
Abstract
Biotransformations of stemofoline (1a), (2′S)-hydroxystemofoline (2a), (11Z)-1′,2′-didehydrostemofoline (3a) and stemocurtisine (4) were studied through fermentation with Cunninghamella elegans TISTR 3370. Three new stemofoline derivatives; (6 R)-hydroxystemofoline (1b), (2′S, 6 R)-dihydroxystemofoline (2b) and (11Z,6R)-1′,2′-didehydro-6-hydroxystemofoline (3b), together with the known compound 1′,2′-didehydrostemofoline-N-oxide (3c), were produced by C-hydroxylation and N-oxidation reactions. Stemocurtisine was not biotransformed under these conditions. The transformed product 1b was four times more potent (IC50 = 11.01 ± 1.49 µM) than its precursor 1a (IC50 = 45.1 ± 5.46 µM) as an inhibitor against acetylcholinesterase.
Graphical Abstract
Stemona alkaloids are the major group of phytochemicals found in the roots of Stemona plants, the root extracts have been widely used as insecticides and enteric helminthes and for the treatment of respiratory diseases [Citation1]. Several different structural classes of Stemona alkaloids have been isolated and identified along with their biological activities [Citation2]. In particular, the stemofoline type alkaloids, including stemofoline 1a, (2′S)-hydroxystemofoline 2a, (11Z)-1′,2′-didehydrostemofoline 3a and the pyrido[1,2-a]azepine alkaloid stemocurtisine 4, have been reported to possess significant insecticidal activities [Citation3–5]. One potential mechanism of this insecticidal activity is the ability to inhibit acetylcholinesterase (AChE). Inhibition of this enzyme is also used in the treatment of patients with Alzheimer’s disease [Citation6,Citation7]. With the view of possibly enhancing the AChE inhibitory activity of these compounds, modification of Stemona alkaloids using a microbial biotransformation process seemed a potential possibility [Citation8]. The microbial transformations of natural products is often carried out under mild conditions, proceeds with high regioselectivities and diastereo- or enantioselectivities and gives rise to derivatives which may otherwise be difficult to prepare using non-enzymatic chemical reactions. An example of this is the biotransformation of the steroidal alkaloid dictyophlebine, a cholinesterase inhibitor, from the plant Sarcococca hookeriana. This alkaloid was transformed by Rhizopus stolonifer into three polar derivatives, one of which revealed higher inhibitory activity than that of the parent compound [Citation9]. Moreover, Lu et al. [Citation10] reported the microbial transformation of vermitaline, a steroidal alkaloid isolated from the roots of Veratrum dahuricum by Cunninghamella echinulata into four metabolites, three of which were new compounds [Citation10]. Additionally, the alkaloid colchicine was transformed by Bacillus spp. into 3-demethylated colchicine, which had improved therapeutic properties as an anti-inflammatory and antitumor drug [Citation11]. The optimisation of process parameters by statistical experimental design was also studied [Citation12]. Furthermore, the process of this biotransformation was enhanced by whole cell immobilisation of recombinant microbial monooxygenase [Citation13].
Herein we report a study of the microbial transformations of the Stemona alkaloids, stemofoline 1a, (2′S)-hydroxystemofoline 2a, (11Z)-1′,2′-didehydrostemofoline 3a and stemocurtisine 4 using the fungus Cunninghamella elegans which have been successfully used as a biocatalyst in the biotransformation of several other compounds [Citation14–16]. Moreover, in our preliminary experiments we screened the fungus Cunninghamella elegans along with the microorganisms Aspergillus niger, Bacillus subtilis, Bacillus safensis A2204, Bacillus altitudinis B1101 and Escherichia coli against these Stemona alkaloids via solid-state fermentation. These screening results showed that only C. elegans was capable of microbial transformation of the stemofoline alkaloids. However, the reactions were not completed after a period of more than 40 days. Faster biotransformation times were obtained through liquid-state fermentation with C. elegans. This work represents the first such study of the microbial transformations of the Stemona alkaloids. In addition, the AChE inhibitory activities of the isolated compounds were evaluated and are reported here.
