
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
To date, several Glucosyltransferase C (GtfC) inhibitors have been identified and experimentally validated. All these inhibitors have been validated at different experimental conditions like degree of purity, animal models, kinetic conditions, experimental environment etc.; and most of these inhibitors (ligands) proved to be quite effective in their respective validation environment. However, due to varied experimental validation conditions, and absence of molecular interaction data, there is no way to prioritize these validated ligands for their inhibition potential against GtfC. The present study is a novel attempt of comparative evaluation of the interaction of the validated ligands on a single platform and under similar conditions with a dual objective, i.e. ligand prioritization for their respective inhibitory potential and elucidation of the involved unknown molecular interactions. Carbohydrate derivatives (6-Deoxy sucrose and Trichloro-galactosucrose) were identified as the most promising GtfC inhibitors. In addition, Asp588, Trp517, and Asn481 amino acid residues of the domain A1 proved vital for the inhibitory effect. The study highlights the importance of the comparative analysis of the validated ligands in order to identify the most promising leads for drug discovery against dental caries.
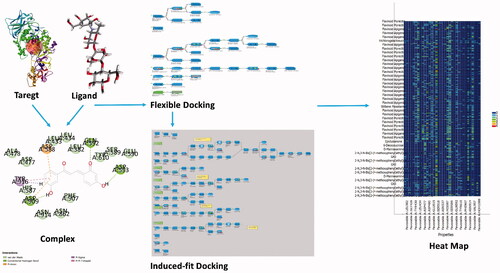
Introduction
Biofilm exopolysaccharides (EPS also termed as glucan) are responsible for the growth and colonization of the cariogenic bacteria. Several Streptococci strains including S. mutans are capable of synthesizing EPS. Streptococci EPS are either homopolysaccharides or heteropolysaccharides. Streptococcus mutans secretes an EPS, which is a homopolysaccharide [Citation1]. Glucosyltransferases (also known as Glycosyltransferases Gtfs; i.e. GtfB, GtfC and GtfD) are known to play central role in biofilm EPS (glucan) generation [Citation2–5]. Streptococcus mutans synthesizes at the minimum 3 genetically different Gtfs, and each of them forms a structurally discrete glucan by using sucrose. The special features of the structural and functional organization of Gtfs have been reported earlier [Citation6,Citation7]. GtfB (generally known as GtfI) produces firstly an insoluble glucan with plenty of 1,3-linkages. Whereas, GtfD (GtfS) produces primarily a soluble glucan [Citation8–10]. Unlike above two Gtfs, GtfC (GtfSI) synthesizes an amalgam of insoluble and soluble (having more α-1,6-linkages) forms of glucans. The reaction showing glucan generation catalyzed by GtfC has been presented in . Earlier, Ooshima et al. [Citation12] reported that the presence of GtfC intensifies the glucan formation in vitro model, which was later endorsed by the report of Tamesada et al. [Citation13]. GtfC is adsorbed into pellicle, where – when exposed to sucrose, and under in situ condition it synthesizes glucan.
Figure 1. GtfC reaction scheme (Brenda enzyme database) [Citation11].
![Figure 1. GtfC reaction scheme (Brenda enzyme database) [Citation11].](/cms/asset/a158be20-b9a0-43bd-b4de-f4db798870ad/ianb_a_1903021_f0001_c.jpg)
The data generated from in situ and in vitro experimental studies suggest that Glucosyltransferase identified in pellicle is predominantly a GtfC from S. mutans (Venkitaraman et al., 1995) [Citation14]. In addition to the above mentioned significant role of GtfC in EPS (glucan) generation, Tsumori and Kuramitsu [Citation15] identified the essential role of GtfC in sucrose‐dependent colonization of tooth surfaces. Knocking out of the GtfC gene (while keeping GtfB and GtfD) radically reduced the adherence to smooth surfaces in quantitative sucrose dependent attachment system. GtfC alone was capable of catalyzing (enough sucrose to EPS) required for adherence to smooth surface and subsequent colonization [Citation15]. These studies identify the unique and central role of GtfC in biofilm (EPS) generation. Sine GtfC is vital for biofilm generation determination and characterization of potential leads against GtfC is a significant step towards potent drug discovery against biofilm generation and associated diseases. Present study attempts to characterize the potent leads against the biofilm drug target, GtfC.
