purchased from American Type Culture Collection (Manassas, VA). HepG2 and Huh7 cells were incubated in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA) containing 10% foetal bovine serum and 0.05% penicillin/streptomycin. For some experiments, HepG2 and Huh7 cells were treated with taccalonolide A (Tauto Biotech, Shanghai, China; 0, 1.25, 2.5, 5, 10, 20 and 40 μM) or sorafenib (Santa Cruz Biotechnology, Santa Cruz, CA; 0, 1, 2, 4, 8 and 16 μM) in the presence or absence of 200 nM SAG (Shh agonist; Santa Cruz Biotechnology, Santa Cruz, CA) for 48 h.
Cell counting kit-8 (CCK-8) assay
Cell viability was evaluated by CCK-8 assay. HepG2 and Huh7 cells (5000 cells/well) were seeded on 96-well plates. After different treatments mentioned above, 10 μL CCK-8 reagent (Beyotime, Shanghai, China) was added to cells and allowed to react for 2 h. The OD value at 490 nm was measured by a Multiskan Spectrum plate reader (Thermo Fisher Scientific, Waltham, MA). The half-maximal inhibitory concentration (IC50) for taccalonolide A was calculated from the dose-dependent curve using GraphPad Prism version 7 (GraphPad Inc., San Diego, CA).
Cell cycle analysis
After treatments, cells (1 × 105/well) were collected and fixed in 70% ethanol at 4 °C overnight. Cells were then treated with RNase A (100 ng/mL; Sigma-Aldrich, St. Louis, MO) at 37 °C for 30 min, followed by staining with propidium iodide (PI) (50 ng/mL) at 37 °C for 20 min. The cell cycle distribution was analyzed with BD FACS Canto II flow cytometry (BD Biosciences, Franklin Lakes, NJ). The percentages of cells in G0/G1, S and G2/M phases were counted and compared.
Apoptosis assay
Apoptosis was examined by flow cytometry. HepG2 and Huh7 cells (1 × 106 cells/well) were seeded on 24-well plates. After different treatments for 48 h, cells were collected by trypsinization and centrifugation. Cells were then washed twice with cold saline, resuspended in 100 μL of binding buffer, co-stained with Annexin V-FITC and PI (Vazyme, Nanjing, China). Apoptosis rate was analyzed by BD FACS Canto II flow cytometry (BD Biosciences). Both early apoptosis (Annexin V-positive, PI-negative) and late apoptosis (double-positive) were considered apoptotic cells.
Caspase-3/7 activity detection
The Caspase-3/7 activity assay kit (Beyotime, Shanghai, China) was employed to detect caspase-3/7 activity. In brief, HepG2 and Huh7 cells (2 × 104 cells/well) were seeded on a 24-well plate for 24 h. After different treatments, the cells were collected by trypsinization and caspase-3/7 activity was detected using the caspase-3/7 activity assay kit. The luminescence was measured by a plate reader (Thermo Fisher Scientific). The caspase-37 activity was normalized to the control group.
Western blot
Western blot assay was conducted under the standard protocol as described previously [Citation19]. Briefly, equal amounts of proteins (20 μg) were separated by 10% SDS/PAGE (Invitrogen, Carlsbad, CA) and transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA), which were blocked in 5% defatted milk for 1 h. The primary antibodies against Bax (ab32503, 1:1000 dilution), Bcl-2 (ab32124, 1:1000 dilution), Shh (ab19897, 1:1000 dilution), Smo (ab236465, 1:1000 dilution), Gli1 (ab49314, 1:1000 dilution) and β-actin (ab8227, 1:2000 dilution) were purchased from Abcam (Cambridge, MA) and HRP-conjugated secondary antibody (ab7090, 1:3000 dilution) was obtained from Sangon (Shanghai, China). β-actin was used as a loading control. Immunoblots were visualized by an enhanced chemiluminescence detection system (Amersham, Piscataway, NJ) and quantified using ImageJ software (NIH, Bethesda, MA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
HepG2 and Huh7 cells were incubated with TRIzol reagent (Invitrogen) to extract total RNA. The cDNA for Shh, Smo and Gli1 were synthesized by All-in-One™ First-Strand cDNA Synthesis Kit (FulenGen, Guangzhou, China). Subsequently, qRT-PCR was performed using SYBR green (Applied Biosystems, Foster City, CA). The expression of Shh, Smo and Gli1 mRNA was normalized to GAPDH. The primers for Shh, Smo, Gli1 and GAPDH were as follows: Shh, (Forward, 5′-CACC GAGC AGTG GATA TGTG-3′; Reverse, 5′-AGTG GCCA GGAG TGAA ACTG-3′); Smo, (Forward, 5′-CTGG TAAG AGGA CGTG CAGA-3′; Reverse, 5′-AGGG TGAA GAGC GTGC ACTG-3′); Gli1, (Forward, 5′-GAAG GTGA AGGT CGGA GT-3′; Reverse, 5′-GTCC AGGC TGGC ATCC GACA-3′); GAPDH, (Forward, 5′-GCAC CGTC AAGG CTGA GAAC-3′; Reverse, 5′-TGGT GAAG ACGC CAGT GGA-3′).