Materials and methods
General experimental procedures
Optical rotations were determined on a on a Jasco P-2000 polarimeter. The Low-resolution mass spectra (ESI) and HRMS (ESI) spectra were recorded on a Micromass Platform LCZ and factory modified Waters QToF Ultima Mass Spectrometer (Wyntheshawe, UK). 1H, 13C and 2 D NMR spectra were recorded on Model DRX 400; 9.3 Tesla NMR, BRUKER, Germany. The IR spectrum was recorded on FT-IR model TENSER 27, BRUKER, Germany. UV spectra were obtained using an Evolution™ 201 UV-visible spectrophotometer. Solvents were distilled prior to use, and spectral grade solvents were applied for spectroscopic measurements. Stationary phases for column chromatography were Sephadex LH-20 and Merck silica gel 60 M (0.04 − 0.063 mm). TLC plates with silica gel F254 HX1 15287 (Merck, Darmstadt, Germany) were used to monitor reactions and chromatograph fractions (CH2Cl2−MeOH mixtures as mobile phase); detection was under UV at 254 and 366 nm or by spraying the plates with Dragendorff’s reagent. AChE inhibitory activity was measured using a BioStack microplate stacker (BioTek Instruments, Inc., Winooski, VT, USA).
Plant material
The roots of Stemona sp. were collected from Phitsanulok Province, Thailand. A voucher specimen was deposited at the Herbarium (number wp5066) of the Department of Biology, Chiang Mai University. Plant material was identified by Asst. Prof. Dr. Angkhana Inta from the Department of Biology, Chiang Mai University. The aerial parts of Stemona aphylla were collected from Lampang Province, Thailand. A voucher specimen was deposited at the Herbarium (number 09-111) of the Department of Biology, Chiang Mai University. Plant material was identified by Mr. James F. Maxwell from the Department of Biology, Chiang Mai University.
Microorganism material
Cunninghamella elegans TISTR3370 was provided by the Thailand Institute of Scientific and Technological Research (TISTR). Microbial cultures were maintained on potato dextrose agar slants at 4 °C and transferred every 6 months to maintain viability. Prior to biotransformation, the fungus was precultivated on PDA in Petri dishes for 3 days at 30 °C.
Extraction and isolation of Stemona alkaloids
The dry, aerial parts of S. aphylla (1.0 kg) were extracted with 95% EtOH (4 × 2000 mL) over 3 days at room temperature. The ethanolic solution was evaporated to give a dark green residue (43.1 g). A portion of this material (4.9 g) was passed through a Sephadex LH20 column using MeOH as the eluent to give the alkaloid fraction (0.862 g). This fraction was chromatographed over silica gel with gradient elution from CH2Cl2 to MeOH/CH2Cl2 (1:1) to give 6 fractions. Fraction 2 was chromatographed over silica gel with gradient elution from CH2Cl2 to MeOH/CH2Cl2 (1:1) to give 6 fractions. Fraction 2.2 was chromatographed over silica gel with gradient elution from EtOAc to MeOH/EtOAc (1:9) to give (2′S)-hydroxystemofoline (2a) [52 mg, eluted with MeOH/EtOAc (1:99))] and stemofoline (1a) [22 mg, eluted with MeOH/EtOAc (2:98)]. The dry powdered root of Stemona sp. from Pitsanulok Province (1.50 kg) was extracted with 95% ethanol (3 days, 3 times) at room temperature. The ethanolic solution was evaporated under reduced pressure to give a dark brown sticky residue (167 g). A portion of the crude extract (1.45 g) was chromatographed on silica gel using gradient elution from CH2Cl2 to MeOH/CH2Cl2 (1:9) as eluent. Ten fractions were collected. Fraction 9 was identified as (11Z)-1′,2′-didehydrostemofoline (3a) (105 mg, eluted with MeOH/CH2Cl2 (1:99). All isolated alkaloids were identified by comparing their 1H NMR spectroscopic data with those reported [Citation3,Citation4].
