
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
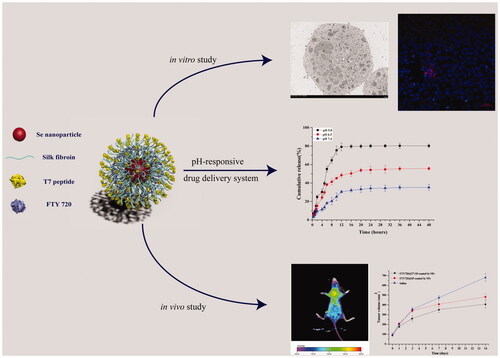
Cancer-targeted drug delivery systems based on nanoparticles (NPs) have been considered promising therapies. In this study, we developed a pH-responsive smart NPs drug delivery system using silk fibroin (SF), selenium nanoparticles (Se NPs), fingolimod (FTY720), and heptapeptide (T7). The prepared FTY720@T7-SF-Se NPs were spheres with an average diameter of 120 nm, which would contribute to the enhanced permeability and retention effects in tumour regions. The encapsulation efficiency (EE) of the FTY720@T7-SF-Se NPs was 71.95 ± 3.81%. The release of FTY720 from the nanocarriers was pH-dependent, and the release of FTY720 was accelerated in an acidic environment. Both in vitro and in vivo studies showed that FTY720@T7-SF-Se NPs had an enhanced cellular uptake selectivity and antitumor activity for thyroid cancer. The bio-distribution study in vivo further demonstrated that FTY720@T7-SF-Se NPs could effectively accumulate in the tumour region, thereby enhancing the ability to kill cancer cells in vivo. In addition, studies of histology and immunohistochemistry showed that FTY720@T7-SF-Se NPs had low toxicity to the major organs of tumour-bearing mice, indicating the prepared NPs has good biocompatibility in vivo. These results suggest that the tumour-targeted NPs delivery system (FTY720@T7-SF-Se NPs) has great potential as a new tool for thyroid cancer therapy.
Graphical Abstract
Introduction
Thyroid cancer (TC) is the most common malignant tumour in the endocrine system [Citation1]. The increased incidence of TC is most likely due to overdiagnosis, radiation expose and obesity [Citation2,Citation3]. Papillary thyroid carcinoma (PTC) is the most common type of TC, accounting for 85–90% of all TC cases [Citation3–5]. Current clinical treatments for TC and other cancers mainly include surgical resection, radiotherapy, and chemotherapy. However, these common cancer treatments usually come with inevitable adverse effects. For example, when tumour tissues are removed, surgical resection may remove part of normal tissues. Chemotherapy can effectively kill tumour cells and inhibit their growth, but it can also impair immune function and cause adverse reactions such as nausea and vomiting. Over the past few decades, the nano-scale anti-tumour drug delivery system has been widely studied as a promising oncotherapy strategy. These nanocarriers are used to load with various anti-tumour agents (e.g. hydrophobic molecules or microRNAs) and deliver them to designated locations for treatment [Citation6,Citation7]. By actively targeting the tumour sites, nanoparticles (NPs) drug delivery system can increase the effective drug concentration, thereby improving the therapeutic efficacy and reducing undesired adverse effects. In this study, we developed a pH-responsive smart NPs drug delivery system using silk fibroin (SF), selenium nanoparticles (Se NPs), fingolimod (FTY720), and heptapeptide (T7).
Silk fibroin (SF) is a natural biomaterial obtained from cocoons of Bombyx mori. Here, we chose SF as a pH-sensitive material to fabricate an intelligent NPs drug delivery system. Due to its low toxicity, good biocompatibility and inherent response to pH changes, it has been used in lysosomotropic delivery systems [Citation8]. In an acidic environment, the carboxyl groups of the SF are protonated and SF will lose its net negative charge. Therefore, drugs loaded into SF through electrostatic or hydrogen bond interactions can be released from SF-based carriers in an acidic environment.
To achieve a more stable drug delivery system with higher therapeutic effect, inorganic NPs have been introduced into the SF-coated drug delivery system [Citation9,Citation10]. Selenium nanoparticles (Se NPs), as one of the potential cancer therapeutic agents and drug carriers in the field of nanomedicine, have attracted increasing attention [Citation11–15]. Selenium is an important trace element that plays a vital role in the growth and function of human living cells. Adequate intake of selenium can promote the production of lymphocytes and maintain high level of blood immunoglobulin, thereby enhancing the activity of T cells and NK cells in the body and improving tumour treatment. Selenium can also trigger apoptosis without the involvement of tumour suppressor gene p53 [Citation16,Citation17]. Selenium compounds are the regulators of oncogene expression, which can promote differentiation, inhibit division, and induce cancer cell programmed death. Liu et al. prepared a Se NPs system with 5-fluorouracil surface decoration, which showed a high synergistic effect on A375 human melanoma cells [Citation11]. By conjugating transferrin (Tf) and loading doxorubicin (DOX), Huang and co-colleagues constructed a Se NPs drug delivery system [Citation12]. The prepared Tf-Se NPs could induce apoptosis through inducing endogenous and exogenous pathways and showed significant cytotoxicity against cancer cells [Citation12]. Se NPs have also been used as a promising vector for siRNA delivery in vitro and in vivo, showing an enhanced siRNA loading and siRNA release efficiency in gene therapy [Citation14,Citation15]. In addition, Se NPs drug carriers with specific cell-targeted peptides or ligands can accurately release drugs in tumour-bearing organs, thereby improving cancer cellular uptake and antitumor efficacy [Citation12,Citation13].
Recent studies have demonstrated that heptapeptide (sequenced HAIYPRH, T7) screened by a phage display system has a strong binding affinity to transferrin receptors (TfRs) [Citation18–20]. It has been reported that TfRs are over-expressed on the surface of most cancer cells, including glioma, breast, lung, prostate and thyroid cancer cells [Citation21–26]. Therefore, T7 peptide has been used as an active targeting agent to improve the delivery efficacy of tumour-specific drugs and reduce off-target effects in cancer-targeted drug delivery systems [Citation22–25,Citation27].
Fingolimod (2-amino-2[2-(4-octylphenyl) ethyl]-1,3-propanediol, FTY720) is synthesised based on the structure of myriocin (a fungal metabolite of the Chinese herb). It is an effective immunosuppressant to inhibit S1P (1-phosphate-neural sheath) activity [Citation28–30]. FTY720 also has been approved by the US FDA in 2010 as a new oral drug to improve the treatments of multiple sclerosis. Furthermore, numerous studies have shown that FTY720 can inhibit the growth of various cancer cells and induce their apoptosis, including renal cell carcinoma, colorectal cancer, breast cancer and pancreatic cancer [Citation28–32].
