
Abstract
Carbon monoxide (CO) is a toxic gas that causes neuropathy. However, CO is endogenously produced in small amounts showing various beneficial effects. We hypothesized that CO-bound haemoglobin-vesicle (HbV) administration would reduce cerebral ischaemia–reperfusion injury without causing neuropathy. Three experiments were conducted. First, rats were exposed to CO inhalation to create a CO-poisoning group, and they were sacrificed on 0, 7, 14, and 21 days after CO exposure. Histopathologically, hippocampal damage was prominent at 14 days. Second, the rats were administered with CO-HbV equivalent to 50 or 25% of circulating blood volume (CO-HbV50 or CO-HbV25 group). Rats were sacrificed 14 days after administration. Third, rats put into haemorrhagic shock by 50% of circulating blood withdrawal were resuscitated using saline, autologous blood, and CO-HbV. They were sacrificed 14 days after resuscitation. Hippocampal damage assessment clarified that almost no necrotic cells were observed in the CO-HbV50 group. Necrotic cells in the CO-HbV25 group were comparable to those found for the control group. In rats resuscitated from haemorrhagic shock, the hippocampal damage in the group using CO-HbV was the mildest. Administration of CO-HbV did not lead to marked hippocampal damage. Furthermore, CO-HbV was effective at preventing cerebral ischaemia-reperfusion injury after haemorrhagic shock.
Introduction
Carbon monoxide (CO), a colourless, tasteless, odourless, and non-irritating toxic gas, produces hypoxia by binding with haemoglobin, thereby reducing the oxygen-carrying capacity of the blood and producing hypoxia in the tissues. Various mechanisms related to CO poisoning are known to cause both tissue hypoxia and direct cellular changes involving immunological or inflammatory damage [Citation1]. Acute poisoning and persistent poisoning are both classifications of CO poisoning. Clinical symptoms of acute CO poisoning are non-specific. They can suggest various common disorders. In addition, diagnostic imaging shows globus pallidus lesions and edoema centred on the white matter of the cerebrum [Citation2,Citation3]. With persistent poisoning, delayed neuropathy occurs days to weeks after the acute symptoms have recovered. Neurological symptoms, such as decreased concentration, learning ability, and Parkinson-like syndrome appear. Furthermore, histopathological and imaging studies show necrosis of the globus pallidus and hippocampus and loss of myelin in the cerebral white matter [Citation4–7].
Actually, CO is produced in small amounts in the body, where it possesses anti-inflammatory antioxidant capabilities. It has attracted interest as a possible clinically viable medical agent [Citation8–11]. The two main modes of CO administration are intratracheal and intravenous. Intratracheal administration is the inhalation of CO gas, which has been reported as effective for lung diseases, such as chronic obstructive pulmonary disease and pneumonia [Citation12–14], endotoxemia [Citation15], and cardiac hypertrophy [Citation16]. For intravenous administration, CO-releasing molecules (CORM), CO-bound red blood cell (CO-RBC), and CO-bound haemoglobin vesicles (CO-HbV) are experimentally tested. Most CORMs are metal carbonyl complexes with CO bound to them. Various specific triggers initiate the release of CO. For example, CO is released by the effects of esterification, phosphorylation, and photochemical external activation by light of various wavelengths, combinations of thermal degradation and ligand replacement, and replacing ligands [Citation17–21]. Reportedly, CO administration using CORM is effective for treating ischaemia–reperfusion injury of kidney and retinal ganglion cells [Citation22,Citation23], inflammatory bowel disease [Citation24], and non-alcoholic steatohepatitis [Citation25]. CO-RBC is produced by isolating RBC from the blood of donor animals and the succeeding exposure to CO gas. It has been reported that CO-RBC improves microvascular function when administered during resuscitation of haemorrhagic shock [Citation26] and reduces hepatic ischaemia-reperfusion injury [Citation27,Citation28].
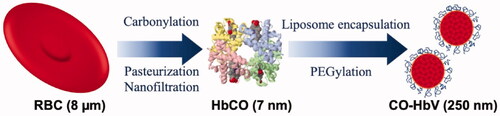
The cellular structure of HbV most closely mimics characteristics of natural red blood cells (RBCs), in which highly concentrated haemoglobin (Hb) is encapsulated within a liposome (). The CO binds easily and stably to Hb in HbV and CO-HbV released CO in blood circulation for 6 h after administration. Until now, animal studies have confirmed various beneficial effects of CO-HbV administration. Reportedly, it exerts protective effects against ischaemic reperfusion injury of the liver, pulmonary fibrosis, acute pancreatitis, and colitis [Citation29–32], but the safety of CO-HbV administration in relation to the cerebral nervous system has not been fully elucidated.
Figure 1. Cellular structure of haemoglobin-vesicles (HbV). A highly concentrated carbonyl haemoglobin (HbCO) is encapsulated within a liposome with polyethylene glycol.
Delayed CO intoxication is associated with learning disabilities, i.e. impaired memory, and cellular damage in the hippocampus has been reported [Citation7,Citation33,Citation34]. Thus, it is necessary to clarify the effects of CO-HbV administration on hippocampal tissue. For this study, we confirmed the safety of CO-HbV administration on the hippocampal tissue and investigated the effectiveness of CO-HbV administration on ischaemia–reperfusion injury in the brain by resuscitating rats in haemorrhagic shock with CO-HbV.