Microbial transformation procedures
The biotransformations were carried out in Czapek medium which consisted of glucose 30 g, sodium nitrate 2.0 g, monopotassium phosphate 1.0 g, magnesium sulphate 5.0 g, potassium chloride 5.0 g, and iron sulphate 0.01 g and distilled water 1 L (final pH 7.3 ± 0.2). The broth was autoclaved in individual Erlenmeyer flasks at 121 °C, 15 psi pressure for 15 min and cooled before incubation. Small scale reactions were conducted using 125 mL flasks containing 25 mL of broth with a loop of spores obtained from a freshly growing agar slant. After that 5.0 mg of stemofoline (1a), (2′S)-hydroxystemofoline (2a) or (11Z)-1′,2′-didehydrostemofoline (3a) were added into each 25 mL of broth. The cultures were incubated on a rotary shaker at 160 rpm and 30 °C according to a one-stage fermentation procedure. The fermentation periods for 1a, 2a and 3a were 15, 35 and 30 days, respectively. After the incubation period, the microorganism was separated from the media by filtration using a Buchner funnel. The filtrate was extracted with CH2Cl2 (x 3). The organic solvent was evaporated under reduced pressure to give the crude product.
AChE inhibitory activity
Acetylthiocholine iodide (ATCI), electric eel AChE (type VI-S), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (St. Louis., MO, USA). The organic solvents and all other reagents were purchased from RCI Labscan (Bangkok, Thailand).
Acetylcholinesterase inhibition assays
AChE inhibitory activity was measured by the spectrophotometric method previously described [Citation17,Citation18]. Briefly, 25 µL of 1.5 mM ATCI, 50 µL of 50 mM Tris–HCl buffer (pH 8.0), 125 µL of 3 mM DTNB, and 25 µL of sample dissolved in buffer containing MeOH (not more than 10%) were added to the wells followed by 25 µL of 0.51 U/mL AChE. The progress of the reaction was read at 405 nm every 17 s for 2 min using a microplate reader. Every experiment was performed in triplicate. Enzyme activity was calculated as a percentage of the velocities of sample compared to the negative control. The inhibitory activity was calculated from one hundred percentage subtracted by the percentage of enzyme activity. The IC50 values were calculated using the software package Prism (Graph Pad Inc, San Diego, CA). In this method, galantamine was used as a positive control.
Results
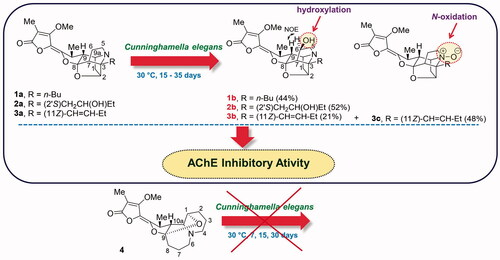
The microbial transformations of the aforementioned Stemona alkaloids, 1a, 2a, 3a and 4, using the fungus C. elegans as a biocatalyst were studied through liquid-state fermentation using TLC analysis to monitor the progress of these biotransformations ( and ). The optimized conditions for these fermentation reactions were incubated at 30 °C for 20, 35 and 15 days, respectively for complete biotransformation of compounds 1a, 2a and 3a. Whereas, the pyrido[1, 2-a]azepine alkaloid 4 could not be transformed by this fungus at 30 °C for 30 days. The biotransformation of 1a gave compound 1b in a yield of 44% after a simple work up by solvent extraction (). Compound 1b was isolated as a yellow gum; Rf 0.42 (CH2Cl2-MeOH, 90:10); [α]31D +201 (c 0.60, MeOH); HRMS (ESI + ve, m/z [M + H+], found 404.2096, calcd for C22H29NO6 found: 404.2096). 1H NMR data (CDCl3, 400 MHz) and 13C NMR data (CDCl3, 100 MHz) are shown in .
All NMR data, HRMS spectrum and the 1D and 2D NMR spectra of 1b are shown in supplemental data (Table S1 and Figures S1-S5)
Figure 1. TLC Chromatograms of the starting materials (1a, 2a and 3a) and transformation products (1b, 2b, 3b and 3c) were sprayed with ammonium molybdate reagent and the mobile phase are 96:4, 93:7 and 90:10 v/v of CH2Cl2 and MeOH.