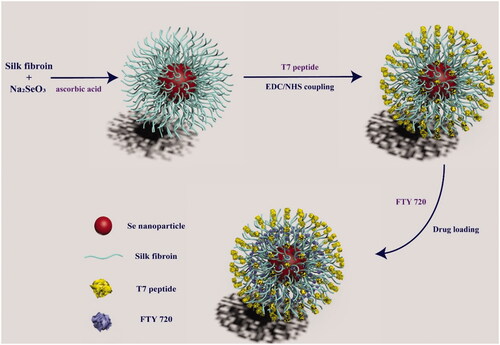
In this study, we constructed a smart tumour-targeted and pH-responsive drug delivery system (FTY720@T7-SF-Se NPs) to improve therapeutic efficiency by achieving tumour targeting and local release of FTY 720. As illustrated in Scheme 1, SF were used as shell materials to cover on Se NPs, then T7 peptides were grafted onto the surface of SF-Se NPs, and FTY720 was loaded into T7-SF-Se NPs. The morphology and size distributions of the prepared SF-Se and T7-SF-Se NPs were characterised using transmission electron microscopy (TEM) and dynamic light scattering (DLS). The drug release behaviour of FTY720@T7-SF-Se NPs under different pH conditions and ionic strength was studied. The cytotoxicity of FTY720@T7-SF-Se NPs against human PTC cell lines (K1 cells) and murine Balb/c 3T3 fibroblast cell lines (3T3 cells) were investigated, followed by the haemolysis analysis of the prepared NPs was performed. We also studied the cellular uptake, and the anti-tumour efficacy for FTY720@T7-SF-Se NPs in vitro and in vivo. These results showed that FTY720@T7-SF-Se NPs could effectively accumulate in tumour area and enhance anti-tumour activities. It may serve as a novel drug delivery system for TC treatment.
Scheme 1. Schematic illustration for preparation of FTY720@T7-SF-Se NPs.
Materials and methods
Materials
Bombyx mori silk cocoons were kindly provided by JINNAN University (Guangdong Province, China). Sodium selenite (Na2SeO3), Vitamin C, dimethyl sulfoxide (DMSO), Cell Counting KIT-8 (CCK-8), 4′,6′-diamidino-2-phenyindole (DAPI), and foetal bovine serum (FBS) were purchased from Sigma-Aldrich Chemical Co. (St Louis, USA). FTY720 (S5002) was purchased from Selleck Chemicals (Shanghai, China). T7 peptide (HAIYPRH, 7 pepitide, MW 892 Da) was synthesised by GL Biochem Ltd. (Shanghai, China). N-Hydroxysuccinimide (NHS) and 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) were purchased from Aladdin reagent co., Ltd. (Shanghai, China). Cy5.5-NHS ester was purchased from GE Healthcare (Buckinghamshire, UK). Dulbecco Eagle Medium (DMEM) was purchased from Capricorn (Hesse, Germany). All reagents and solvents were analytical grade and used without further purification.
Preparations of the silk fibroin (SF)
Silk fibroin was prepared as described previously [Citation33]. Briefly, 10 g of Bombyx mori silk cocoons were boiled in Na2CO3 (0.05 M) solution for 45 min and then washed three times with sterile distilled water. The degummed silk was dehydrated in oven at 60 °C overnight. The obtained silk fibroin was dissolved in the LiBr (9.2 M) solution at 60 °C for 6 h. Afterward, the SF solution was dialysed against deionised water for 3 days (MWCO 3500 Da), the dialysed SF solution was collected and freeze dried.
Preparations of SF-Se NPs and T7-SF-Se NPs
SF-Se NPs were prepared as previously reported with slight modifications [Citation11,Citation34–36]. Briefly, 500 mg of SF was dissolved in deionised water to obtain SF solution (10 mg/mL). 20 ml of the prepared SF solution was mixed with 10 ml of Na2SeO3 (0.1 M) solution by stirring at room temperature for 1 h. Then, 10 ml of ascorbic acid solution (0.1 M) was added dropwise to the solution, and the resulted solution was stirred for 12 h at room temperature. The obtained solution was dialysed (MWCO 3500 Da) against deionised water for 3 days to remove the uncoated SF. The dialysed solution was collected and freeze dried to obtain the SF-Se NPs.
EDC-NHS coupling was used for the connection of T7 peptides to SF-Se NPs. Briefly, 50 mg SF-Se NPs was dispersed in 10 ml deionised water, followed by adding 50 mg EDC and 25 mg NHS into the solution. The mixture was stirred at room temperature for 1 h. Then, 100 μL of T7 peptide (1 M) was added into the solution, mixed well and shook gently for 4 h. The reaction mixture was dialysed against deionised water for 24 h to remove the remaining reactants. The dialysate was collected and freeze dried to obtain the T7-SF-Se NPs.
Preparation of FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs
To prepare the drug-loaded nanoparticles, FTY720 solution (2 mg/mL) was drop-wisely added into the SF-Se NPs solution (10 mg/mL) and T7-SF-Se NPs (10 mg/mL), respectively. The solution was stirred at room temperature for 6 h and then dialysed against deionised water for 2 days to remove unbound FTY720. The dialysate was collected and freeze dried to obtain the drug-loaded NPs.
Characterisations of SF-Se NPs and T7-SF-Se NPs
The morphology of SF-Se NPs and T7-SF-Se NPs were examined by a transmission electron microscope (TEM) (H-800 model, Hitachi, Japan). Briefly, a sample solution (10 mg/mL) was diluted to 0.1 mg/mL with deionised water, and then place on a 300-mesh carbon-coated copper grid and dried under ambient conditions. The particle size of samples was determined by dynamic light scattering (DLS) using Nano Zetasizer (Malvern Instruments Ltd, UK).
Drug encapsulation efficiency and in vitro drug release behaviour
To investigate the encapsulation efficiency (EE) of FTY720 in the T7-SF-SE NPs, a certain amount of FTY720@T7-SF-SE NPs was dispersed in PBS solution. The solution was centrifuged at 10,000 rpm for 30 min. The supernatant was collected for absorbance detection using an ultraviolet-visible spectrophotometer at the wavelength of 220 nm (UH5300, Hitachi, Japan). The concentrations of FTY720 in various samples were determine with the standard curve. The EE was calculated as:
(1)
(1)
The release profile of FTY720 from FTY720@T7-SF-Se NPs was evaluated using a dialysis method. 10 mg of FTY720@T7-SF-Se NPs was dispersed in 5 ml phosphate buffer saline (PBS) solution and then placed in a dialysis bag with a molecular weight cut-off of 3500 Da. The sealed dialysis bag was immersed in 40 ml PBS buffer solutions of pH 5.0, 6.5 and 7.4, respectively, and shaken at 100 rpm at 37 °C. At different time points, 1 ml of sample was collected, and an equal volume of PBS was replenished into the solution. The concentration of FTY720 was determined by the UV spectrophotometer at the wavelength of 220 nm.