There are at least 2 subsites, i.e. glucosyl and fructosyl available on GtfC that might be the target for inhibitors [Citation16–18]. The site includes 3 aspartic acid residues that has been discovered as the location for glucose conjugation. Whereas, Zn2+ has been shown to bind to the fructose site [Citation19]. Also, the enzyme has numerous separate/unrelated glucan-binding sites, including one present on a 60kD domain of the C-terminus [Citation20]. Distinctly, there are some additional sites present on Gtfs to which potential inhibitors could attach. Various chemical compounds have been used to inhibit GtfC. Initially, quaternary ammonium compounds, aliphatic amines and antibodies were found efficacious in inhibiting Gtfs to a certain extent in the solution.
Later, oleanic and oleic acids were found potential inhibitors of Gtfs activity in a solution of perborate, metaperiodate, sodium, metal ions and naturally available substances [Citation21–23]. The activity of GtfB in a solution is drastically affected (reduced) by many commercially available agents, for e.g. alexidine dihydrochloride, cetylpyridinium chloride or triclosan. 4′,5,7-trihydroxyflavone (commonly known as Apigenin) is extremely efficient inhibitor (non-competitive inhibition) of Gtfs even at the concentrations of micromoles. Interestingly, only few agents had any effect on GtfC. GtfC has several hydrophobic domains especially at the C-terminus [Citation24] and these hydrophobic domains possibly block the access through hydrophilic inhibitors to the active site that may describe its comparative resistance towards inhibitory agents.
In this study, an advanced Quantum Mechanical Molecular Mechanical (QM/MM) technique has been employed for the optimization of the conformations of the potential leads as many studies published in recent times have shown that there are significant advantages of QM/MM methods compared to other empirical molecular mechanics techniques [Citation25].
Structural details of GtfC
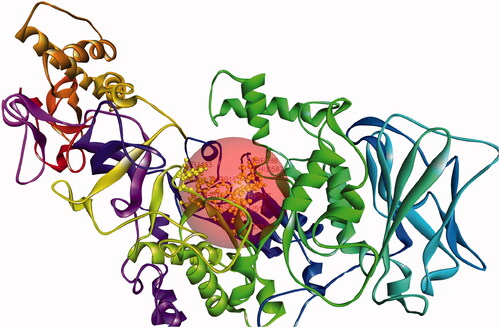
In the crystal, GtfC is organized as tetramer, the outcomes of size-exclusion chromatography suggested that GtfC is probably a monomer in a solution (). Also, the structures of the individual tetramer members were found nearly identical. The present model refined at 2.1Å resolution comprises amino acid residues 244–1087, as the entire protein could not be crystallized. The structure of GtfC encompasses four different domains, i.e. A, B, C and IV, and except domain C, each domain was made up of two separate regions of the polypeptide chain. Domain IV consisted of IV1 (residues 244–372) and IV2 (residues 1057–1087); domain A consisted of A1 (residues 443–692) and A2 (residues 829–961); and domain B consisted of B1 (residues 373–442) and B2 (residues 962–1056). Domain C was formed of a single stretch of amino acid (693–828) residues. Domain A composed the core of the GTF-SI catalytic domains [Citation19].
Figure 2. GtfC monomer (A-chain) with active site.
Materials and methods
Structural details of validated ligands/inhibitors of GtfC
The ligands/inhibitors subjected to prioritization during this study are presented in . All the ligands were derived from references provided in . The structural details of these inhibitors are provided in the Supplementary Information (Figure SI L1–L15). The structural .sdf files of these inhibitors were obtained from PubChem database. Some structures (Flavonoid new, G16, G43 and Flavonoid (Z)-3-(2,4-Dihydroxyphenyl)-1-(3-hydroxyphenyl)-2-propen-1-one) were designed in Discovery Studio Visualizer. IUPAC names were generated using BIOVIA Draw, 2019. All the ligands were prepared following the ligand protocol of BIOVIA Discovery Studio (Accelrys@). The ligands were typed with CHARMM36 force field [Citation32].