Statistical analysis
All the data are presented as means ± standard deviation (SD). Statistical analysis was carried out by GraphPad Prism version 7 (GraphPad Inc.). Difference comparison was carried out using Student’s t-test or one-way analysis of variance (ANOVA) following with posthoc tests. A p-value of less than .05 was considered statistically significant.
Results
Taccalonolide A suppressed the viability of HCC cells
The influence of taccalonolide A on HCC cell viability was evaluated by CCK-8 assay. As illustrated in , taccalonolide A (0, 1.25, 2.5, 5, 10, 20 and 40 μM) repressed cell viability of HepG2 and Huh7 cells with IC50 values for HepG2 and Huh7 cells were 11.9 μM and 16.8 μM, respectively. These data indicated that taccalonolide A inhibited cell viability in a concentration-dependent manner. The IC50 values were employed for the following experiments.
Figure 1. Taccalonolide A inhibited the viability of HCC cells. HepG2 (A) and Huh7 cells (B) were treated with different concentrations of taccalonolide A (0, 1.25, 2.5, 5, 10, 20 and 40 μM) for 48 h. Cell viability was evaluated by CCK-8 assay and IC50 was calculated. *p < .05 vs vehicle control. 0.1% dimethyl sulfoxide (DMSO) was used as vehicle control. All experiments were repeated three times in triplicate. Significance was analyzed using one-way ANOVA.
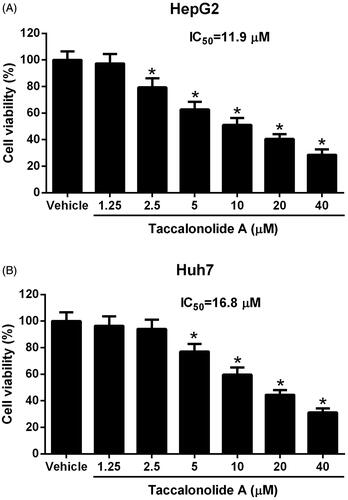
Taccalonolide A accelerated apoptosis of HCC cells
We further assessed the effect of taccalonolide A on HCC cell apoptosis using flow cytometry, a caspase-3/7 activity detection kit and western blot assays. HepG2 and Huh7 cells were treated with 11.9 and 16.8 μM taccalonolide A for 48 h, respectively. As displayed in , apoptosis was enhanced by taccalonolide A treatment. Meanwhile, caspase-3/7 activity was increased after taccalonolide A treatment (). The expression of the apoptosis-associated protein Bax was up-regulated whereas Bcl-2 was down-regulated in HepG2 and Huh7 cells treated with taccalonolide A (). Altogether, we concluded that taccalonolide A promoted apoptosis of HCC cells.
Figure 2. Taccalonolide A induced apoptosis of HCC cells. HepG2 and Huh7 cells were treated with 11.9 μM and 16.8 μM taccalonolide A for 48 h, respectively. (A) Apoptosis of HepG2 and Huh7 cells was determined by flow cytometry. (B,C) Caspase-3/7 activity of treated HepG2 and Huh7 cells was assessed by a Caspase-3/7 activity assay kit. (D, E) Protein expression of Bax and Bcl-2 in treated HepG2 and Huh7 cells was detected by western blot assay. β-actin was used as a loading control. *p < .05 vs control group. All experiments were repeated three times. Significance was analyzed using Student’s t-test.