To investigate the ionic strength effects on the drug release profile of FTY720@T7-SF-Se NPs in vitro, 10 mg of FTY720@T7-SF-Se NPs was dispersed in 5 ml PBS buffer (pH7.4), and then placed in a dialysis bag with a molecular weight cut-off of 3500 Da. The sealed dialysis bag was immersed in different 40 ml PBS buffers (pH7.4) contained 0 M, 0.1 M and 0.3 M NaCl, respectively. A beaker containing a sample was shaken at 100 rpm at 25 °C. At different time points, 1 ml of sample was collected, and an equal volume of sodium chloride solutions was replenished into the solution. The concentration of FTY720 was determined by the UV spectrophotometer at a wavelength of 220 nm.
Cell lines for human PTC cell and murine Balb/c 3T3 fibroblast cell
Human TPC cell lines (K1 cells) and murine Balb/c 3T3 fibroblast cell lines (3T3 cells) were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM medium with 10% FBS and 100 U/mL penicillin-streptomycin at 37 °C in a humidified atmosphere containing 5% CO2.
In vitro cell viability
The cytotoxicity of SF-Se NPs, FTY720@SF-Se NPs, and FTY720@T7-SF-Se NPs at different concentrations (10, 25, 50 or 100 μg/mL) against 3T3 or K1 cells were determined using CCK-8 assay as previously reported [Citation37]. Briefly, K1 or 3T3 cells were seeded in 96-well culture plates at a density of 5 × 103 cells/well for 24 h at 37 °C. The cells were then incubated with PBS or different concentrations of NPs for 24 h at 37 °C. These cells were treated with CCK-8 solution for 2 h at 37 °C. The cells with PBS treatment (i.e. without NPs) were used as a blank control. The absorbance of each sample well was measured at 450 nm using a microplate reader (Multiscan MK3, Thermo Fisher Scientific, USA).
The viability of K1 cells was measured using a Live/Dead staining assay (Live/Dead Viability/Cytotoxicity Kit, Thermo Fisher Scientific, USA). According to the protocol, 400 μL of Live/Dead staining stock solution was added to PBS, FTY720@ SF-Se NPs, and FTY720@T7-SF-Se NPs treated cells, respectively. The stained samples were incubated for 30 min at room temperature and then imaged using a fluorescence microscope (MVX10, Olympus, Japan). Live cells were indicated in green and dead cells were in red.
In vitro haemolysis study
The haemolysis assay was used to investigate the effect of the prepared NPs on rat red blood cells (RBCs). Fresh rat blood was collected and centrifuged at 1500 rpm for 10 min. The supernatant was removed, and the remaining blood cells were washed with same volume of saline (0.9%, w/v). RBCs suspension was obtained by repeating the above process three times and then diluted with 9 times volume of saline (0.9%, w/v). The obtained RBCs dilution (0.5 ml) were incubated with the prepared NPs at various concentrations at 37 °C for 1 h. PBS solution and Triton X-100 (1%) were also incubated with RBCs under same conditions, and set as negative control (NC) and positive control (PC), respectively. The treated RBCs samples were then subjected to centrifugation, and the supernatants were collected for absorbance detection using a microplate reader (Multiscan MK3, Thermo Fisher Scientific, USA) at the wavelength of 540 nm. The percentage of haemolysis rate (Haemolysis %) for NPs samples at various concentration was calculated as follows:
(2)
(2)
where Asample, ANC, and APC were the absorbance of sample, positive control, and negative control, respectively.
Intercellular uptake and lysosome-targeted destruction of FTY720@T7-Se NPs
The intercellular uptakes of FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs by K1 cells were evaluated by TEM (H-800 model, Hitachi, Japan). The K1 cells were seeded at a density of 4 × 105 cells per well into a six-well plate and incubated at 37 °C for 24 h. The cells were then replaced with the fresh medium containing FTY720@SF-Se NPs or FTY720@T7-SF-Se NPs (50 μg/mL). The samples were incubated at 37 °C for 6 h, and then fixed with 3% glutaraldehyde, and dehydrated with ethanol. After washing with PBS buffer three times, the embedded cells were sectioned (75 nm) and mounted onto 200 mesh copper grids.
Confocal-laser scanning microscope (CLSM) (LSM710, CarlZeiss, Germany) was used to investigate the intercellular uptake of FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs by K1 cells. Briefly, K1 cells were seeded on a glass-bottomed dish at a density of 1 × 105 cells per well and incubated at 37 °C for 24 h. Then, the cells were replaced with the fresh medium containing Cy5.5-labelled FTY720@SF-Se NPs or Cy5.5-labelled FTY720@T7-SF-Se NPs. The samples were incubated at 37 °C for 6 h in dark and then washed with PBS three times, followed by fixation with 4% paraformaldehyde at room temperature for 15 min. DAPI was used to stain cell nuclei for 15 min in dark. The cell samples were washed three times with PBS and visualised by CLSM. The nucleuses of cells were indicated in blue while the NPs were in red.
In vivo antitumor efficacy
Female BALB/c nude mice (5 weeks old, 16–22 g) were purchased from Southern Medical University Animal Centre (Guangzhou, China). All animal experiments (No. 20170310104715) were approved by the Laboratory Animal Ethics Committee Jinan University. Harvested K1 cells (1 × 106 cells per 100 μL PBS) were subcutaneously injected into the abdomens of mice. The mice were randomly divided into three groups (n = 5) when the tumour volume reached about 80–100 mm3. For administration, the mice were given with SF-Se NPs, FTY720@SF-Se NPs or FTY720@T7-SF-Se NPs at a dosage of 5 mg/kg through intravenously injection every other day. The tumour size was monitored, and the tumour volume was calculated as:
(3)
(3)
In vivo real-time fluorescence imaging
The biodistribution and tumour-targeted efficacy of FTY720@T7-Se NPs or FTY720@T7-Se NPs in tumour-bearing BALB/c nude mice were investigated using IVIS imaging system (Xenogen, Alameda, USA) as previously reported [Citation24,Citation38]. Briefly, the mice were anaesthetised with an injection of 5% chloral hydrate, and then injected intraperitoneally with Cy5.5-labelled FTY720@Se or FTY720@T7-Se NPs. The distribution behaviour of NPs in the mice was monitored at different time points.
Histology and immunohistochemistry
For the histological assay, tissue samples of tumour and organ extracted from nude mice were fixed with 4% paraformaldehyde. After dehydration in an alcohol solution, the sample tissue was embedded in paraffin and cut into sections (5 mm). The fixed sections were stained with haematoxylin and eosin (HE), and histological observation was performed using an optical microcopy (DMi8 digital microscope, Leica Microsystems, Germany).