Table 1. Details of the validated ligands: the compound name refers to the compound used in the respective study.
Target structure and active site
The structural coordinates of GtfC was retrieved from RCSB-PDB (3AIC). This .pdb file represents the crystal structure of Glucansucrase from Streptococcus mutans in complex with D-Acarbose [Citation19]. This structure was deciphered by x-ray diffraction @3.11 Å resolution. The native state of GtfC is known to be a monomer [Citation19]. A GtfC monomer complexed with alpha D-Acarbose, 2-(n-morpholino)-ethanesulfonic acid and calcium was selected from the crystallised octamer .pdb file. The target structure was free of ligands (D-Acarbose and 2-(n-morpholino)-ethanesulfonic acid) but include the water molecules found to be linked with the crystal structure. The target GtfC monomer was prepared according to the protein preparation protocol of BIOVIA Discovery Studio. The input parameters have been given in the Supplementary Information (SI) file. The target protein monomer was typed with CHARMM36 force field [Citation32]. The active site was defined based on pdb site record (). Includes Glu515, Asp477, Asp588, Arg475, His587, Tyr916, Tyr430, Leu433, Asn481, and Trp517.
Customized flexible docking protocol
An exhaustive flexible docking protocol was implemented (Supplementary Information: Figure SI 1) on BIOVIA Discovery Studio platform. Briefly, this protocol permits for receptor flexibility during the docking of flexible ligands up to some extent [Citation33]. The side-chains of specified amino acids (in this case Glu515, Asp477, Asp588, and Asn481) were allowed to move during the docking process. This movement allows the receptor to adapt to different ligands in an induced-fit model (Supplementary Information: Figure SI 2). The protocol employs a combination of components from other protocols to perform the docking, and is based on methods within CHARMm [Citation34] to sample side-chain and ligand conformations. The detailed input parameters are given in Supplementary Information (SI) file. Flexible docking protocol performs the following steps: (i) Calculation of receptor side-chain conformations: Initially, the protocol creates protein side-chain conformations using the ChiFlex algorithm [Citation35], (ii) Create ligand conformations: Low energy ligand conformations are created for the docking process using Generate Conformations protocol of BIOVIA Discovery Studio platform, (iii) Perform initial placement of the ligand conformations: The ligand conformations are docked into the active site of each receptor side-chain conformation using LibDock [Citation36], (iv) Clustering to remove similar ligand poses: Poses are clustered irrespective of the protein conformation as the protein conformations are rebuilt during the next step, (v) Refine side-chains: In the presence of the ligand, the specified receptor side-chain residues are refined using the ChiRotor algorithm [Citation35], and (vi) Refine docking: A final simulated annealing and energy minimization of each ligand pose was done by using CDOCKER [Citation37]
Scoring ligand poses
BIOVIA Discovery Studio implementation of the empirical scoring functions LigScore1, LigScore2 [Citation38] was used to score the ligand poses including the reference ligand (D-Acarbose). Table SI1 represents detailed scores of the ligands on the two scales (Supplementary Information)
ADMET properties calculation
ADMET properties were calculated on Discovery studio using ADMET Descriptor predictor tool. Table SI3 and SI3E (supplementary Information) represents the summarized ADMET properties of the Ligands.
Results
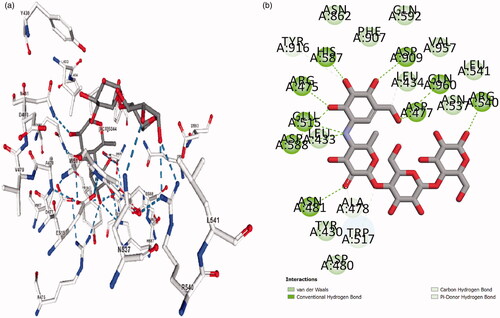
The active site of GtfC () was derived from the pseudo-tetrasaccharide, αD-Acarbose, binding site of the crystallized GtfC-α D-Acarbose complex (Active site BC8: A chain, 3AIC.pdb). αD-Acarbose was removed from the complex and subjected to same docking protocol (as discussed above, ) while keeping the crystallized (3AIC.pdb) protein conformation as constant. compares the interaction profile of αD-Acarbose in the crystallized complex (3AIC) with highest scoring docked pose of αD-Acarbose at the active site. The root mean square deviation (RMSD) of the redocked highest scoring acarbose pose was determined by aligning it to the bound D-acarbose in the experimental PDB structure (3AIC). A low RMSD value of 0.6536 for the redocked D-acarbose underscores the efficiency of the applied docking protocol.