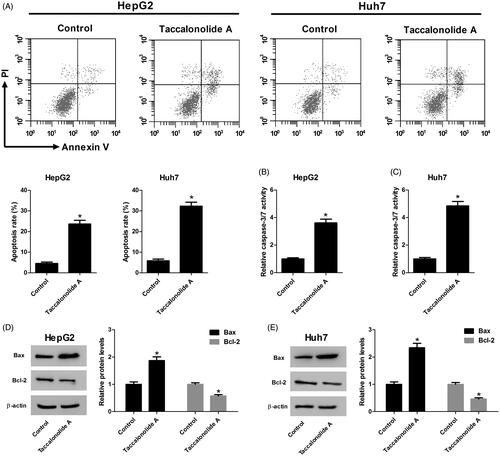
Taccalonolide A enhanced sorafenib cytotoxicity in HCC cells
We hypothesized that taccalonolide A enhanced sorafenib cytotoxicity in HCC cells. It is obvious that sorafenib (0, 1, 2, 4, 8 and 16 μM) attenuated cell proliferation in a concentration-dependent manner (). To confirm the hypothesis, HepG2 cells were treated with 2.5 μM taccalonolide A and/or 2 μM sorafenib and Huh7 cells were treated with 5 μM taccalonolide A and/or 2 μM sorafenib for 48 h. As exhibited in , taccalonolide A and sorafenib individually inhibited cell viability to a certain extent. However, taccalonolide A improved sorafenib-induced cytotoxicity in HCC cells.
Figure 3. Taccalonolide A enhanced sorafenib cytotoxicity in HCC cells. (A,B) HepG2 and Huh7 cells were treated with different concentrations of sorafenib (0, 1, 2, 4, 8 and 16 μM) for 48 h. Cell viability was evaluated by CCK-8 assay. *p < .05 vs vehicle control. 0.1% DMSO was used as vehicle control. (C,D) HepG2 cells were treated with 2.5 μM taccalonolide A and/or 2 μM sorafenib and Huh7 cells were treated with 5 μM taccalonolide A and/or 2 μM sorafenib for 48 h. Cell viability was evaluated by CCK-8 assay. *p < .05. 0.1% DMSO was used as vehicle control. All experiments were repeated three times in triplicate. Significance was analyzed using one-way ANOVA.
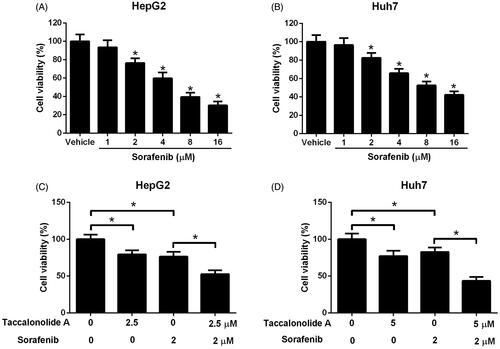
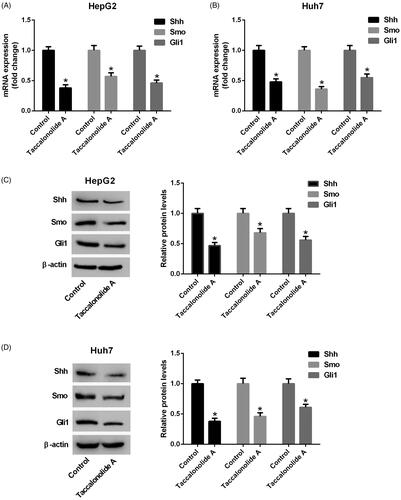
Taccalonolide A restricted the activation of Shh pathway in HCC cells
To illuminate the molecular mechanism by which taccalonolide A suppresses HCC progression, the expression of Shh, Smo and Gli1 in HepG2 and Huh7 cells treated with 11.9 and 16.8 μM taccalonolide A, respectively, was measured by qRT-PCR and western blot assays. Reduction of Shh, Smo and Gli1 mRNA expression was observed in taccalonolide A treatment group compared with the control group (). Consistently, protein expression levels of Shh, Smo and Gli1 were decreased in HepG2 and Huh7 cells treated with taccalonolide A (). Taking together, we concluded that taccalonolide A inhibited the Shh pathway in HCC cells.