Statistical analysis
All experiments were carried out at least in triplicates. The experimental results were presented as mean ± SD and analysed using Student’s t-test. A p-value of <.05 was considered statistically significant.
Results and discussion
The functional tumour-targeted drug delivery system of FTY720@T7-SF-Se NPs was prepared according to previous literatures and modified [Citation11–14,Citation34–36]. As illustrated in Scheme 1, T7 peptide was conjugated to SF-Se NPs via EDC-NHS coupling, and FTY720 was loaded into T7-SF-Se NPs. The TEM images of SF-Se NPs and T7-SF-Se NPs showed that they were spheres with an average diameter of 120 nm. The conjugation of T7 peptide did not obviously affect the morphology of SF-Se NPs ( and Figure S1). Previous studies have shown that due to enhanced permeability and retention (EPR) effects in tumour regions, NPs with a diameter of 10–200 nm are expected to become a promising drug carrier in biomedical applications [Citation39,Citation40]. The zeta potentials of SF-Se and T7-SF-Se NPs were −23.4 mV and −11.2 mV, respectively (), suggesting that the conjugation of T7 peptide affected the zeta potential of the NPs. Dynamic scattering spectra showed that the average diameter of SF-Se NPs was 80 nm and the size distribution was narrow (), while the average diameter of T7-SF-Se NPs was 100 nm (). Furthermore, in the PBS solution (pH7.4) at room temperature, the size of SF-Se and T7-SF-Se NPs increased slightly in the first four days and remained stable for the next three days. This result showed that both SF-Se NPs and T7-SF-Se NPs were well dispersed in the PBS solution at room temperature ().
Figure 1. Characterisations of the prepared NPs: TEM images of SF-Se NPs (A) and T7-SF-Se NPs (B); Zeta potential of the SF-Se NPs and T7-SF-Se NPs (C); The hydrodynamic size distribution graphs of SF-Se NPs (D) and T7-SF-Se NPs (E); Stability of SF-Se NPs and T7-SF-Se NPs at room temperature (F).
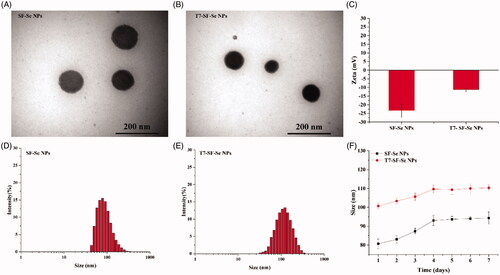
After loading FTY720, the average zeta potential of the drug carrier (FTY720@T7-SF-Se NPs) was −5.17 ± 0.25 mV (). At pH7.4, the surface charge of the T7-SF-Se NPs was negative (), while the net charge of FTY720 was positive. Therefore, FTY720 can be loaded on the T7-SF-Se NPs via electrostatic attraction, and the average zeta potential of the prepared NPs would change. The average diameter of FTY720@T7-SF-Se NPs was 100–150 nm, which was similar to the average diameter of T7-SF-Se NPs (Figure S2). The encapsulation efficiency (EE) of T7-SF-Se NPs loaded with FTY720 was 71.95 ± 3.81% ().
Table 1. Characterisations of FTY720@T7-SF-Se NPs at 25 °C.
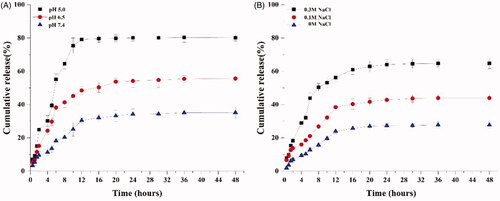
The effects of pH on the in vitro drug release of FTY720@T7-SF-Se NPs were investigated in PBS solution at 37 °C, as shown in . The PBS solution (pH at 5.0) was used to simulate the microenvironment pH of endosomes and lysosomes in cancer cells. The PBS solution (pH at 6.5) was used to mimic the extracellular environment in tumour tissues, and the PBS solution (pH at 7.4) was used to mimic the blood plasma environment. The nano-drug carrier of T7-SF-Se NPs was sensitive to pH changes, as the release rate of FTY720 was the fastest in the solution at pH 5.0, followed by pH 6.5, and the slowest at pH 7.4. At pH 5.0, the release of FTY720 reached its maximum within 12 h, releasing 79.1% of the total drug load. At pH 6.5, approximately 50.2% of the loaded FTY720 was released into the buffer within 24 h. At pH 7.4, the drug release peaked at 20 h, and 30.5% of the loaded FTY720 was found in the buffer. These data indicated that the release of FTY720 from FTY720@T7-SF-Se NPs was pH-dependent. This phenomenon might be related to the changes of electrostatic interaction between the T7-SF-Se NPs and FTY720. The prepared drug delivery system (FTY720@T7-SF-Se NPs) was constructed by electrostatic interactions between the amino groups of FTY720 and the carboxyl groups of SF side chains. In an acidic environment, the carboxyl protonation of T7-SF-Se NPs would reduce the electrostatic interactions between FTY720 and T7-SF-Se NPs. This would cause FTY720@T7-SF-Se NPs to swell and promote the accelerated release rate of FTY720 from T7-SF-Se NPs in acidic environments. The effect of ionic strength on the release profile of FTY720@T7-SF-Se was also investigated at room temperature and at pH7.4 (). The release amount and rate of FTY720 increased with the increase of NaCl concentration. In the absence of additional NaCl, approximately 27.77% of FTY720 was released within 48 h, while in the presence of additional 0.1 M and 0.3 M NaCl, approximately 49.93% and 64.83% of the drug were released into the buffer, respectively. This might be due to the presence of excess sodium ions around FTY720@T7-SF-Se NPs, which would weak the interactions between the amino groups of FTY720 and carboxyl groups on the surface of T7-SF-Se NPs. Therefore, the stability of FTY720@T7-SF-Se NPs was reduced, resulting in more and faster drug released into the buffer.
Figure 2. In vitro release profiles of FTY720 from FTY720@T7-SF-Se NPs under different conditions: (A) Samples were measured in different pH environments at 37 °C; (B) Samples were measured at different ionic strengths at 25 °C, pH7.4.