Materials and methods
Preparation of HbV and CO-HbV
HbV was prepared under sterile conditions, as reported earlier [Citation29,Citation35]. The Hb was purified from outdated donated blood that had been provided by the Japanese Red Cross Society (Tokyo, Japan). First, Hb was converted to carbonyl haemoglobin (HbCO) for stabilization. It was pasteurized (60 °C for 12 h) and nano-filtrated to inactivate and remove the virus. After the obtained HbCO solution was dialyzed and concentrated by ultrafiltration to 40 g/dL, it was encapsulated in vesicles using the kneading method with a liposomal membrane comprising biocompatible four lipids: 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine, cholesterol, 1,5-O-dihexadecyl-N-succinyl-L-glutamate, and 1,2-distearoyl-sn-glycerol-3-phosphatidylethanolamine-N-PEG5000 to obtain CO-HbV. Finally, CO-HbV was adjusted with saline (Otsuka Pharmaceutical Co., Ltd., Tokyo, Japan) to provide the Hb concentration of 10 g/dL.
Animals
All experimental protocols were reviewed by the Committee on the Ethics of Animal Experiments at Nara Medical University (approval number: 11691, 11797, and 12438) and were conducted in accordance with the Guidelines for Animal Experiments issued by the University and the European Directive 2010/63/EU. The ethical guidelines conformed to guiding principles issued by the National Academy of Sciences.
Male Wistar rats aged 7–10 weeks were purchased from Oriental Bioservice, Inc. (Kyoto, Japan). They were housed in cages with a bed of cellulose paper in a ventilated, temperature-controlled, specific-pathogen-free environment with a 12-h light-dark cycle. The animals were provided with free access to food and water. The following three experiments were conducted using these rats.
Exposure by CO gas
This experiment was conducted to create a model for CO poisoning using 16 rats. Each rat was anaesthetized using 1.5% isoflurane (Mylan Seiyaku Ltd., Tokyo, Japan)-mixed air (1 L/min) inhalation using a vaporizer (Forawick; Muraco Medical Co., Ltd., Tokyo, Japan) throughout the experiment (fraction of inspired O2: FiO2 = 21%) while spontaneous breathing was maintained. A polyethylene catheter (SP31; Natsume Seisakusho Co., Ltd., Tokyo, Japan) filled with heparinized normal saline was inserted into the femoral artery. Next, the carrier gas for isoflurane was changed from air to 3000 ppm CO (Sumitomo Seika Chemicals Co., Ltd., Osaka, Japan) and was exposed to CO for 60 min. Arterial blood gas analysis was conducted before exposure and every 10 min during CO exposure and 30 min after the end of the exposure. The rats were sacrificed for purposes of histopathological examination on the day of CO exposure (n = 4) and at 7 days (n = 4), 14 days (n = 4), and 21 days (n = 4) after exposure.
Administration of CO-HbV to healthy rats
Rats were anaesthetized and catheterized in the same manner. Each rat received overdose administration of CO-HbV equivalent to 50% (n = 4) or 25% (n = 4) of circulating blood volume from the femoral artery (CO-HbV50 group or CO-HbV25 group) at a rate of 1 ml/min using a syringe (Terumo Corp., Tokyo, Japan). The systemic blood volume was estimated as 56 ml/kg body weight. Arterial blood gas analysis was performed before and after administration. Then the catheter was removed from the femoral artery. The wound was closed. Then the rats were brought out of anaesthesia and were returned to their respective cages. At 14 days after CO-HbV administration, the rats were sacrificed for purposes of histopathological examination.
Resuscitation from hemorrhagic shock with CO-HbV
Rats were anaesthetized and catheterized similarly using 12 rats. Haemorrhagic shock was induced by withdrawing 50% of the circulating blood volume (28 ml/kg, 1 ml/min) from the femoral artery using a heparinized syringe. After 15 min, the rats were resuscitated by infusion of CO-HbV (n = 4), autologous whole blood (WB) (n = 4), and normal saline solution (n = 4) at a rate of 1 ml/min. The volume of the infused resuscitative fluid was identical to the shed volume: 50% of the blood volume at baseline. The CO-HbV used for resuscitation (8.6 ml) was mixed with human serum albumin (HSA; 25%, 1.4 ml; Japan Blood Products Organization, Tokyo, Japan) to regulate [HSA] in the suspending medium to 5 g/dL and the colloid osmotic pressure to ∼20 Torr. Arterial blood gas analysis was performed before and after withdrawing blood and after administration of resuscitation fluid. The blood pressure of the femoral artery was monitored continuously (PROPAQ 204 EL; Welch Allyn Inc., NY, USA). Then the catheter was removed from the femoral artery. The wound was closed. The rats were brought out of anaesthesia and were returned to the cage. For purposes of histopathological examination, the rats were sacrificed 14 days after resuscitation.
The shock state severity was confirmed from the assessment of four rats that received no resuscitative fluid. The survival rate decreased in a few hours. All the rats died within 24 h.
Control group
Rats were anaesthetized and catheterized in the same manner (n = 4). After 15 min, the catheter was removed from the femoral artery and the wound was closed. The rats were brought out of anaesthesia and were returned to the cage. After 14 days, the rats were sacrificed for purposes of histopathological examination.
Arterial blood gas analysis
All arterial blood gas analyses conducted for this study were performed using a portable blood gas analyzer (ABL80 FLEX-CO-OX; Radiometer Medical ApS Co. Ltd., Tokyo, Japan). The measurement items were HbCO to evaluate the amount of CO taken into the body, arterial partial pressure of carbon dioxide (PaCO2), arterial partial pressure of oxygen (PaO2) to evaluate the changes in respiratory status because of CO inhalation, and pH to evaluate the acid–base balance.