Figure 4. Taccalonolide A attenuated the activation of Shh pathway in HCC cells. HepG2 and Huh7 cells were treated with 11.9 and 16.8 μM taccalonolide A for 48 h, respectively. (A,B) The expression of Shh, Smo and Gli1 mRNA in treated HepG2 and Huh7 cells was determined by qRT-PCR. (C,D) Protein expression of Shh, Smo and Gli1 in treated HepG2 and Huh7 cells was detected by western blot assay. β-actin was used as a loading control. *p < .05 vs control group. All experiments were repeated three times. Significance was analyzed using Student’s t-test.
Activation of Shh pathway inhibited taccalonolide A-induced cytotoxicity and cell cycle arrest in HCC cells
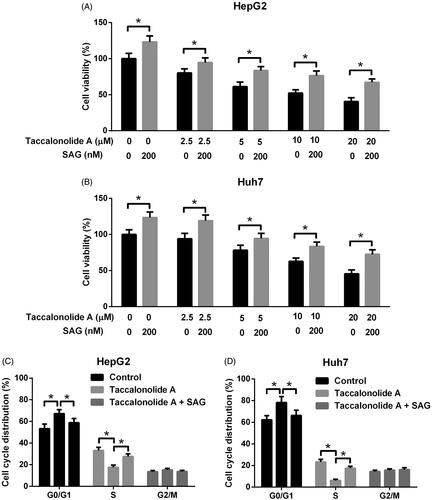
The effects of the Shh pathway on taccalonolide A-induced cytotoxicity were determined by CCK-8 assay. HepG2 and Huh7 cells were treated with 0, 2.5, 5, 10 and 20 μM taccalonolide A in the presence or absence of 200 nM SAG for 48 h. As displayed in , cell viability was reduced dramatically after taccalonolide A treatment, but the trend was reversed by activation of the Shh pathway. After treatment with taccalonolide A in the presence or absence of SAG for 48 h, cell cycle distribution was analyzed by flow cytometry. Results showed that taccalonolide A treatment induced cell cycle arrest at G0/G1 phase in HepG2 and Huh7 cells, whereas activation of the Shh pathway attenuated this effect of taccalonolide A (). All the data revealed that activation of the Shh pathway abolished taccalonolide A-induced cytotoxicity and cell cycle arrest in HCC cells.
Figure 5. Activation of Shh pathway suppressed taccalonolide A-induced cytotoxicity and cell cycle arrest in HCC cells. (A,B) HepG2 and Huh7 cells were treated with 0, 2.5, 5, 10 and 20 μM taccalonolide A in the presence or absence of 200 nM SAG for 48 h. Cell viability was evaluated by CCK-8 assay. *p < .05. 0.1% DMSO was used as vehicle control. All experiments were repeated three times in triplicate. Significance was analyzed using Student’s t-test. (C,D) HepG2 and Huh7 cells were treated with 11.9 and 16.8 μM taccalonolide A, respectively, in the presence or absence of 200 nM SAG for 48 h. Cell cycle distribution was analyzed by flow cytometry. *p < .05. 0.1% DMSO was used as vehicle control. All experiments were repeated three times. Significance was analysed using one-way ANOVA.
Activation of Shh pathway counteracted taccalonolide A-induced apoptosis in HCC cells
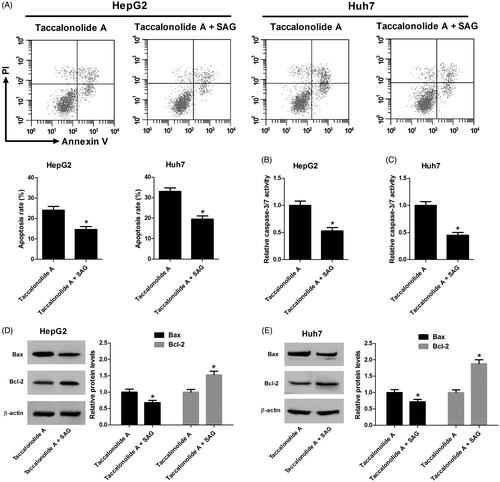
Subsequently, we evaluated the impact of the Shh pathway on taccalonolide A-induced apoptosis in HCC cells. As shown in , SAG treatment inversed taccalonolide A-induced promotive effect on HCC cell apoptosis. Similarly, caspase-3/7 activity was enhanced by taccalonolide A and activation of the Shh pathway inversed the trend (). Activation of Shh pathway counteracted taccalonolide A-induced promotion on the expression of Bax and Bcl-2 (). Collectively, activation of the Shh pathway alleviated taccalonolide A-induced apoptosis of HCC cells.