To investigate the cytotoxicity of the prepared NPs, we measured the viability of normal fibroblast cells (3T3 cells) and thyroid cancer cells (K1 cells) treated with various NPs at different concentrations. As shown in , when the prepared NPs was administered at a concentration of 10 to 50 mg/mL, the cell viability of 3T3 cells was approximately 80%. This result indicated that SF-Se NPs with or without FTY720-loaded showed low cytotoxicity to 3T3 cells. When cocultured with SF-Se NPs, FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs at 100 mg/mL, the viability of 3T3 cells decreased to 61.1%, 62.7% and 69.7%, respectively. Compared to the control group, SF-Se, FTY720@SF-Se, and FTY720@T7-SF-Se NPs exhibited slight cytotoxic effects to 3T3 cells. As shown in , FTY720@T7-SF-Se NPs had the strongest dose-dependent inhibition on K1 cell proliferation when compared to other groups. When the concentration of FTY720@T7-SF-Se NPs was set at 10, 25, 50 and 100 μg/mL, the cell viability of K1 cells were 74.2%, 70.7%, 28.7% and 19.4%, respectively. FTY720@Se NPs also showed a similar dosage-dependent inhibition on the K1 cell proliferation. In addition, FTY720@T7-SF-Se NPs had the lowest half inhibitory concentration (Table S1) and strongest antitumor efficacy compared with SF-Se NPs and FTY720@SF-Se NPs. This might be due to the conjugation of T7 peptide to the nano-drug carrier, leading to a higher cellular uptake efficiency of FTY720@T7-SF-Se NPs, which in turn triggered the increase of FTY720 release.
Figure 3. Cell viability of (A) 3T3 cells and (B) K1 cells cocultured with various NPs at different concentration. (*p < .05, **p < .01); Haemolysis study of various prepared NPs (C).
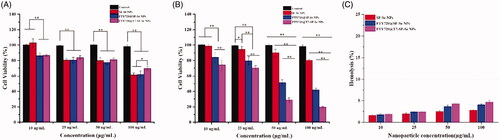
According to the effect of drug concentration on cell viability (), FTY720@T7-SF-Se NPs at 50 μg/mL showed good compatibility to 3T3 cells, 81.3%; while the cell viability of K1 cells treated with FTY720@T7-SF-Se NPs (at 50 μg/mL) was only 28.7%. This result indicated that FTY720@T7-SF-Se NPs at 50 μg/mL showed low cytotoxic effect on the normal cells and still maintained strong anticancer activity to the tumour cells. Therefore, for the following antitumor studies, the effective concentration of FTY720@T7-SF-Se NPs was selected to be 50 μg/mL. The studies on the cell viability of 3T3 and K1 cells demonstrated that the prepared FTY720@T7-SF-Se NPs had a strong selectivity between tumour cells and normal cells, which might be served as a potential antitumor drug delivery system in the therapeutic applications. In addition, SF-Se NPs, FTY720@SF-Se NPs, and FTY720@T7-SF-Se NPs exerted very low haemolysis rate (). All the prepared NPs had excellent blood-compatibility since their haemolysis rate were small than 5.0%.
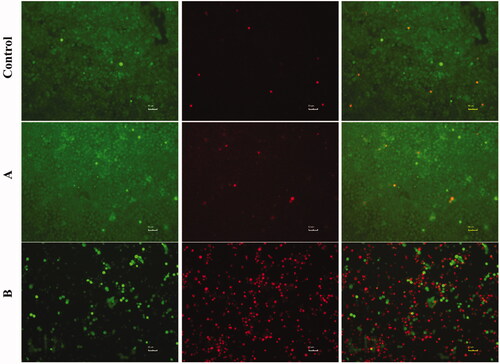
Fluorescence stained live/dead cell assay further revealed the anti-tumour efficacy of FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs against K1 cells (). After 24 h, only a small amount of red fluorescence signal was observed in the control group, indicating that there was no remarkable cell death. Compared with the control group, the amount of red fluorescence of K1 cells treated with FTY720@SF-Se NPs was enhanced, indicating that partial K1 cells were killed by FTY720@SF-Se NPs. When K1 cells were treated with FTY720@T7-SF-Se NPs, the number of viable cells (indicated in green) decreased sharply comparing to the other two groups; while the number of dead cells (indicated in red) were the highest among all groups. This finding was consistent with the results of cell viability (), suggesting that FTY720@T7-SF-SE NPs had the highest anti-tumour activity compared with the control group and FTY720@SF-SE NPs.
Figure 4. Live and dead cell staining of K1 cells after co-cultured with PBS (Control), FTY720@SF-SeNPs (A), and FTY720@T7-SF-SeNPs (B) for 24 h. Red colour indicates dead cells and green for live cells.
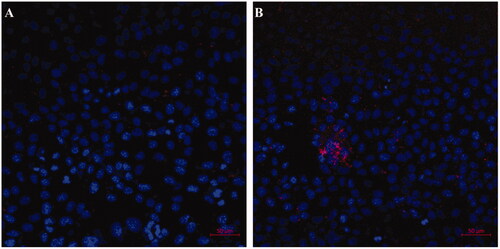
Effective intercellular uptake of drug-loaded NPs is essential to improve their therapeutic efficacy against cancer cells. The cellular uptakes of FTY720@SF-Se and FTY720@T7-SF-Se NPs were investigated using TEM and CLSM. After 6 h of incubation with K1 cells, FTY720@SF-Se NPs (i.e. the black spots) were scattered within K1 cells (). This result indicated that FTY720@SF-Se NPs were internalised by K1 cell and accumulated on cell membrane. For the group treated with FTY720@T7-SF-Se NPs (), there were more black spots clustered in the vicinity of cell membrane and in the nucleus. This result suggested that under the same conditions, the uptake of FTY720@T7-SF-Se NPs by K1 cells was higher than that of FTY720@SF-Se NPs in the endocytosis process. The confocal images () showed a similar trend. The group of K1 cells treated with FTY720@T7-SF-Se NPs had stronger red florescence intense than the group of K1 cells treated with FTY720@SF-Se NPs.
Figure 5. TEM images of K1 cells incubated with FTY720@SF-Se NPs (A) and FTY720@T7-SF-Se NPs (B) after 8 h.
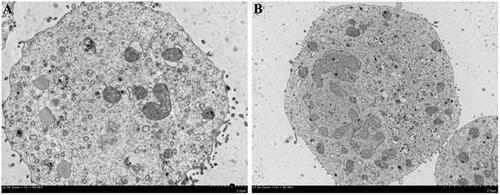
Figure 6. Confocal images of K1 cells incubated with the prepared NPs (blue colour indicates cell nucleus and red for the Cy 5.5 labelled nanoparticles): (A) Cy5.5-labelled FTY720@SF-Se NPs, and (B) Cy5.5-labelled FTY720@T7-SF-Se NPs (B) after 6 h.
Previous studies have shown that T7 peptide exhibit a high binding affinity to transferrin receptors, which is usually over-expressed in various tumour cells, including glioma, breast, lung, and prostate cancer cells [Citation22–25]. This explained our findings that the FTY720@T7-SF-Se NPs had a greater selectivity of cellular uptake compared to the FTY720@SF-Se NPs. In this study, compared with FTY720@SF-Se NPs, FTY720@T7-SF-Se NPs had higher cancer cell-targeted efficiency through T7 modification. The higher the efficiency of FTY720@T7-SF-Se NPs in targeting K1 cells, the higher the anti-tumour activity ( and ).