Histopathological examination
Sacrificed rats had their brains perfused with saline and 10% formalin neutral buffer (Sigma-Aldrich Japan K.K., Tokyo, Japan). After decapitation, the brains were fixed in formalin. Then paraffin sections of the hippocampus were stained with haematoxylin/eosin (HE) to assess the cellular structure. Immunohistochemical analyses were done to detect 8-hydroxy-2′-deoxyguanosine (8-OHdG): the most direct indication of oxidative damage. Paraffin sections of the same area used for HE stains were reacted with anti-8-OHdG antibody (Abcam plc., Tokyo, Japan) and Histostar™ (Ms + Rb) for rat Tissue (Medical & Biological Laboratories Co., Ltd., Tokyo, Japan) and were then stained (Super Sensitive™ DAB; Medical & Biological Laboratories Co., Ltd., Tokyo, Japan). Meyer haematoxylin (Muto Pure Chemicals Co., Ltd., Tokyo, Japan) was used for nuclear staining. Phosphate-buffered saline (LSI Medience Corp., Tokyo, Japan) was used for washing after each procedure.
All cells (about 1000–2000 cells) in the hippocampal area on the right or left side of these specimens were evaluated to determine the ratio of normal to necrotic cells. Necrotic cells were defined as those that did not retain their cell shape and whose nuclei could not be identified.
Statistical analyses
All datasets were analyzed using software (BellCurve for Excel; Social Survey Research Information Co., Ltd., Tokyo, Japan). The p-values correspond to two-tailed tests for which significance inferred for p < .05. Means ± standard deviation are reported for all measurements unless otherwise specified.
Results
CO poisoning
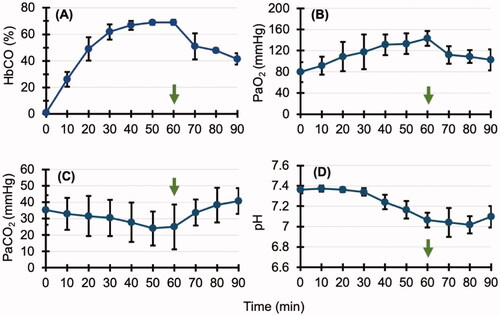
The HbCO concentration in the blood was 0.9 ± 0.4% before the start of CO inhalation. It increased gradually and eventually reached 69.0 ± 1.9% (). After the end of CO inhalation, it decreased gradually and reached 41.4 ± 4.3 after 30 min. Before CO inhalation, PaO2 was 79.6 ± 18.7 mmHg; it increased gradually after the start of inhalation to a maximum of 143.3 ± 14.0 mmHg after 60 min and decreased gradually after the end of CO inhalation to a final value of 102.4 ± 19.9 mmHg. Also, PaCO2 decreased gradually from 35.3 ± 9.01 to 24.9 ± 13.6 mmHg, but it increased after the end of CO inhalation. It reached 40.7 ± 7.8 mmHg 30 min after the end of inhalation. The pH decreased from 7.36 ± 0.03 to 7.02 ± 0.08; it increased to 7.1 ± 0.11 30 min after the end of CO inhalation.
Figure 2. Arterial blood gas analysis in the carbon monoxide (CO) poisoning group. Arrows indicate when CO inhalation is complete. (A,B) The carbonyl haemoglobin (HbCO) and arterial partial pressure of oxygen (PaO2) concentration in the blood increased gradually. After the end of CO inhalation, it decreased gradually. (C,D) Arterial partial pressure of carbon dioxide (PaCO2) and pH decreased gradually and then increased after CO inhalation ended. Collectively, these results suggest that the increased respiratory rate because of CO inhalation caused an increase in blood oxygen (O2) and a decrease in blood carbon dioxide (CO2), but it also caused an O2 deficiency in peripheral tissues, resulting in acidosis.
The percentages of necrotic cells in hippocampal tissue specimens (HE stains) after 0, 7, 14, and 21 days of CO exposure were, respectively, 7.0 ± 2.2, 15.3 ± 4.2, 28.0 ± 7.2, and 17.7 ± 7.6% (). The percentages of necrotic cells in 8-OHdG-stained tissue specimens after 0, 7, 14, and 21 days of exposure were, respectively, 12.0 ± 3.3, 20.4 ± 2.7, 34.0 ± 6.1, and 15.7 ± 3.9%, which were similar profiles to those of HE-stained specimens. Figures 4(A,B) are HE and 8-OHdG stained hippocampal specimens of the control group. Figures 4(A,B) are HE and 8-OHdG stained hippocampal specimens of the control group. Tissue samples at 14 days after CO exposure, when necrotic cells were most abundant, are shown in .
Figure 3. Percentage of necrotic cells in carbon monoxide (CO) poisoning group: (A) haematoxylin/eosin (HE) stained and (B) 8-hydroxy-2′-deoxyguanosine (8-OHdG) stained. *p < .05, **p < .01.