Figure 2. TLC Chromatograms of the starting materials (1a, 2a and 3a) and transformation products (1b, 2b, 3b and 3c) were sprayed with Dragendroff’s reagent and the mobile phase are 96:4, 93:7 and 90:10 v/v of CH2Cl2 and MeOH.

Figure 3. (a) The microbial transformations of 1a, 2a, 3a and 4 using Cunninghamella elegans and (b) 5 (the C-6 epimer of 1b).
Table 1. 1H and 13C NMR spectroscopic data of compounds 1b–3b in CDCl3.
The biotransformation of 2a gave compound 2b in a yield of 52% after a simple work up by solvent extraction (). Compound 2b was isolated as a light yellow gum; Rf 0.30 (CH2Cl2-MeOH, 93:7); [α]31D +120 (c 0.30, MeOH); HRMS (ESI + ve, m/z [M + H+], found 420.2006, calcd for C22H29NO7 found: 420.2005); 1H NMR data (CDCl3, 400 MHz) and 13C NMR data (CDCl3, 100 MHz) are shown in .
All NMR data, HRMS spectrum and the 1D and 2D NMR spectra of 2b are shown in supplemental data (Table S2 and Figures S6-S11).
The biotransformation of 3a gave compound 3b in a yield of 21% and the known compound 1′, 2′-didehydrostemofoline-N-oxide (3c) in a yield of 48% (). Compound 3b was isolated as a yellow gum; Rf 0.28 (CH2Cl2-MeOH, 96:4); [α]31D +75 (c 0.20, MeOH); HRMS (ESI + ve, m/z [M + H+], found 402.1934, calcd for C22H27NO6 found: 402.1933); 1H NMR data (CDCl3, 400 MHz) and 13C NMR data (CDCl3, 100 MHz) are shown in . All NMR data, HRMS spectrum and the 1D and 2D NMR specra of 3b are shown in supplemental data (Table S3 and Figures S12-S17). The known compound 3c was isolated as a yellow brown gum; Rf 0.14 (CH2Cl2-MeOH, 96:4). 1H NMR (CDCl3, 400 MHz) and 13C NMR data (CDCl3, 100 MHz) were consistent with those previously reported [Citation19].
The precursor compounds (1a, 2a and 3a) and their biotransformed products (1b, 2b, 3b and 3c) were evaluated for their AChE (from electric eel) inhibitory activities. The results are shown in . The transformed product 1b was four times more potent (IC50 = 11.0 ± 1.49 µM) than its precursor 1a (IC50 = 45.1 ± 5.46 µM) as an inhibitor against acetylcholinesterase. Whereas, 2b was two times more potent (IC50 = 40.4 ± 4.17 µM) than its starting material 2a (IC50 = 90.4 ± 3.15 µM). However, 3c (IC50 = 41.6 ± 4.42 µM) showed a similar activity to 3a (IC50 = 44.6 ± 3.89 µM), while 3b (IC50 = 178 ± 14.55 µM) showed reduced AChE activity when compared to its starting material 3a.
Table 2. The IC50 values (µM) for acetylcholinesterase inhibitory of activities of the starting materials (1a, 2a and 3a) and biotransformation products (1b, 2b, 3b and 3c).