Figure 3. Interaction profile of GtfC-D-Acarbose. (a) From 3AIC.pdb structure. (b) Docked complex.
The binding efficiency, of the docked poses in the order of their inhibitory potential, determined as per the scoring algorithm (Scoring ligand poses as discussed above) is listed in . The binding score and interaction profile of αD-Acarbose in complex with GtfC serve as reference (, ). represents interacting chemical fragments of the top 3 Ligands w.r.t Acarbose. Table SI2 represents interacting chemical fragments of the rest of the ligands w.r.t Acarbose (Supplementary Information). The ADMET properties of all the ligands discussed in this study are reported in Tables SI3 and SI3E (Supplementary Information).
Table 2. Summary of ligand interactions.
Table 3. Summary of Interacting chemical fragments.
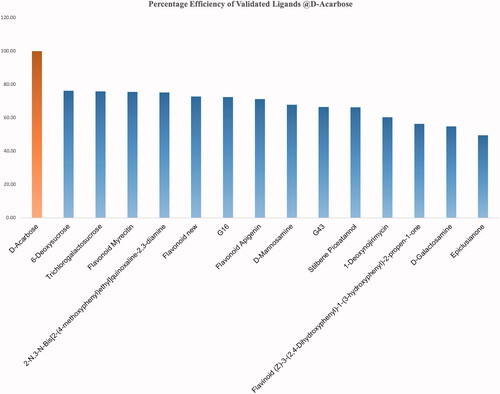
A uniform scoring algorithm, Score ligand poses, was applied to both the reference (αD-Acarbose) and the selected ligands. and Citation3 summarizes the interaction profile of the top scoring ligand pose representing a ligand group (ligand group represents all the docked poses of a single ligand) along with Average ligand Interaction Score (ALIS). summarizes the percentage efficiency of the validated ligands @αD-Acarbose. None of the validated ligands could match the inhibitory potential of αD-Acarbose.
Figure 4. Percentage efficiency of the validated ligands @αD-Acarbose.
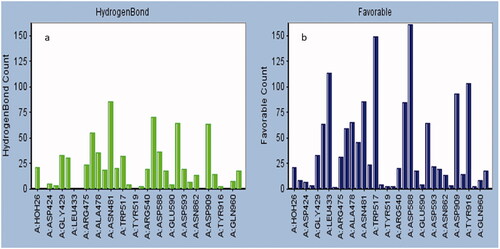
The detailed interaction profile of the top four validated ligands having binding efficiency within 75%@αD-Acarbose is summarized in . The interaction profile of the rest of the ligands is shown in the Supplementary Information (Figure SI C1–C10). The amino acid residues interacting with the ligands were classified on the basis of the type and frequency of the interaction. All the ligand poses were analyzed to determine the amino acid residues frequently involved in hydrogen bonding (). The amino acid residues responsible for maximum favourable interactions are summarized in and also as favourable heat map in the Supplementary Information (Figure SI H1) file. The amino acids playing central role in hydrophobic and ionic interactions are summarized in the histogram (Figure SI AI, and Figure SI AH) in Supplementary Information.
Figure 5. GtfC-interaction profile. (a) 6-Deoxysucrose, (b) trichloro-galactosucrose, (c) flavonoid myrecitin, (d) 2-N,3-N-Bis(2-(4-methoxyphenyl) ethyl) quinoxaline-2,3-diamine.