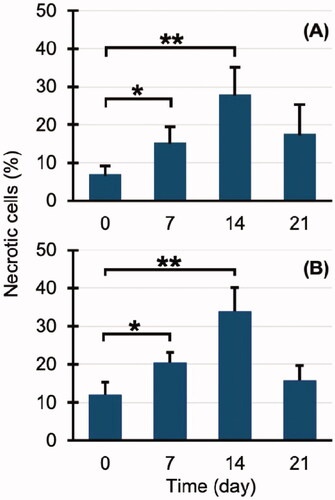
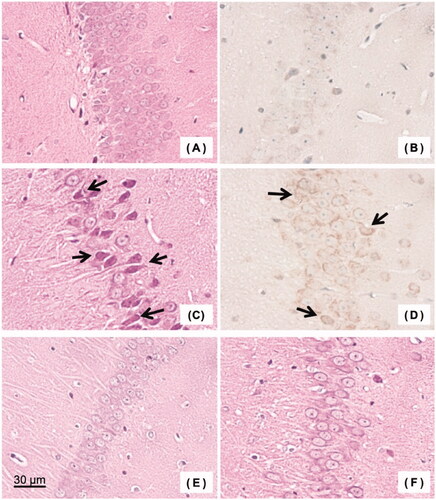
Figure 4. Histopathological examination of brain (hippocampus): (A) Haematoxylin/eosin (HE) stained specimen of the control group, (B) 8-hydroxy-2′-deoxyguanosine (8-OHdG) stained specimen of the control group, (C) HE stained specimen of the carbon monoxide (CO) poisoning group, (D) 8-OHdG stained specimen of the CO poisoning group, (E) HE stained specimen of the CO-bound haemoglobin vesicles (CO-HbV) 50 group, and (F) HE stained specimen of the CO-HbV25 group. Arrows indicate some representative necrotic cells.
Safety of CO-HbV administration
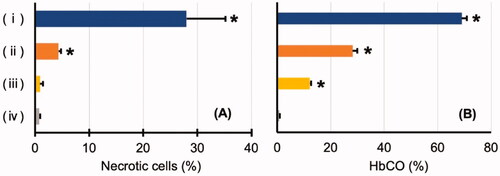
In hippocampal tissue specimens, almost no necrotic cells were found in the CO-HbV50 and CO-HbV25 groups (). The percentages of necrotic cells were the small numbers of 4.4 ± 0.4% in the CO-HbV50 group and 0.9 ± 0.5% in the CO-HbV25 group, which were comparable to 0.8 ± 0.2% found for the control group (). In the CO poisoning group, the level of HbCO after CO inhalation was as high as about 70%, whereas, in the CO-HbV50 group, the level of HbCO after administration was 28.2 ± 1.8%. In the CO-HbV25 group, it was as low as 12.2 ± 0.5% ().
Figure 5. (A) Comparison of the degree of hippocampal cell damage. (B) Level of carbonyl haemoglobin (HbCO). (i) Carbon monoxide (CO) poisoning group after CO inhalation, (ii) CO-bound haemoglobin vesicles (CO-HbV) 50 group after CO-HbV administration, (iii) CO-HbV25 group after CO-HbV administration, and (iv) control group. *Significant difference compared to the control group (p < .05).
Resuscitation from hemorrhagic shock
The blood pressure decreased after bleeding. The average blood pressure became <30 mmHg. After administration of the resuscitation fluid, all groups quickly recovered to higher 60 mmHg as reported in the previous study [Citation26]. The blood HbCO concentration increased to only 25.6 ± 1.1% after resuscitation in the CO-HbV resuscitation group ().
Figure 6. (A) Level of carbonyl haemoglobin (HbCO). (B) Degree of hippocampal cell damage 14 days after resuscitation. (CO-HbV) The group resuscitated using carbon monoxide-bound haemoglobin vesicles, (WB) The group resuscitated using the autologous whole blood, (NS) The group resuscitated using the normal saline. *p < .05, **p < .01.
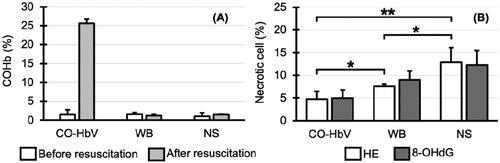
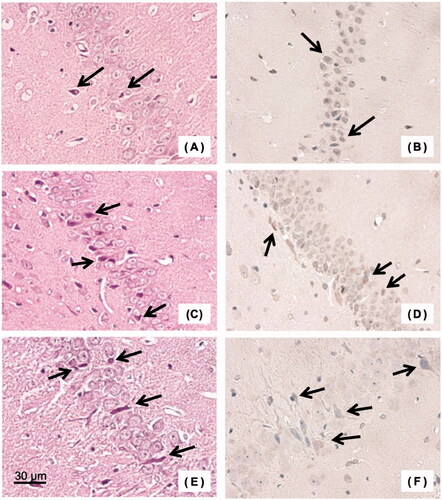
At 14 days after resuscitation, hippocampal tissue samples showed necrotic cells in all groups (). The lowest percentage of necrotic cells in the tissue specimens (HE stains) was in the group resuscitated with CO-HbV (4.8 ± 1.5%). The next lowest was 7.6 ± 0.5% in the group resuscitated with WB, and the highest was 12.9 ± 3.2% in the group resuscitated with saline. There was a significant difference in the percentage of necrotic cells among the three groups (p < .05), especially between the group resuscitated with CO-HbV and the group resuscitated with saline (p < .01). The percentages of necrotic cells in 8-OHdG-stained tissue specimens 14 days after resuscitation were 5.0 ± 1.8% in the group resuscitated with CO-HbV, 9.0 ± 2.0% in the group resuscitated with autologous blood, and 12.3 ± 3.1% in the group resuscitated with saline (). These results were similar to those obtained for HE-stained specimens.