Discussion
The structures of the transformed products 1b, 2b, 3b and 3c were identified using NMR spectroscopy and mass spectrometry with comparisons of the data made with the starting materials 1a, 2a and 3a. When the NMR spectroscopic data of 1b were compared to those of stemofoline (1a), the chemical shifts of the resonances for H-6/C-6 [δH 4.29 (m, 1H)/ δC 73.0 (CH)] and H2-5/C-5 [δH 3.09 (m, 1H); 3.47 (m, 1H)/ δC 58.7 (CH2)] were significantly deshielded (), consistent with hydroxylation of 1a having occurred at C-6. Interestingly, (6S)-hydroxystemofoline (5), the C-6 epimer of 1b (), has been isolated from the extracts of the leaves and stems of Stemona japonica [Citation20] which had similar, but not the same NMR spectroscopic data as 1b (). For example, in the 1H NMR spectrum of 1b, the H-9 resonance (δH 1.52) was shielded relative to that found in 1a (δH 1.88), while this resonance was significantly deshielded in 5 (δH 2.80) (). The chemical shift in the latter spectrum was consistent with the exo-configuration of the C-6 hydroxy group and thus the originally assigned (6S)-configuration of 5. Furthermore, the 13C NMR chemical shifts for C-7 in compounds 1b (δC 58.4) and 5 (δC 53.7) were also significantly different, consistent with their different configurations at C-6. While the ROESY spectrum of 5 showed a correlation between H-6 and H2-1′ of the n-butyl C-3 side chain (), the NOESY spectrum of 1b showed a correlation between H-6 and H-9 which indicated the endo-configuration of the C-6 hydroxy group and thus the (6 R)-configuration. This key correlation was consistent with a molecular modelling study of 1b () which indicated the distance between H-6 and H-9 was 2.1 Å. Thus 1b and 5 are C-6 epimers. The (6 R)-configuration of 1b may be expected as hydroxylation of 1a would most likely occur from the less hindered (exo approach of “oxygen”) side of the caged ring structure. Perhaps the C-6 configuration of natural product 5 arises from reduction of hypothetical 6-oxostemofoline from the sterically less hindered exo-face. The biotransformation of 2a, according to the 1 D and 2 D NMR spectroscopic data revealed that hydroxylation had occurred at the C-6 position of compound 2a. The oxymethine carbon resonance at δC 72.7 (C-6) was observed in the 13C NMR and DETP135 spectra. The position of this new hydroxy group was evident from the HMBC correlations of H-6 to C-5 and the COSY correlations between the resonance at δH 3.22 (H-5) and δH 4.31 (H-6). The (6 R)-configuration of 2b was assigned from the NOESY correlation between H-6 and H-9 which indicated the OH group had also been delivered to the less hindered exo-position of the pyrrolidine ring of 2a. Thus, the structure of 2b was elucidated as (2′S,6R)-dihydroxystemofoline. The biotransformation of 3a gave compound 3b and 1′,2′-didehydrostemofoline-N-oxide (3c). The 1H NMR spectroscopic data of 3b revealed an additional low-field signal at δH 4.22 (H-6). While a new oxymethine carbon resonance at δC 73.7 (C-6) was observed in the 13C NMR and DETP135 spectra. The position of this new hydroxy group was evident from the HMBC correlations of H-6 to C-5. The (6 R)-configuration of 3b was assigned from the NOESY correlation between H-6 and H-9 which indicated the OH group had been delivered to the less hindered exo-position of the pyrrolidine ring of 3a. Based on these data, the structure of 3b was determined as (11Z, 6 R)-1′,2′-didehydro-6-hydroxystemofoline. While the structure of the known compound 3c was identified by comparing its 1H and 13C NMR spectroscopic data with those reported [Citation19]. It was significant to note that the biohydroxylation reactions of the stemofolines alkaloids (1a, 2a and 3a) occurred at a relatively unactivated site (C-6) on these caged substrates rather than at more chemically activated sites (for example, sites α to N or O or at an allylic position). Thus indicating hydroxylation is specifically directed at C-6 by substrate orientation in the enzyme active site. Interestingly, in plants the pyrido[1,2-a]azepine alkaloid stemocurtisine (4) gives rise to other natural products most likely via hydroxylation and ring opening reactions of the azepine ring at C-6 [Citation21,Citation22]. However, it was found to be inert to biotransformation by C. elegans. The stemofoline alkaloids 1a-3a have a common caged heterocyclic ring system which is clearly recognized by the hydroxylation enzyme and undergoes hydroxylation at C-6. It appears that stemocurtisine having a different pyrido[1,2-a]azepine core structure is not recognized by this enzyme and does not undergo the biotransformation process.
Figure 4. Molecular model of compound 1b generated in SPATAN 18 using DFT calculations (ωB97X-D/6-31G*) of the equilibrium geometry (gas phase).