Figure 6. Activation of Shh pathway attenuated taccalonolide A-mediated apoptosis in HCC cells. HepG2 and Huh7 cells were treated with 11.9 μM and 16.8 μM taccalonolide A, respectively, in the presence or absence of 200 nM SAG for 48 h. (A) Apoptosis of HepG2 and Huh7 cells was determined by flow cytometry. (B,C) Caspase-3/7 activity of treated HepG2 and Huh7 cells was assessed by a Caspase-3/7 activity assay kit. (D,E) Protein expression of Bax and Bcl-2 in treated HepG2 and Huh7 cells was detected by western blot assay. β-actin was used as a loading control. *p < .05 vs cells treated with taccalonolide A only. All experiments were repeated three times. Significance was analyzed using Student’s t-test.
Activation of Shh pathway restored taccalonolide A-mediated acceleration on sorafenib cytotoxicity in HCC cells
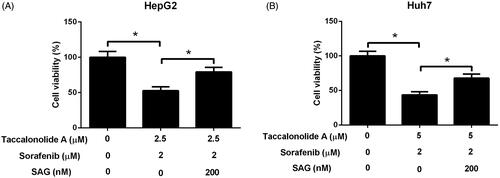
HepG2 cells were treated with 2.5 μM taccalonolide A and 2 μM sorafenib and Huh7 cells were treated with 5 μM taccalonolide A and 2 μM sorafenib in the presence or absence of 200 nM SAG for 48 h. We discovered that activation of Shh pathway by SAG restricted taccalonolide A-induced promotion on sorafenib cytotoxicity in HCC cells (). Thus, we suggested that taccalonolide A enhanced sorafenib cytotoxicity by inactivation of Shh pathway in HCC cells.
Figure 7. Activation of Shh pathway abrogated the promotion of taccalonolide A on sorafenib cytotoxicity in HCC cells. (A,B) HepG2 cells were treated with 2.5 μM taccalonolide A and 2 μM sorafenib and Huh7 cells were treated with 5 μM taccalonolide A and 2 μM sorafenib in the presence or absence of 200 nM SAG for 48 h. Cell viability was evaluated by CCK-8 assay. *p < .05. All experiments were repeated three times in triplicate. Significance was analysed using one-way ANOVA.
Discussion
Taccalonolides are highly acetylated pentacyclic steroids and they participate in the interruption of mitotic progression and initiation of apoptosis in multiple types of cancer [Citation20]. Du et al. identified that C-6 was a novel linker conjugation site on the taccalonolide backbone [Citation21]. Risinger et al. analyzed the pharmacokinetic as well as anti-tumour efficiency of taccalonolides and found that taccalonolides could covalently bind to an effective anticancer target tubulin and exert prolonged anti-proliferative function in vivo [Citation22]. Modification of taccalonolides by epoxidization at C-22,23 improved the biological activity, increased microtubule stabilizing potency and facilitated tumour therapy efficiency [Citation23].
Previous studies have revealed that the Shh signalling pathway is activated after binding to Patched1, causing the relief of Smoothened (Smo) and regulation of the target gene Gli1 and vascular endothelial growth factor [Citation24]. The Shh signalling pathway is associated with oncogenesis, cell proliferation, differentiation and angiogenesis [Citation25,Citation26]. For example, signal peptide-CUB-EGF domain-containing protein 2 (SCUBE2) was reported to attenuate cell proliferation, migration and invasion by regulating Shh signalling pathway in human non-small cell lung cancer [Citation27]. In addition, liver kinase B1 inhibited lung cancer cell survival while baicalein suppressed pancreatic cancer cell progression through the Shh signalling pathway [Citation28,Citation29]. Interestingly, epigenetic silencing of Shh induced an antitumor immune response in metastatic spine tumour by inhibition of the Shh signalling pathway [Citation30].