Figure 7. Histopathological examination of the brain (hippocampus) in the group of resuscitation from haemorrhagic shock. (A) Haematoxylin/eosin (HE) stained specimen of carbon monoxide-bound haemoglobin vesicles (CO-HbV) resuscitation group, (B) 8-hydroxy-2′-deoxyguanosine (8-OHdG) stained specimen of the CO-HbV resuscitation group, (C) HE stained specimen of the autologous whole blood (WB) resuscitation group, (D) 8-OHdG stained specimen of the WB resuscitation group, (E) HE stained specimen of the normal saline (NS) resuscitation group, and (F) 8-OHdG stained specimen of the NS resuscitation group. Arrows indicate some representative necrotic cells.
Discussion
The main finding of this study is that CO administration with HbV is not only safe for the hippocampal tissue when administered to normal rats. It is also effective at reducing ischaemia–reperfusion injury after haemorrhagic shock.
Although CO is a toxic gas, trace amounts of CO are also produced endogenously in living organisms. The main source of endogenous CO is haem degradation by haem oxygenase, but non-haem sources, such as lipid peroxidation, photooxidation, and metabolic activity of intestinal bacteria also exist [Citation36–39]. The endogenous CO produced in this way is regarded as a messenger molecule in vascular and neurologic tissues [Citation40–43]. However, the amount and the source of haem as a substrate and the amount of CO produced remain unclear. These observations engender the concept of using exogenous, not endogenous, CO for therapeutic purposes.
Many reports have described the various benefits of CO administration. Regarding the administration method, various methods, such as trans-respiratory administration, such as inhalation of CO gas and intravenous administration using HbV and CORM have been tried [Citation44,Citation45]. For CO administration via the respiratory tract, it was difficult to adjust the dosage because CO exposure varied with changes in breathing patterns. Nevertheless, it is now possible to maintain a constant CO gas concentration in the ventilatory circuit by adjusting changes in the ventilation rate and flow rate [Citation13]. A phase I clinical trial of inhaled CO is also underway to treat acute respiratory distress syndrome because of sepsis using this system [Citation46].
Most of the CORM is metal carbonyl complexes. Side effects caused by the contained metals pose some difficulty. Therefore, CORM using various transition metals and main group elements have been investigated [Citation19,Citation47,Citation48]. In addition, because CORM releases CO rapidly, one must continue administering CORM to maintain the therapeutic effect. Therefore, delivery systems and triggers for CO release are being investigated to prevent CO release until the CORM reaches the target organ [Citation17,Citation49–57]. The biological activity of the reaction products after the release of CO is also important [Citation58]. Further studies are needed.
Because HbCO is thermally and chemically stable in CO-HbV, it can be stored for long periods of time; it can continue to release CO in blood circulation for 6 h after administration [Citation29,Citation45]. After administration of CO-HbV in blood circulation and dissociation of CO, the resulting HbV starts to bind molecular oxygen (O2) reversibly. It is thereby transformed into an O2 carrier. The CO-HbV administration effectiveness has been reported in animal models of pulmonary fibrosis, acute pancreatitis, and colitis [Citation30–32,Citation59], but its effects on the cranial nerve system have not been fully investigated.
Considering that CO poisoning causes dysmnesia, one must examine the effects of CO administration on the hippocampal tissue for clinical application of CO-HbV administration. During this study, many necrotic cells were observed in the hippocampal tissue specimens of the CO poisoning group, although few necrotic cells were observed in the CO-HbV50 group. In the CO-HbV25 group, the proportion of necrotic cells was similar to that of the control group. In other words, CO-HbV administration is considered to have almost no adverse effects on the cranial nerve system. Two possible causes for this can be inferred. The first point is the amount of CO administered to rats. Although direct comparison is difficult because of different administration methods, the level of HbCO after CO inhalation was 69.0 ± 1.9%, whereas the HbCO level after CO-HbV50 administration was 28.2 ± 1.8%; after CO-HbV25 administration, it was 12.2 ± 0.5%. Therefore, one must consider that the group of CO poisoning was exposed to more CO. The second point is the difference in administration methods. Some earlier reports have described inhaled CO as much more toxic than HbCO or CO gas transfused by intraperitoneal injection [Citation60,Citation61]. Actually, CO is administered intravenously as CO-HbV binds tightly to Hb. Therefore, less CO is transferred to the tissue. However, CO administered via the respiratory tract can be expected to produce high tension in the alveoli and increase dissolved CO in the blood, resulting in increased amounts of CO transferred to brain tissue, which is likely to cause CO poisoning. In addition to this study, we are carefully examining the neurological effect of CO-HbV on the central nervous system by functional observational battery (FOB). The results will be presented elsewhere.
Excitotoxicity, oxidative stress, inflammation, and apoptosis have been well-established as major pathobiological mechanisms of ischaemia reperfusion injury [Citation62,Citation63]. Among these factors, oxidative stress plays a central role in cerebral ischaemia reperfusion injury [Citation64]. Four main pathways for the production of reactive oxygen species (ROS) that cause oxidative stress have been identified [Citation65–67].
The first is a pathway involving xanthine oxidase, which is produced when the tissue becomes ischaemic and generates ROS from oxygen after reperfusion. The second is a pathway involving nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is normally inactivated by division into a core subunit integrated into the cell membrane and the enzyme in the cytosol. Various factors and inflammatory cytokines are produced when cells become ischaemic and activate NADPH oxidase. Activated NADPH oxidase produces superoxide from oxygen. The third is a pathway involving mitochondrial disorders. Mitochondria most likely generate ROS during normal oxidative metabolism. These ROS are processed by the antioxidant system in mitochondria. When the mitochondrial microstructure and function are impaired by ischaemia, even if the metabolism is partially restored during reperfusion, the antioxidant system is not rebuilt, resulting in increased ROS production. The fourth is a pathway involving endothelial nitric oxide synthase (NOS). Under normal conditions, NOS synthesizes nitric oxide (NO); after ischaemia–reperfusion, NO reacts with superoxide to convert to highly reactive peroxynitrite.