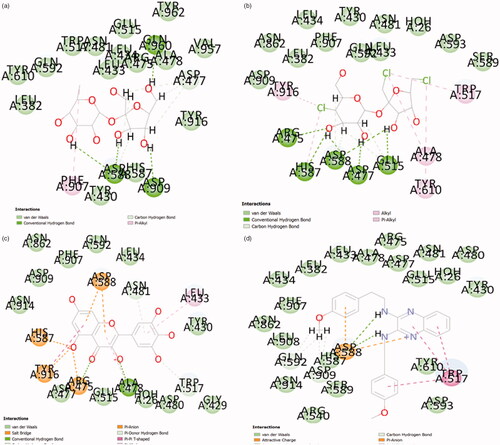
Figure 6. Frequently interacting residues. (a) Hydrogen bonding; (b) favourable count.
Discussion
GtfC is an established drug target; chemically diverse inhibitors have been tested/validated on GtfC from Streptococcus mutans in recent years. These inhibitors may be broadly classified as oligosaccharide derivatives (D-Acarbose, 6-Deoxysucrose and Trichloro-galactosucrose), Amino-monosaccharide derivatives (Galactosamine, Mannosamine), Flavonoids (Apigenin, Myrecitin, Flavonoid new: 6Z)-3-hydroxy-6-((Z)-3-hydroxy-3-(3-hydroxyphenyl)prop-2-enylidene)cyclohexa-2,4-dien-1-on, Flavonoid: (Z)-3-(2,4-Dihydroxyphenyl)-1-(3-hydroxyphenyl)-2-propen-1-one), Hydroxylated phenyls (Stilbene), Quinolaxine diamine deivatives (2-N,3-N-Bis(2-(4-methoxyphenyl)ethyl)quinoxaline-2,3-diamine), Prenyl benzophenone (Epiclusianone), Heterocyclic amine polyhydroxylated derivatives (Deoxynojirimycin), Carboximidic acid derivative (G43: (2Z)-N-(2-carbamoylphenyl)-5-nitro-benzothiophene-2-carboximidic acid), Pheny methyl fluoro cyclohexanes (G16:6E)-3-(dihydroxymethyl)-6-(5-(4-((4-fluorophenyl)-hydroxy-methylene)cyclohexa-2,5-dien-1-ylidene)-4-phenyl-imidazol-2-ylidene)cyclohexa-2,4-dien-1-one) etc.
These inhibitors have been tested or validated either as impure extracts or under diverse experimental conditions (). Owing to the dearth of uniform experimental conditions it is impossible to prioritize these validated ligands with respect to (w.r.t) each other. The knowledge of the efficiency of inhibitors w.r.t each other assist in the discovery of the specific pharmacophore properties, which will determine the foundation for screening exhaustive ligand libraries for superior leads. Also, the majority of the validation studies performed for these inhibitors failed to report any information regarding the type and degree of chemical interaction between the ligand and the receptor (i.e. target protein). Present study is a comprehensive analysis of validated ligands of GtfC. This study compares the efficiency of the active ligands under uniform experimental conditions and to characterize and explore their interaction. The binding efficiency and interaction details of the reference ligand D-Acarbose (from GtfC-D-Acarbose crystallized complex:3AIC) was found to be highest when compared with any of the validated ligands considered in this study (ALIS = 7.66, and Citation3]. No unfavourable interactions were absorbed alongside high degree of hydrogen bonding and other interactions (). Amino acids at GtfC subsite-1 (Arg475, Asp477, Glu515, His587, Asp588) establish hydrogen bonds with oxygen and nitrogen atoms of D-Acarbose, Leu434, Trp517 and Tyr916 are involved in weak van der Waals, carbon hydrogen and Pi-Donor hydrogen bond with Acarbose. The inhibition efficiency of D-Acarbose may be specifically attributed to its stable interaction with Glu515, Asp477, Asp588, Leu433, Trp517, and Asn481. For the natural substrate (sucrose), Asp477 undergoes a nucleophilic substitution reaction thereby establishing a covalent interaction with the positively charged glycosyl moiety resulting in a stable intermediate. Glu 515 placed above the substrate protonates the leaving group, fructose, at subsite + 1 [Citation19]. Asp588 further stabilizes the covalently bonded Asp477-glycosyl intermediate. The glutamate anion (Glu515) attracts out the proton from the second sucrose molecule at Subsite +1, generating a hydroxyl anion attacking the glucosyl intermediate producing glucan at Subsite −1. D-Acarbose is involved in stable hydrogen bonding and weak Pi-Donor hydrogen bonding with Leu433 and Trp517, respectively at Subsite +1. Since, Ca2+ binding site Asn481 side chain hydrogen bonding with the acceptor maltose/sucrose plays a key role in the stabilization of the acceptor [Citation19], hydrogen bonding of D-Acarbose with the Asn481 side chain defines its inhibitory potential. 