The Shh pathway has been reported to be involved in the development of HCC. A previous report showed that expression of Shh pathway-associated moleculars Ptch1 and Gli1 mRNA in HCC tissues indicated a risk of postresection recurrence of HCC [Citation31]. A recent study suggested that knockdown of Shh suppressed the proliferation of HCC cells [Citation32]. Zhao et al. reported that total alkaloids of Rubus alceifolius inhibited migration and invasion via suppression of the Shh pathway [Citation33]. Nitidine chloride, a natural benzophenanthridine alkaloid, was found to inhibit hepatic cancer growth partly by suppression of the Shh pathway [Citation34]. Whether the anti-hepatoma effect of taccalonolide A is mediated by the Shh pathway is unknown. In the present study, we discovered that taccalonolide A repressed proliferation and boosted apoptosis in HCC cells. Interestingly, taccalonolide A enhanced sorafenib cytotoxicity in HCC cells. By detecting the expression of Shh pathway-associated moleculars Shh, Smo and Gli1, we illuminated that the suppression of taccalonolide A on HCC cell progression was mediated by the blockage of the Shh pathway. More importantly, activation of the Shh pathway was found to attenuate taccalonolide A-induced inhibition on cell viability and promotion on apoptosis in HCC cells. Furthermore, we revealed that activation of the Shh pathway counteracted the promotive effect of taccalonolide A on sorafenib cytotoxicity in HCC cells.
In summary, we clarified that taccalonolide A inhibited cell proliferation, accelerated apoptosis and enhanced sorafenib cytotoxicity through suppression of the Shh pathway in HCC cells. Therefore, our study might present a novel method for HCC therapy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Huang S, Zhu P, Sun B, et al. Modulation of yrdc promotes hepatocellular carcinoma progression via mek/erk signaling pathway. Biomed Pharmacother. 2019;114:108859.
- Shimada S, Mogushi K, Akiyama Y, et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine. 2019;40:457–470.
- de Bono JS, Ashworth A. Translating cancer research into targeted therapeutics. Nature. 2010;467(7315):543–549.
- Okuda T, Hayashi N, Takahashi M, et al. Clinical outcomes of brain metastases from hepatocellular carcinoma: a multicenter retrospective study and a literature review. Int J Clin Oncol. 2018;23(6):1095–1100.
- Sun X, Zhuo XB, Hu YP, et al. A novel matrine derivative wm622 inhibits hepatocellular carcinoma by inhibiting pi3k/akt signaling pathways. Mol Cell Biochem. 2018;449(1–2):47–54.
- Hughes A, Dhoot GK. Dysregulated cancer cell transdifferentiation into erythrocytes is an additional metabolic stress in hepatocellular carcinoma. Tumour Biol. 2018;40(11):1010428318811467.
- Dike VT, Vihiior B, Bosha JA, et al. Antitrypanosomal activity of a novel taccalonolide from the tubers of tacca leontopetaloides. Phytochem Anal. 2016;27(3–4):217–221.
- Tinley TL, Randall-Hlubek DA, Leal R, et al. Taccalonolides E and A: plant-derived steroids with microtubule-stabilizing activity. Cancer Res. 2003;63:3211–3220.
- Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. 2014;5:316.
- Risinger AL, Mooberry SL. Taccalonolides: novel microtubule stabilizers with clinical potential. Cancer Lett. 2010;291(1):14–19.
- Peng J, Risinger AL, Fest GA, et al. Identification and biological activities of new taccalonolide microtubule stabilizers. J Med Chem. 2011;54(17):6117–6124.
- Risinger AL, Jackson EM, Polin LA, et al. The taccalonolides: microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res. 2008;68(21):8881–8888.
- Lv T, Shen L, Yang L, et al. Polydatin ameliorates dextran sulfate sodium-induced colitis by decreasing oxidative stress and apoptosis partially via sonic hedgehog signaling pathway. Int Immunopharmacol. 2018;64:256–263.
- Liu F, Feng XX, Zhu SL, et al. Sonic hedgehog signaling pathway mediates proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis via mapk/erk signaling pathway. Front Immunol. 2018;9:2847.