The ROS produced by the processes described above can damage vascular endothelial cells, resulting in microcirculatory disturbances that eventually engender organ damage. When CO-HbV is administered, CO suppresses ROS production and reduces ischaemia–reperfusion injury by suppressing NADPH oxidase, inhibiting inflammatory cytokine production, and improving mitochondrial function [Citation23,Citation25,Citation28,Citation31,Citation68].
This study revealed that cerebral ischaemia resulted from hypotension and hypovolemia by removing 50% of the circulating blood volume. The ischaemic region was re-perfused by administering a resuscitation solution 15 min later. In the cerebral ischaemia–reperfusion injury model thus prepared, hippocampal necrosis in the group resuscitated using CO-HbV was the mildest. This is true probably because the earlier reported reduction of ischaemia–reperfusion injury in the kidney, lung, liver, etc. by CO-administration also occurred in the brain. After releasing CO, the resulting HbV starts to bind O2 reversibly to become an O2 carrier, which is advantageous in comparison to the other CO-releasing molecules.
Conclusion
Administration of CO-HbV increased blood HbCO concentrations but did not cause hippocampal necrosis, such as CO poisoning. Results also show that CO-HbV was effective at cerebral ischaemia–reperfusion injury after haemorrhagic shock.
Acknowledgements
The authors acknowledge Dr. Takashi Matsuhira, and Dr. Tomoko Kure (Department of Chemistry, Nara Medical University), and Dr. Masahiko Kawaguchi and Dr. Yusuke Naito (Department of Anesthesiology, Nara Medical University) for their assistance with experiments.
Disclosure statement
The authors declare the following competing financial interests: H.S. is an inventor holding some patents related to the production and utilization of Hb-vesicles.
Data availability statement
All data is included in the submission/manuscript file.
Additional information
Funding
References
- Roderique JD, Josef CS, Feldman MJ, et al. A modern literature review of carbon monoxide poisoning theories, therapies, and potential targets for therapy advancement. Toxicology. 2015;334:45–58.
- Prockop LD, Chichkova RI. Carbon monoxide intoxication: an updated review. J Neurol Sci. 2007;262(1–2):122–130.
- Jeon S-B, Sohn CH, Seo D-W, et al. Acute brain lesions on magnetic resonance imaging and delayed neurological sequelae in carbon monoxide poisoning. JAMA Neurol. 2018;75(4):436–443.
- Fan D, Hu H, Sun Q, et al. Neuroprotective effects of exogenous methane in a rat model of acute carbon monoxide poisoning. Brain Res. 2016;1633:62–72.
- Chang KH, Han MH, Kim HS, et al. Delayed encephalopathy after acute carbon monoxide intoxication: MR imaging features and distribution of cerebral white matter lesions. Radiology. 1992;184(1):117–122.
- Piantadosi CA, Zhang J, Levin ED, et al. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp Neurol. 1997;147(1):103–114.
- Beppu T. The role of MR imaging in assessment of brain damage from carbon monoxide poisoning: a review of the literature. Am J Neuroradiol. 2014;35(4):625–631.
- Vreman HJ, Wong RJ, Kadotani T, et al. Determination of carbon monoxide (CO) in rodent tissue: effect of heme administration and environmental CO exposure. Anal Biochem. 2005;341(2):280–289.
- Liu Y, Wang X, Xu X, et al. Carbon monoxide releasing molecule-2 (CORM-2)-liberated CO ameliorates acute pancreatitis. Mol Med Rep. 2019;19:5142–5152.
- Foresti R, Bani-Hani MG, Motterlini R. Use of carbon monoxide as a therapeutic agent: promises and challenges. Intensive Care Med. 2008;34(4):649–658.
- Taguchi K, Yamasaki K, Sakai H, et al. The use of hemoglobin vesicles for delivering medicinal gas for the treatment of intractable disorders. J Pharm Sci. 2017;106(9):2392–2400.
- Bathoorn E, Slebos D-J, Postma DS, et al. Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J. 2007;30(6):1131–1137.
- Fredenburgh LE, Kraft BD, Hess DR, et al. Effects of inhaled CO administration on acute lung injury in baboons with pneumococcal pneumonia. Am J Physiol Lung Cell Mol Physiol. 2015;309(8):L834–L846.
- Dalli J, Kraft BD, Colas RA, et al. The regulation of proresolving lipid mediator profiles in baboon pneumonia by inhaled carbon monoxide. Am J Respir Cell Mol Biol. 2015;53(3):314–325.
- Mayr FB, Spiel A, Leitner J, et al. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med. 2005;171(4):354–360.
- Kobayashi A, Ishikawa K, Matsumoto H, et al. Synergetic antioxidant and vasodilatory action of carbon monoxide in angiotensin II–induced cardiac hypertrophy. Hypertension. 2007;50(6):1040–1048.
- Romanski S, Kraus B, Guttentag M, et al. Acyloxybutadiene tricarbonyl iron complexes as enzyme-triggered CO-releasing molecules (ET-CORMs): a structure-activity relationship study. Dalton Trans. 2012;41(45):13862–13875.
- Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9(9):728–743.