6-Deoxysucrose was found to be the most efficient ligand (, ALIS score: 5.84, %Efficiency@D-Acarbose: 76.24, , ). The inhibitory effect of this ligand may be attributed to its multiple hydrogen bonding with Asp588 and highly conserved Asp909 [Citation19] and multiple weak van der Waals and carbon hydrogen interactions with Asp477 at Subsite −1 (). Trichloro-galactosucrose appear to be establish an almost equally stable interaction with GtfC (, ALIS score: 5.81, %Efficiency@D-Acarbose: 75.88, , ). This halogenated disaccharide appears to be the closest analogue of the natural substrate (i.e. sucrose). Multiple hydrogen bonding with the Subsite −1 amino acids including Glu515, Asp477, Arg475, His587, and Asp588 along with weak Pi-alkyl interactions with Tyr916 and Trp517 (at Subsite +1) stabilizes the inhibition complex of GtfC-Trichloro-galactosucrose. The inhibitory potential of the Flavonoid Myrecitin appears to closely follow Tricloro-galactosucrose. The stability of Myrecitin-GtfC complex may be especially ascribed to its multiple salt bridge and Pi-Anion interactions with Subsite −1 amino acids: Asp588, Arg475, His587 and Tyr916 (, ALIS score: 5.79, %Efficiency@D-Acarbose: 75.79, , ). Weak van der Waals and Pi-alkyl interactions are also observed with Asn481 and Subsite +1 (Leu433 and Trp517). A quinolaxine diamine derivative also exhibits an inhibitory effect >¾ of D-Acarbose (, ALIS score: 5.75, %Efficiency@D-Acarbose: 75.10, , Table SI2). The stability of diamine derivative-GtfC complex is based on multiple interaction with Asp588 and Trp517. The Aspartate anion is attracted towards the aromatic rings of the quinoxaline diamine derivatives due to Pi-Anion interactions and possible hydrogen bonding amino hydrogen atoms. Trp517 undergoes Pi-Pi T shaped interactions with the aromatic rings of the ligand. The ADMET properties of the top three Ligands (6-Deoxysucrose, Trichlorogalactosucrose and Flavonoid Myrecitin) have favourable aqueous solubility, low blood brain barrier penetration, almost null Cytochrome P450 Inhihibitory potential, almost zero hepatotoxicity, null plasma protein binding and moderate to poor Intestinal absorption. The inhibitory potential of the rest of the inhibitors is <¾ of D-Acarbose (, Supplementary Information: Figure SI-6a–14a, Table SI2). The abstraction of the collective interaction of all the validated ligands considered in this study reveals the identity of the critically placed amino acid residues responsible for the inhibitory effect of the ligands. The amino acids residues at GtfC Subsite −1 (Arg475, 7, Glu515, His587, Asp588, Tyr 916), Subsite +1 (Leu433, Trp517) and Ca2+ binding site Asn481 were observed to have most frequent favourable interaction (including hydrogen bonding) with the validated ligands. Subsite −1, Asp588, and subsite +1, Trp517, were observed to have the highest count of favourable interaction. Asp588 and Trp517 were also found to have highest counts of charged and hydrophobic interactions (Supplementary Information: Figure SI AI and Figure SI AH). Asp 588, Trp517 and Asn481 belong to the domain A1 of GtfC [Citation19]. Since, these amino acids play a vital role in the catalysis of the natural substrate (sucrose), any favourable interaction (including hydrogen) successfully interrupts its native catalytic role (discussed above) thereby inhibiting the enzyme.