- Lin Z, Sheng H, You C, et al. Inhibition of the cyclind1 promoter in response to sonic hedgehog signaling pathway transduction is mediated by gli1. Exp Ther Med. 2017;13(1):307–314.
- Carballo GB, Honorato JR, de Lopes GPF, et al. A highlight on sonic hedgehog pathway. Cell Commun Signal. 2018;16(1):11.
- Dayer D, Tabar MH, Moghimipour E, et al. Sonic hedgehog pathway suppression and reactivation accelerates differentiation of rat adipose-derived mesenchymal stromal cells toward insulin-producing cells. Cytotherapy. 2017;19(8):937–946.
- Sharma A, De R, Javed S, et al. Sonic hedgehog pathway activation regulates cervical cancer stem cell characteristics during epithelial to mesenchymal transition. J Cell Physiol. 2019;234(9):15726–15741.
- Stolzenburg LR, Wachtel S, Dang H, et al. Mir-1343 attenuates pathways of fibrosis by targeting the TGF-β receptors. Biochem J. 2016;473(3):245–256.
- Risinger AL, Mooberry SL. Cellular studies reveal mechanistic differences between taccalonolide A and paclitaxel. Cell Cycle. 2011;10(13):2162–2171.
- Du L, Risinger AL, Yee SS, et al. Identification of c-6 as a new site for linker conjugation to the taccalonolide microtubule stabilizers. J Nat Prod. 2019;82(3):583–588.
- Risinger AL, Li J, Du L, et al. Pharmacokinetic analysis and in vivo antitumor efficacy of taccalonolides AF and AJ. J Nat Prod. 2017;80(2):409–414.
- Peng J, Risinger AL, Li J, et al. Synthetic reactions with rare taccalonolides reveal the value of c-22,23 epoxidation for microtubule stabilizing potency. J Med Chem. 2014;57(14):6141–6149.
- Barakat MT, Humke EW, Scott MP. Learning from jekyll to control hyde: hedgehog signaling in development and cancer. Trends Mol Med. 2010;16(8):337–348.
- Hooper JE, Scott MP. Communicating with hedgehogs. Nat Rev Mol Cell Biol. 2005;6(4):306–317.
- He Y, Guo Q, Cheng Y, et al. Abnormal activation of the sonic hedgehog signaling pathway in endometriosis and its diagnostic potency. Fertil Steril. 2018;110(1):128.e2–136.e2.
- Yang B, Miao S, Li Y. Scube2 inhibits the proliferation, migration and invasion of human non-small cell lung cancer cells through regulation of the sonic hedgehog signaling pathway. Gene. 2018;672:143–149.
- Song L, Chen X, Wang P, et al. Effects of baicalein on pancreatic cancer stem cells via modulation of sonic hedgehog pathway. Acta Biochim Biophys Sin. 2018;50(6):586–596.
- Song K, Zheng G, Zhao Y. Liver kinase b1 suppresses growth of lung cancer cells through sonic hedgehog signaling pathway. Cell Biol Int. 2018;42(8):994–1005.
- Shi G, Zhang H, Yu Q, et al. Epigenetic silencing of sonic hedgehog elicits antitumor immune response and suppresses tumor growth by inhibiting the hedgehog signaling pathway in metastatic spine tumors in sprague-dawley rats. J Cell Biochem. 2018;119(11):9591–9603.
- Jeng KS, Sheen IS, Jeng WJ, et al. High expression of patched homolog-1 messenger rna and glioma-associated oncogene-1 messenger rna of sonic hedgehog signaling pathway indicates a risk of postresection recurrence of hepatocellular carcinoma. Ann Surg Oncol. 2013;20(2):464–473.
- Chen Y, Zhu W. Knockdown of the sonic hedgehog (shh) gene inhibits proliferation of hep3b and smmc-7721 hepatocellular carcinoma cells via the pi3k/akt/pck1 signaling pathway. Med Sci Monit. 2019;25:6023–6033.
- Zhao J, Liu L, Wan Y, et al. Inhibition of hepatocellular carcinoma by total alkaloids of rubus alceifolius poir involves suppression of hedgehog signaling. Integr Cancer Ther. 2015;14(4):394–401.
- Lin J, Shen A, Chen H, et al. Nitidine chloride inhibits hepatic cancer growth via modulation of multiple signaling pathways. BMC Cancer. 2014;14:729.