- Motterlini R, Mann BE, Foresti R. Therapeutic applications of carbon monoxide-releasing molecules. Expert Opin Investig Drugs. 2005;14(11):1305–1318.
- Kretschmer R, Gessner G, Görls H, et al. Dicarbonyl-bis(cysteamine)iron(II): a light induced carbon monoxide releasing molecule based on iron (CORM-S1). J Inorg Biochem. 2011;105(1):6–9.
- Bannenberg GL, Vieira HLA. Therapeutic applications of the gaseous mediators carbon monoxide and hydrogen sulfide. Expert Opin Ther Pat. 2009;19(5):663–682.
- Ulbrich F, Kaufmann KB, Meske A, et al. The CORM ALF-186 mediates anti-Apoptotic signaling via an activation of the p38 MAPK after ischemia and reperfusion injury in retinal ganglion cells. PLOS One. 2016;11(10):e0165182.
- Kim DK, Shin S-J, Lee J, et al. Carbon monoxide-releasing molecule-3: amelioration of renal ischemia reperfusion injury in a rat model. Investig Clin Urol. 2020;61(4):441–451.
- Yin H, Fang J, Liao L, et al. Styrene-maleic acid copolymer-encapsulated CORM2, a water-soluble carbon monoxide (CO) donor with a constant CO-releasing property, exhibits therapeutic potential for inflammatory bowel disease. J Control Release. 2014;187:14–21.
- Upadhyay KK, Jadeja RN, Vyas HS, et al. Carbon monoxide releasing molecule-A1 improves nonalcoholic steatohepatitis via Nrf2 activation mediated improvement in oxidative stress and mitochondrial function. Redox Biol. 2020;28:101314.
- Cabrales P, Tsai AG, Intaglietta M. Hemorrhagic shock resuscitation with carbon monoxide saturated blood. Resuscitation. 2007;72(2):306–318.
- Ogaki S, Taguchi K, Maeda H, et al. Kupffer cell inactivation by carbon monoxide bound to red blood cells preserves hepatic cytochrome P450 via anti-oxidant and anti-inflammatory effects exerted through the HMGB1/TLR-4 pathway during resuscitation from hemorrhagic shock. Biochem Pharmacol. 2015;97(3):310–319.
- Ogaki S, Taguchi K, Watanabe H, et al. Carbon monoxide-bound red blood cell resuscitation ameliorates hepatic injury induced by massive hemorrhage and red blood cell resuscitation via hepatic cytochrome P450 protection in hemorrhagic shock rats. J Pharm Sci. 2014;103(7):2199–2206.
- Sakai H, Horinouchi H, Tsuchida E, et al. Hemoglobin vesicles and red blood cells as carriers of carbon monoxide prior to oxygen for resuscitation after hemorrhagic shock in a rat model. Shock. 2009;31(5):507–514.
- Nagao S, Taguchi K, Sakai H, et al. Carbon monoxide-bound hemoglobin-vesicles for the treatment of bleomycin-induced pulmonary fibrosis. Biomaterials. 2014;35(24):6553–6562.
- Nagao S, Taguchi K, Sakai H, et al. Carbon monoxide-bound hemoglobin vesicles ameliorate multiorgan injuries induced by severe acute pancreatitis in mice by their anti-inflammatory and antioxidant properties. Int J Nanomedicine. 2016;11:5611–5620.
- Nagao S, Taguchi K, Miyazaki Y, et al. Evaluation of a new type of nano-sized carbon monoxide donor on treating mice with experimentally induced colitis. J Control Release. 2016;234:49–58.
- Nabeshima T, Katoh A, Ishimaru H, et al. Carbon monoxide-induced delayed amnesia, delayed neuronal death and change in acetylcholine concentration in mice. J Pharmacol Exp Ther. 1991;256(1):378–384.
- Qingsong W, Yeming G, Xuechun L, et al. The free radical scavenger, edaravone, ameliorates delayed neuropsychological sequelae after acute carbon monoxide poisoning in rabbits. Undersea Hyperb Med J Undersea Hyperb Med Soc Inc. 2013;40:223–229.
- Kure T, Sakai H. Preparation of artificial red blood cells (hemoglobin vesicles) using the rotation-revolution mixer for high encapsulation efficiency. ACS Biomater Sci Eng. 2021;7(6):2835–2844.
- Vreman HJ, Wong RJ, Sanesi CA, et al. Simultaneous production of carbon monoxide and thiobarbituric acid reactive substances in rat tissue preparations by an iron-ascorbate system. Can J Physiol Pharmacol. 1998;76(12):1057–1065.
- Vreman HJ, Gillman MJ, Downum KR, et al. In vitro generation of carbon monoxide from organic molecules and synthetic metalloporphyrins mediated by light. Dev Pharmacol Ther. 1990;15(2):112–124.
- Engel RR, Matsen JM, Chapman SS, et al. Carbon monoxide production from heme compounds by bacteria. J Bacteriol. 1972;112(3):1310–1315.
- Engel RR, Modler S, Matsen JM, et al. Carbon monoxide production from hydroxocobalamin by bacteria. Biochim Biophys Acta. 1973;313(1):150–155.
- Ryter SW, Otterbein LE, Morse D, et al. Heme oxygenase/carbon monoxide signaling path-ways: regulation and functional significance. In: Vallyathan V, Shi X, Castranova V, editors. Oxygen/nitrogen radicals: cell injury and disease. Boston (MA): Springer US; 2002 [cited 2021 Aug 11]. p. 249–263. Available from: http://link.springer.com/10.1007/978-1-4615-1087-1_29
- Johnson RA, Colombari E, Colombari DSA, et al. Role of endogenous carbon monoxide in central regulation of arterial pressure. Hypertension. 1997;30(4):962–967.