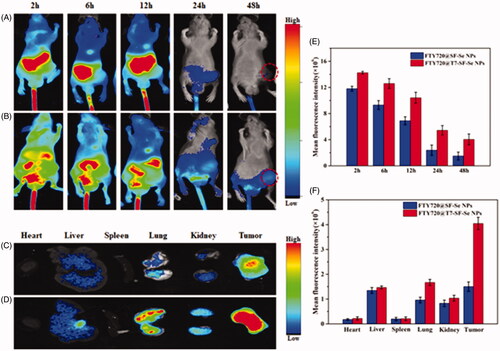
As an effective drug delivery system targeting cancer, the carrier should be able to release the anticancer drugs in the tumour organ regions to achieve tumour-targeted therapy. The bio-distribution study of FTY720@T7-SF-Se NPs was carried out by tracking the fluorescence of Cy5.5-labled NPs injected intravenously in BALB/c nude mice model. showed in vivo fluorescence images of tumour-bearing mice treated with Cy5.5-labelled FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs after 2, 6, 12, 24 and 48 h, respectively. As time passed, the fluorescence intensity of the subjects gradually decreased (). Compare with the mice treated with FTY720@T7-SF-Se NPs, the fluorescence intensity of mice treated with FTY720@SF-Se NPs became much weaker in the tumour area after 48 h. The ex vivo images of major organs and tumours collected at 48 h after injection showed that the fluorescence intensity of FTY720@T7-SF-Se NPs was stronger in tumour tissues than FTY720@SF-Se NPs (). These findings indicated that the T7-decorated NPs could effectively target tumour tissues and accumulate in the lesions. Combining the drug release profile () and the results of cellular uptake ( and ), we concluded that FTY720@T7-SF-Se NPs could effectively accumulate in the tumour area and release FTY720 to kill cancer cells in acidic microenvironments. Compared with FTY720@SF-Se NPs, the prepared smart bio-responsive FTY720@T7-SF-Se NPs showed higher tumour-targeted efficiency and anticancer activity in vitro and in vivo.
Figure 7. In vivo near-infrared fluorescence images of K1 tumour-bearing nude mice after injection with Cy5.5-labled SF-Se NPs FTY720@ SF-Se NPs (A), or FTY720@T7-SF-Se NPs (B) at 2, 6, 12, 24 and 48 h; ex vivo images of major organs and tumours collected at 48 h post-injection with Cy5.5-labled FTY720@ SF-Se NPs (C), or FTY720@T7-SF-Se NPs (D); fluorescence intensities measured from in vivo images of nude mice (E) and ex vivo images of major organs and tumours (F).
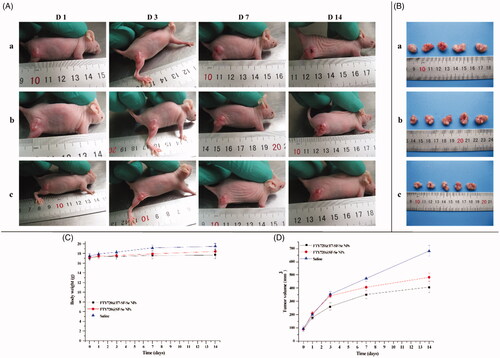
The in vivo therapeutic effects of FTY720@T7-SF-Se NPs were investigated by using a tumour model of BALB/c nude mice pre-inoculated with K1 cells. The tumour-bearing mice were randomly divided into three groups, and saline, FTY720@ SF-Se NPs, and FTY720@T7-SF-Se NPs were injected intravenously every other day. showed the appearance of tumours of the experimental subjects at different time points. The tumour volume of all administration groups increased over time (). The control group (i.e. treated with saline) had the largest tumour, followed by the group treated with FTY720@SF-Se NPs, and finally the group treated with FTY720@T7-SF-Se NPs. The body weight of the control group (mice treated with saline) increased slightly, while the weight change of the other groups (i.e. treated with FTY720@SF-Se NPs or FTY720@T7-SF-Se NPs) was negligible (). In addition, the tumour growth rate was the fastest in the saline control group, followed by the group treated with FTY720@SF-Se NPs and the group treated with FTY720 @T7-SF-Se NPs (). After 12 days of treatment, the tumour volume of the control group was approximately 1.5 or 2 times higher than the groups treated with FTY720@SF-Se NPs and FTY720@T7-SF-Se NPs, respectively. These results suggested that the non-targeted drug delivery NPs (FTY720@SF-Se NPs) had an inhibitory effect on tumour growth, while the active cancer-targeted drug delivery NPs (FTY720@T7-SF-SeNPs) had a more remarkable anti-tumour effect. This might be due the fact that T7 peptide conjugation helped the drug-loaded NPs effectively accumulate in tumour lesions (as shown in ), thereby enhancing the anti-tumour activities of NPs.
Figure 8. In vivo antitumor therapeutic effects of FTY720@T7-SF-Se NPs. The K1 tumour-bearing nude mice were intravenously injected with saline as the (a) control group, (b)FTY720@SF-Se NPs, and (c)FTY720@T7-SF-Se NPs: the images of tumour growth in vivo (A); the images of dissected tumours from the mice after last injection with the prepared NPs (B); the changes in body weight (C) and the tumour volume (D) of K1 tumour-bearing nude mice. Data were shown as mean ± SD.
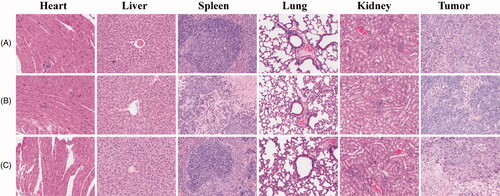
We further evaluated the potential toxicity of FTY720@SF-Se and FTY720@T7-SF-Se NPs to major organs in vivo. The mice were sacrificed on the 14th day after injection of the prepared NPs. The tumour tissues and important organs were collected for pathological analysis. As shown in , there were no histological changes or tumour metastasis observed in any organ. These findings indicated that both FTY720@SF-Se and FTY720@T7-SF-Se NPs had good biocompatibilities with normal tissues at the dose used, which was consistent with the findings of 3T3 cell viability in vitro ().
Figure 9. HE staining images of major organs and tumour tissues of mice. The tumour bearing mice were treated with saline (A), FTY720@SF-Se NPs (B), and FTY720@T7-SF-Se NPs (C), respectively.
The incidence of thyroid cancer is a common endocrine malignancy. It has developed rapidly in recent years and its death toll is more than all other endocrine cancers [Citation1]. Currently, the conventional treatment methods for thyroid cancer include surgical treatment, radioiodine therapy, and chemotherapy. One of the major obstacles of thyroid cancer chemotherapy is the specific delivery of the therapeutic drug to the thyroid gland [Citation3–5].