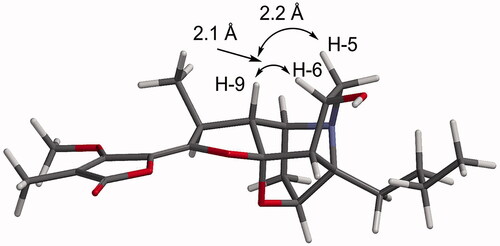
Table 3. Selected 1H and 13C NMR spectroscopic data of compounds 1a, 1b and 5 in CDCl3.
The results of this research demonstrated that the fungus C. elegans TISTR3370 can metabolize compounds 1a, 2a and 3a via liquid-state fermentation. The biotransformed compounds 1b, 2b and 3b were produced by hydroxylation at the methylene carbon C-6 of 1a, 2a and 3a, respectively, while direct oxidation of the nitrogen of 1c afforded the N-oxide of 3c. The biotransformation reaction of 1c to 3c is similar to the biotransformation reaction of the Cinchona alkaloids by the endophytic Xylaria sp. which produced N-oxide derivatives [Citation23]. Moreover, the hydroxylated metabolites may be formed from a cytochrome P450 (CYP450) mediated process. The CYP450 enzyme superfamily is one of the most important groups of metabolising enzymes responsible for metabolising drugs. The reactions catalysed by CYP450 include aliphatic and aromatic carbon hydroxylations, N-, O- and S- dealkylations, via an initial hydroxylated intermediate, as well as the oxidations of heteroatoms [Citation24].
The results of AChE inhibition studies of the stemofoline alkaloids and their transformed products indicated that the transformed products 1b and 2b, which were obtained from the oxidative biotransformations of 1a and 2a, had enhanced AChE inhibitory activities over their respective precursors. These increased AChE inhibitory activities could indicate that the extra hydroxy group at C6 may bind in the active site of AChE through H-bonding similar to that found in galantamine as described by Felipe et al. [Citation25]. In contrast compounds 3b and 3c, showed no increase in inhibitory activities over their respective precursors. In the case of compound 3c, this may be due to the more rigid alkene side chain at C-3 which may inhibit H-bonding of the C-6 hydroxy group.
Conclusions
This research resulted in the successful application of the microbial transformation process for the modification of the stemofoline alkaloids 1a, 2a and 3a using C. elegans TISTR3370 to produce the oxidized compounds; 1b, 2b, 3b, and 3c. The process was carried out under mild conditions, with high selectivity. In contrast, the pyrido[1,2-a]azepine alkaloid stemocurtisine (4) was not transformed under this process, most likely due to its different heterocyclic ring structure. The AChE inhibitory activity of the substrates and their transformed products was investigated revealing that the C-hydroxylation products 1b and 2b were more active inhibitors than their corresponding substrates. Whereas, the C-hydroxylation product 3b and the N-oxidation product 3c, that contain an alkene side chain, had very similar AChE inhibitory activities to their precursor substrates.
Supplemental Material
Download MS Word (3.2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The supporting data in this research is included within the article, and the data are available from the corresponding author upon request.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Pilli RA, Rosso GB, de Oliveira M. d C F. The chemistry of Stemona alkaloids: an update. Nat Prod Rep. 2010;27(12):1908–1937.
- Greger H. Structural classification and biological activities of Stemona alkaloids. Phytochem Rev. 2019;18(2):463–493.
- Brem B, Seger C, Pacher T, et al. Feeding deterrence and contact toxicity of Stemona alkaloids-a source of potent natural insecticides. J Agric Food Chem. 2002;50(22):6383–6388.
- Jiwajinda S, Hirai N, Watanabe K, et al. Occurence of the insecticidal 16,17-didehydro-16(E)-stemofoline in Stemona collinsae. Phytochemistry. 2001;56(7):693–695.
- Mungkornasawakul P, Pyne SG, Jatisatienr A, et al. Stemocurtisine, the first pyrido[1,2-a]azapine Stemona alkaloid. J Nat Prod. 2003;66(7):980–982.