- Meffert MK, Haley JE, Schuman EM, et al. Inhibition of hippocampal heme oxygenase, nitric oxide synthase, and long-term potentiation by metalloporphyrins. Neuron. 1994;13(5):1225–1233.
- Leinders-Zufall T, Shepherd GM, Zufall F. Regulation of cyclic nucleotide-gated channels and membrane excitability in olfactory receptor cells by carbon monoxide. J Neurophysiol. 1995;74(4):1498–1508.
- Adach W, Błaszczyk M, Olas B. Carbon monoxide and its donors – chemical and biological properties. Chem Biol Interact. 2020;318:108973.
- Sakai H. Overview of potential clinical applications of hemoglobin vesicles (HbV) as artificial red cells, evidenced by preclinical studies of the academic research consortium. JFB. 2017;8(1):10.
- Fredenburgh LE, Perrella MA, Barragan-Bradford D, et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI Insight. 2018;3(23):e124039.
- Romão CC, Blättler WA, Seixas JD, et al. Developing drug molecules for therapy with carbon monoxide. Chem Soc Rev. 2012;41(9):3571–3583.
- Zobi F. CO and CO-releasing molecules in medicinal chemistry. Future Med Chem. 2013;5(2):175–188.
- Johnson TR, Mann BE, Teasdale IP, et al. Metal carbonyls as pharmaceuticals? [Ru(CO)3Cl(glycinate)], a CO-releasing molecule with an extensive aqueous solution chemistry. Dalton Trans. 2007;(15):1500–1508.
- Santos-Silva T, Mukhopadhyay A, Seixas JD, et al. CORM-3 reactivity toward proteins: the crystal structure of a Ru(II) dicarbonyl-lysozyme complex. J Am Chem Soc. 2011;133(5):1192–1195.
- Santos MFA, Seixas JD, Coelho AC, et al. New insights into the chemistry of fac-[Ru(CO)3]2+ fragments in biologically relevant conditions: the CO releasing activity of [Ru(CO)3Cl2(1,3-thiazole)], and the X-ray crystal structure of its adduct with lysozyme. J Inorg Biochem. 2012;117:285–291.
- Pierri AE, Pallaoro A, Wu G, et al. A luminescent and biocompatible PhotoCORM. J Am Chem Soc. 2012;134(44):18197–18200.
- Antony LAP, Slanina T, Šebej P, et al. Fluorescein analogue xanthene-9-carboxylic acid: a transition-metal-free CO releasing molecule activated by green light. Org Lett. 2013;15(17):4552–4555.
- Peng P, Wang C, Shi Z, et al. Visible-light activatable organic CO-releasing molecules (PhotoCORMs) that simultaneously generate fluorophores. Org Biomol Chem. 2013;11(39):6671–6674.
- Kunz PC, Meyer H, Barthel J, et al. Metal carbonyls supported on iron oxide nanoparticles to trigger the CO-gasotransmitter release by magnetic heating. Chem Commun. 2013;49(43):4896–4898.
- Zobi F, Blacque O, Jacobs RA, et al. 17 e− Rhenium dicarbonyl CO-releasing molecules on a cobalamin scaffold for biological application. Dalton Trans. 2012;41(2):370–378.
- Ma M, Noei H, Mienert B, et al. Iron metal-organic frameworks MIL-88B and NH2-MIL-88B for the loading and delivery of the gasotransmitter carbon monoxide. Chemistry. 2013;19(21):6785–6790.
- Seixas JD, Mukhopadhyay A, Santos-Silva T, et al. Characterization of a versatile organometallic pro-drug (CORM) for experimental CO based therapeutics. Dalton Trans. 2013;42(17):5985–5998.
- Taguchi K, Nagao S, Maeda H, et al. Biomimetic carbon monoxide delivery based on hemoglobin vesicles ameliorates acute pancreatitis in mice via the regulation of macrophage and neutrophil activity. Drug Deliv. 2018;25(1):1266–1274.
- Goldbaum LR, Ramirez RG, Absalon KB. What is the mechanism of carbon monoxide toxicity? Aviat Space Environ Med. 1975;46(10):1289–1291.
- Goldbaum LR, Orellano T, Dergal E. Mechanism of the toxic action of carbon monoxide. Ann Clin Lab Sci. 1976;6(4):372–376.
- Özbal S, Erbil G, Koçdor H, et al. The effects of selenium against cerebral ischemia-reperfusion injury in rats. Neurosci Lett. 2008;438(3):265–269.
- Dong S, Tong X, Li J, et al. Total flavonoid of Litsea coreana leve exerts anti-oxidative effects and alleviates focal cerebral ischemia/reperfusion injury. Neural Regen Res. 2013;8(34):3193–3202.
- Buch P, Patel V, Ranpariya V, et al. Neuroprotective activity of Cymbopogon martinii against cerebral ischemia/reperfusion-induced oxidative stress in rats. J Ethnopharmacol. 2012;142(1):35–40.
- McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312(3):159–163.
- Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551.
- Crabtree MJ, Hale AB, Channon KM. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic Biol Med. 2011;50(11):1639–1646.
- Sahara H, Shimizu A, Setoyama K, et al. Carbon monoxide reduces pulmonary ischemia-reperfusion injury in miniature swine. J Thorac Cardiovasc Surg. 2010;139(6):1594–1601.