Here, we construct a smart tumour targeting NPs platform (FTY720@T7-SF-Se NPs), which can effectively deliver FTY720 in vitro and in vivo to the thyroid cancer tumour, and continuously release FTY720 though pH response. Different from the previous pH-sensitive NPs, we use natural biological macromolecule SF as the pH sensitive shell coating on Se NPs to construct a pH-responsive drug delivery platform. In this way, we overcome several key limitations of the inorganic NPs drug delivery platform, such as complex multistep syntheses and purification, poor biocompatibility, and negative effects on the activity of FTY720.
FTY720 is a synthetic analog of sphingosine and is currently undergoing multiple clinical trials, for instance, in the prevention of kidney graft rejection and treatment of relapsing multiple sclerosis. FTY720-induced immunosuppression is mainly attributed to its phosphorylated state, in which it binds to sphingosine 1-phosphate (S1P) receptors and causes its effects on lymphocyte recirculation and immune effector cell functions. In addition to the immunosuppressive effects, FTY720 has also shown preclinical antitumor efficacy in various cancer models, including breast, bladder, prostate, lung and liver, and haematopoietic malignancies [Citation41–45]. The antitumor activity of FTY720 is attributed to its cytotoxicity towards cancer cells through both caspase-dependent and caspase-independent apoptotic pathways [Citation46]. To the best of our knowledge, FTY720 has not been reported as a therapeutic agent for thyroid cancer in any NPs drug delivery system.
Our results showed that FTY720@T7-SF-Se NPs could efficiently carry FTY720 (encapsulation efficiency up to 71.95% ± 3.81%), has a good targeting effect both in vitro and in vivo. The pH response of FTY720@T7-SF-Se NPs enabled it to release more FTY720 in an acidic environment. The cytotoxicity study demonstrated that equipped with T7 peptide, FTY720@T7-SF-Se NPs could effetely target K1 cells and the released FTY720 would induce K1 Cell death. For normal health cells, the prepared FTY720@T7-SF-Se NPs showed little cytotoxicity on 3T3 cells. This result might be explained for two reasons. First, the prepared NPs might not able to be internalised by 3T3 cells. Second, our preliminary result (data not included) showed that FTY720 alone at low does (i.e. at 10 μg/mL) did not cause a remarkable reduction in 3T3 cells. In addition, a recent study suggested that FTY720 (20 mmol/L, i.e. 6.88 μg/mL) has a slight effect on the viability of normal dermal fibroblasts (NFs), causing only 20% cell death at 24 h [Citation47]. Guo et al. prepared RGD-conjugated nanobubbles loaded with FTY720 and superparamagnetic iron oxide nanoparticles. Their results indicated that the cell viability of 3T3 cells treated with the prepared NBs (100 μg/mL) were higher than 80% [Citation48]. In addition, SF-Se NPs, FTY720@SF-Se NPs, and FTY720@T7-SF-Se NPs exerted very low haemolysis rate (below 5%) in vitro (), indicating all the prepared NPs had excellent blood-compatibility. Taking these results together, FTY720@T7-SF-SeNPs could be served as a safe and effective antitumor drug delivery system in the thyroid therapy applications.
Conclusions
In this study, we successfully designed an effective tumour-targeted drug delivery system FTY720@T7-SF-Se NPs for the treatment of thyroid cancer. The in vitro drug release behaviour showed that the nano-drug carrier could efficiently release FTY720 in an acidic microenvironment, which indicated that FTY720@T7-SF-Se NPs was expected to be a pH-mediated cancer therapeutics. The in vitro and in vivo studies showed that, compared with FTY720@SF-Se NPs, FTY720@T7-SF-Se NPs had higher cellular uptake selectivity and antitumor activity for thyroid cancer. Studies on cytotoxicity and haemolysis showed that the prepared NPs had good biocompatibility and blood-compatibility in vitro. The HE analyses further demonstrated that the major organs of tumour-bearing mice treated with FTY720@T7-SF-Se NPs had low damage, indicating the prepared NPs had good biocompatibility of in vivo. We envisage the active tumour-targeted delivery system (FTY720@T7-SF-Se NPs) proposed in this study could represent a promising strategy for the treatment of thyroid cancer.
supplementary_information.docx
Download MS Word (159.4 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Carty S, Cooper D, Doherty G, et al. Consensus statement on the terminology and classification of central neck dissection for thyroid cancer. Thyroid. 2009;19(11):1153–1158.
- Vaccarella S, Dal Maso L, Laversanne M, et al. The impact of diagnostic changes on the rise in thyroid cancer incidence: a population-based study in selected high-resource countries. Thyroid. 2015;25(10):1127–1136.
- Cabanillas M, McFadden D, Durante C. Thyroid cancer. Lancet. 2016;388(10061):2783–2795.
- Xing M, Haugen B, Schlumberger M. Progress in molecular-based management of differentiated thyroid cancer. Lancet. 2013;381(9871):1058–1069.
- Kim T, Kim Y, Kim H, et al. Prognostic value of the eighth edition AJCC TNM classification for differentiated thyroid carcinoma. Oral Oncol. 2017;71:81–86.
- Da Silva C, Rueda F, Löwik C, et al. Combinatorial prospects of nano-targeted chemoimmunotherapy. Biomaterials. 2016;83:308–320.
- Farrell D, Alper J, Ptak K, et al. Recent advances from the National Cancer Institute Alliance for Nanotechnology in Cancer. ACS Nano. 2010;4(2):589–594.
- Mehrotra S, Chouhan D, Konwarh R, et al. Comprehensive review on silk at nanoscale for regenerative medicine and allied applications. ACS Biomater Sci Eng. 2019;5(5):2054–2078.
- Wani S, Veerabhadrappa G. Silk fibroin based drug delivery applications: promises and challenges. Curr Drug Targets. 2018;19(10):1177–1190.
- Crivelli B, Perteghella S, Bari E, et al. Silk nanoparticles: from inert supports to bioactive natural carriers for drug delivery. Soft Matter. 2018;14(4):546–557.
- Liu W, Li X, Wong Y, et al. Selenium nanoparticles as a carrier of 5-fluorouracil to achieve anticancer synergism. ACS Nano. 2012;6(8):6578–6591.
- Huang Y, He L, Liu W, et al. Selective cellular uptake and induction of apoptosis of cancer-targeted selenium nanoparticles. Biomaterials. 2013;34(29):7106–7116.
- Fu X, Yang Y, Li X, et al. RGD peptide-conjugated selenium nanoparticles: antiangiogenesis by suppressing VEGF-VEGFR2-ERK/AKT pathway. Nanomedicine. 2016;12(6):1627–1639.