- Chaiyong S, Jatisatienr A, Mungkornasawakul P, et al. Phytochemical investigations of Stemona curtisii and synthetic studies on stemocurtisine alkaloids. J Nat Prod. 2010;73(11):1833–1838.
- Kongkiatpaiboon S, Mikulicic S, Keeratinijakal V, et al. HPLC simultaneous analysis for quality assessment of Stemona curtisii roots and determination oftheir insecticidal activities. Ind Crops Prod. 2013;43:648–653.
- Abraham WR, Spassov G. Biotransformation of alkaloids: a challenge. Heterocycles. 2002;56(1–2):711–741.
- Devkota KP, Choudhary MI, Nawaz SA, et al. Microbial transformation of the steroidal alkaloid dictyophlebine by Rhizopus stolonifer. Chem Pharm Bull. 2007;55(4):682–684.
- Lu YF, Chen KY, Li HL, et al. Biotransformation of vermitaline by Cunninghamella echinulata. Helv Chim Acta. 2008;91(5):819–824.
- Dubey KK, Haque S, Jawed A, et al. Construction of recombinant E. coli for enhanced bioconversion of colchicine into 3-demethylated colchicine at 70-l bioreactor level. Process Biochem. 2010;45(7):1036–1042.,
- Dubey KK, Behera BK. Statistical optimization of process variables for the production of an anticancer drug (colchicine derivatives) through fermentation: at scale-up level. N Biotechnol. 2011;28(1):79–85.
- Dubey KK, Jawed A, Haque S. Enhanced bioconversion of colchicine by whole cell immobilization of recombinant E. coli harboring P450 BM-3 gene. Process Biochem. 2013;48(8):1151–1158.
- Chang JC, Daniel RD, Carl EC. Biotransformation of malachite green by the fungus Cunninghamella elegans. Appl Environ Microbiol. 2001;67(9):4358–4360.
- Jessica A, Katherine G, Cormac DM. Biotransformation of flurbiprofen by Cunninghamella Species. Appl Environ Microbiol. 2010;76(18):6299–6303.
- Moody JD, Zhang D, Heinze TM, et al. Transformation of amoxapine by Cunninghamella elegans. Appl Environ Microbiol P. 2000;66(8):3646–3649.
- Ellman GL, Courtney KD, Andres V, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95.
- Ingkaninan K, Temkitthawon P, Chuenchom K, et al. Screening for acetylcholinesterase inhibitory activity in plants used in Thai traditional rejuvenating and neurotonic remedies. J Ethnopharmacol. 2003;89(2–3):261–264.
- Sastraruji T, Jatisatienr A, Pyne SG, et al. Phytochemical studies on Stemona plants: isolation of stemofoline alkaloids. J Nat Prod. 2005;68(12):1763–1767.
- Tang CP, Chen T, Velten R, et al. Alkaloids from stems and leaves of Stemona japonica and their insecticidal activities. J Nat Prod. 2008;71(1):112–116.
- Chalom S, Panyakaew J, Phaya M, et al. Cytotoxic and larvicidal activities of Stemona alkaloids from the aerial parts and roots of Stemona curtisii Hook.f. Nat Prod Res. 2020. InPress.Doi:https://doi.org/10.1080/14786419.2019.1709188
- Mungkornasawakul P, Pyne SG, Willis AC, et al. 6-Hydroxy-5,6-seco-stemocurtisine: a novel seco-stemocurtisine-type alkaloid. Phytochemistry Lett. 2013;6(4):602–605.
- Hirotaka S, Chinami K, Shoji M, et al. Transformation of cinchona alkaloids into 1-N-oxide derivatives by endophytic Xylaria sp. Isolated from Cinchona pubescens. Chem Pharm Bull. 2003;51(1):71–74.
- Nelson DR, Kamataki T, Waxman DJ, et al. The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes, and nomenclature. DNA Cell Biol. 1993;12(1):1–51.
- Felipe MN, ClaudiaJara RG, PatricioIturriaga-Vásquez HV, et al. Rhodolirium andicola: a new renewable source of alkaloids with acetylcholinesterase inhibitory activity, a study from natureto molecular docking. Revista Brasileira de Farmacognosia. 2018;28(1):34–43.