Conclusion
The present study successfully prioritizes and characterizes the binding efficiency of the validated ligands. This study also focuses on the chemical interactions specifically responsible for the inhibitory effect of the potent inhibitors. Structural subdomain of GtfC vital for the inhibitory effect of were revealed. The structural interaction details shared in this study may be employed to determine better suited leads for combating biofilm generation and associated diseases of the oral cavity.
Author contributions
Conceived and designed the study and experiments: HAH SK MK PS MAM RKM SH. Performed the experiments: HAH SK MK PS MAM RKM SH. Analysed the data: HAH SK MK PS. Contributed reagents/materials/analysis tools: MAM RKM SH. Wrote the paper: SK MK MAM SH. All authors reviewed the manuscript.
ianb_a_1903021_sm8665.pdf
Download PDF (2.6 MB)Acknowledgements
The authors are grateful to Hail University, Saudi Arabia for providing the access of the Saudi Digital Library and other software related help for this research study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All the data required is available in supplementary Information and on request.
Additional information
Funding
References
- Nishimura J, Saito T, Yoneyama H, et al. Biofilm formation by Streptococcus mutans and related bacteria. AiM. 2012; 02(03):208–215.
- Bowen WH, Koo H. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 2011;45(1):69–86.
- Lis M, Kuramitsu HK. The stress-responsive dgk gene from Streptococcus mutans encodes a putative undecaprenol kinase activity. Infect Immun. 2003;71(4):1938–1943.
- Matsumoto-Nakano M. Role of Streptococcus mutans surface proteins for biofilm formation. Jpn Dent Sci Rev. 2018;54(1):22–29.
- Yoshida A, Kuramitsu HK. Multiple Streptococcus mutans genes are involved in biofilm formation. Appl Environ Microbiol. 2002;68(12):6283–6291.
- Monchois V, Arguello-Morales M, Russell RR. Isolation of an active catalytic core of Streptococcus downei MFe28 GTF-I glucosyltransferase. J Bacteriol. 1999;181(7):2290–2292.
- Van Hijum SA, Kralj S, Ozimek LK, et al. Structure-function relationships of glucansucrase and fructansucrase enzymes from lactic acid bacteria. Microbiol Mol Biol Rev. 2006;70(1):157–176.
- Aoki H, Shiroza T, Hayakawa M, et al. Cloning of a Streptococcus mutans glucosyltransferase gene coding for insoluble glucan synthesis. Infect Immun. 1986;53(3):587–594.
- Hanada N, Kuramitsu HK. Isolation and characterization of the Streptococcus mutans gtfC gene, coding for synthesis of both soluble and insoluble glucans. Infect Immun. 1988;56(8):1999–2005.
- Hanada N, Kuramitsu HK. Isolation and characterization of the Streptococcus mutans gtfD gene, coding for primer-dependent soluble glucan synthesis. Infect Immun. 1989;57(7):2079–2085.
- Barthelmes J, Ebeling C, Chang A, et al. BRENDA, AMENDA and FRENDA: the enzyme information system in 2007. Nucleic Acids Res. 2007;35(Database issue):D511–4.
- Ooshima T, Matsumura M, Hoshino T, et al. Contributions of three glycosyltransferases to sucrose-dependent adherence of Streptococcus mutans. J Dent Res. 2001;80(7):1672–1677.
- Tamesada M, Kawabata S, Fujiwara T, et al. Synergistic effects of streptococcal glucosyltransferases on adhesive biofilm formation. J Dent Res. 2004;83(11):874–879.
- Venkitaraman AR, Vacca-Smith AM, Kopec LK, et al. Characterization of glucosyltransferase B, gtfC, and gtfD in solution and on the surface of hydroxyapatite. J Dent Res. 1995;74(10):1695–1701.
- Tsumori H, Kuramitsu H. The role of the Streptococcus mutans glucosyltransferases in the sucrose-dependent attachment to smooth surfaces: essential role of the GtfC enzyme. Oral Microbiol Immunol. 1997;12(5):274–280.
- Devulapalle KS, Mooser G. Subsite specificity of the active site of glucosultransferases from Streptococcus sobrinus. J Biol Chem. 1994;269(16):11967–11971.