- Yu Q, Liu Y, Cao C, et al. The use of pH-sensitive functional selenium nanoparticles shows enhanced in vivo VEGF-siRNA silencing and fluorescence imaging. Nanoscale. 2014;6(15):9279–9292.
- Chen Q, Yu Q, Liu Y, et al. Multifunctional selenium nanoparticles: chiral selectivity of delivering MDR-siRNA for reversal of multidrug resistance and real-time biofluorescence imaging. Nanomedicine. 2015;11(7):1773–1784.
- Zakharia Y, Bhattacharya A, Rustum Y. Selenium targets resistance biomarkers enhancing efficacy while reducing toxicity of anti-cancer drugs: preclinical and clinical development. Oncotarget. 2018;9(12):10765–10783.
- Miao Q, Xu J, Lin A, et al. Recent advances for the synthesis of selenium-containing small molecules as potent antitumor agents. Curr Med Chem. 2018;25(17):2009–2033.
- Lee J, Engler J, Collawn J, et al. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur J Biochem. 2001;268(7):2004–2012.
- Han L, Huang R, Liu S, et al. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol Pharm. 2010;7(6):2156–2165.
- Du W, Fan Y, He B, et al. Bionano interactions of mcf-7 breast tumor cells with a transferrin receptor targeted nanoparticle. Mol Pharm. 2015;12(5):1467–1476.
- Keer H, Kozlowski J, Tsai Y, et al. Elevated transferrin receptor content in human prostate cancer cell lines assessed in vitro and in vivo. J Urol. 1990;143(2):381–385.
- Liang M, Gao C, Wang Y, et al. Enhanced blood-brain barrier penetration and glioma therapy mediated by T7 peptide-modified low-density lipoprotein particles. Drug Deliv. 2018;25(1):1652–1663.
- Yu M, Pang W, Yang T, et al. Systemic delivery of siRNA by T7 peptide modified core-shell nanoparticles for targeted therapy of breast cancer. Eur J Pharm Sci. 2016;92:39–48.
- Riaz M, Zhang X, Wong K, et al. Pulmonary delivery of transferrin receptors targeting peptide surface-functionalized liposomes augments the chemotherapeutic effect of quercetin in lung cancer therapy. IJN. 2019;14:2879–2902.
- Lu Y, Jiang W, Wu X, et al. Peptide T7-modified polypeptide with disulfide bonds for targeted delivery of plasmid DNA for gene therapy of prostate cancer. Int J Nanomedicine. 2018;13:6913–6927.
- Magro G, Cataldo I, Amico P, et al. Aberrant expression of TfR1/CD71 in thyroid carcinomas identifies a novel potential diagnostic marker and therapeutic target. Thyroid. 2011;21(3):267–277.
- Wang R, Hu Y, Yang Y, et al. Using PEGylated iron oxide nanoparticles with ultrahigh relaxivity for MR imaging of an orthotopic model of human hepatocellular carcinoma. J Nanopart Res. 2017;19(2):12.
- Tonelli F, Lim K, Loveridge C, et al. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell Signal. 2010;22(10):1536–1542.
- Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: recent findings and new perspectives. Pharmacol Ther. 2018;185:34–49.
- Nagahashi M, Yamada A, Katsuta E, et al. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation, and breast cancer metastasis. Cancer Res. 2018;78(7):1713–1725.
- Oaks J, Santhanam R, Walker C, et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood. 2013;122(11):1923–1934.
- Azuma H, Takahara S, Horie S, et al. Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J Urol. 2003;169(6):2372–2377.
- Huang X, Liang H, Li Z, et al. Monodisperse phase transfer and surface bioengineering of metal nanoparticles via a silk fibroin protein corona. Nanoscale. 2017;9(8):2695–2700.
- Zhang Y, Wang J, Zhang L. Creation of highly stable selenium nanoparticles capped with hyperbranched polysaccharide in water. Langmuir. 2010;26(22):17617–17623.
- Zhang J, Zhou X, Yu Q, et al. Epigallocatechin-3-gallate (EGCG)-stabilized selenium nanoparticles coated with Tet-1 peptide to reduce amyloid-β aggregation and cytotoxicity. ACS Appl Mater Interfaces. 2014;6(11):8475–8487.
- Yin T, Yang L, Liu Y, et al. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood-brain barrier permeability peptide-B6 peptide for potential use in Alzheimer's disease. Acta Biomater. 2015;25:172–183.
- Long X, Zhang Z, Han S, et al. Structural mediation on polycation nanoparticles by sulfadiazine to enhance DNA transfection efficiency and reduce toxicity. ACS Appl Mater Interfaces. 2015;7(14):7542–7551.
- Zhang X, Lin C, Lu A, et al. Liposomes equipped with cell penetrating peptide BR2 enhances chemotherapeutic effects of cantharidin against hepatocellular carcinoma. Drug Deliv. 2017;24(1):986–998.
- Tahmasbi R, Chen C, Aresh W, et al. Combinational effects of active targeting, shape, and enhanced permeability and retention for cancer theranostic nanocarriers. ACS Appl Mater Interfaces. 2019;11(11):10505–10519.
- Shim M, Park J, Yoon H, et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J Control Release. 2019;294:376–389.
- Azuma H, Takahara S, Ichimaru N, et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62(5):1410–1419.
- Chua C, Lee D, Ling M, et al. FTY720, a fungus metabolite, inhibits in vivo growth of androgen-independent prostate cancer. Int J Cancer. 2005;117(6):1039–1048.
- Schmid G, Guba M, Papyan A, et al. FTY720 inhibits tumor growth and angiogenesis. Transplant Proc. 2005;37(1):110–111.
- Lee T, Man K, Ho J, et al. FTY720 induces apoptosis of human hepatoma cell lines through PI3-K-mediated Akt dephosphorylation. Carcinogenesis. 2004;25(12):2397–2405.
- Hung J, Lu Y, Wang Y, et al. FTY720 induces apoptosis in hepatocellular carcinoma cells through activation of protein kinase C delta signaling. Cancer Res. 2008;68(4):1204–1212.
- Kalhori V, Magnusson M, Asghar M, et al. FTY720 (Fingolimod) attenuates basal and sphingosine-1-phosphate-evoked thyroid cancer cell invasion. Endocr Relat Cancer. 2016;23(5):457–468.
- Shi F, Cao X, Hu Z, et al. Pleiotropic FTY720 is a specific and potent therapy for hypertrophic scars. J Invest Dermatol. 2017;137(7):1552–1561.
- Guo X, Chen J, Zeng B, et al. Ultrasound-mediated delivery of RGD-conjugated nanobubbles loaded with fingolimod and superparamagnetic iron oxide nanoparticles: targeting hepatocellular carcinoma and enhancing magnetic resonance imaging. RSC Adv. 2020;10(64):39348–39358.