- Devulapalle KS, Mooser G. Subsite specificity of divalent metal ions to glucosyltransferase. J Craniofac Genet Dev Biol. 2000;20(2):107–108.
- Devulapalle KS, Mooser G. Glucosyltransferase inactivation reduces dental caries. J Dent Res. 2001;80(2):466–469.
- Ito K, Ito S, Shimamura T, et al. Crystal structure of glucansucrase from the dental caries pathogen Streptococcus mutans. J Mol Biol. 2011;408(2):177–186.
- Mooser G, Wong C. Isolation of a glucan-binding domain of glucosyltransferase (1,6-alpha-glucan synthase) from Streptococcus sobrinus. Infect Immun. 1988;56(4):880–884.
- Koo H, Rosalen PL, Cury JA, et al. Effects of compounds found in propolis on Streptococcus mutans growth and on glucosyltransferase activity. Antimicrob Agents Chemother. 2002;46(5):1302–1309.
- Vacca-Smith AM, Bowen WH. Effect of some antiplaque agents on the activity of glucosyltransferases of Streptococcus mutans adsorbed onto saliva-coated hydroxyapatite and in solution. Biofilms. 19971:1–19
- Wunder D, Bowen WH. Action of agents on glucosyltransferases from Streptococcus mutans in solution and adsorbed to experimental pellicle. Arch Oral Biol. 1999;44(3):203–214.
- Ueda S, Shiroza T, Kuramitsu HK. Sequence analysis of the gtfC gene from Streptococcus mutans GS-5. Gene. 1988;69(1):101–109.
- Gleeson MP, Gleeson D. QM/MM calculations in drug discovery: a useful method for studying binding phenomena? J Chem Inf Model. 2009;49(3):670–677.
- Binder TP, Robyt JF. Inhibition of Streptococcus mutans 6715 glucosyltransferases by sucrose analogs modified at positions 6 and 6'′. Carbohydr Res. 1985;140(1):9–20.
- Nijampatnam B, Casals L, Zheng R, et al. Hydroxychalcone inhibitors of Streptococcus mutans glucosyl transferases and biofilms as potential anticaries agents. Bioorg Med Chem Lett. 2016;26(15):3508–3513.
- Ren Z, Chen L, Li J, et al. Inhibition of Streptococcus mutans polysaccharide synthesis by molecules targeting glycosyltransferase activity. J Oral Microbiol. 2016;8(1):31095.
- Lee CG, Park JK. Comparison of inhibitory activity of bioactive molecules on the dextransucrase from Streptococcus mutans. Appl Microbiol Biotechnol. 2015;99(18):7495–7503.
- Nijampatnam B, Zhang H, Cai X, et al. Inhibition of Streptococcus mutans biofilms by the natural stilbene piceatannol through the inhibition of glucosyltransferases. ACS Omega. 2018;3(7):8378–8385.
- Zhang Q, Nijampatnam B, Hua Z, et al. Structure-based discovery of small molecule inhibitors of cariogenic virulence. Sci Rep. 2017;7(1):1–0.
- Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields . J Comput Chem. 2010;31(4):671–690.
- Koska J, Spassov VZ, Maynard AJ, et al. Fully automated molecular mechanics based induced fit protein-ligand docking method. J Chem Inf Model. 2008; 48(10):1965–1973.
- Brooks BR, Bruccoleri RE, Olafson BD, et al. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4(2):187–217.
- Spassov VZ, Yan L, Flook PK. The dominant role of side-chain backbone interactions in structural realization of amino acid code. ChiRotor: a side-chain prediction algorithm based on side-chain backbone interactions . Protein Sci. 2007;16(3):494–506.
- Diller DJ, Merz ML. III., High throughput docking for library design and library prioritization. Proteins. 2001;43(2):113–124.
- Wu G, Robertson DH, Brooks CL, III, et al. Detailed analysis of grid-based molecular docking: a case study of CDOCKER – a CHARMm-based MD docking algorithm. J Comput Chem. 2003;24(13):1549–1562.
- Krammer A, Kirchhoff PD, Jiang X, et al. LigScore: a novel scoring function for predicting binding affinities. J Mol Graph Model. 2005;23(5):395–407.