
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Computational modelling is a technique for modelling and solving real-world problems by utilising computing to provide solutions. This paper presents a novel predictive model of cell survival/death-related effects of Extracellular Signal-Regulated Kinase Protein. The computational model was designed using Neural Networks and fuzzy system. Three hundred ERK samples were examined using ten different concentrations of three input proteins: EGF, TNF, and insulin. Based on the different concentrations of input proteins and different samples of ERK protein, adjustment Anderson darling (AD) statistics for multiple distribution functions were computed considering different test such as visual test, Pearson correlation coefficient, and uniformity tests. The results reveal that utilising different concentrations and samples, values such as 7.55 AD and 18.4 AD were obtained using the Weibull distribution function for 0 ng/ml of TNF, 100 ng/ml of EGF, and 0 ng/mL of insulin concentrations. The model was validated by predicting the various ERK protein values that fall within the observed range. The proposed model agrees with the deterministic model, which was developed using difference equations.
Introduction
System biology is the study of the properties of complex biological systems that develop from the interactions of several proteins. Systems biology aims to expand organisms' behavior by utilizing scientific and technological knowledge so they can carry out new tasks. Deoxyribonucleic acid (DNA), ribonucleic acid (RNA), proteins, and metabolites (including carbohydrates, lipids, amino acids, and nucleotides) from the newly constructed behaviour’s bottom-up hierarchy are a few examples of biological systems. Biological systems have recently been compared to electronic systems, which contain a bottom-up layer consisting of a physical layer, a device layer, and a module layer [Citation1]. Transistors, capacitors, and resistors form the physical layer, while all the computations performed in the computer by the electrical circuits form the device layer, and components like the integrated circuits form the module layer [Citation2,Citation3].
Engineering problems require mathematical solutions. Some problems are difficult to solve, while others necessitate precise answers that are not determinable [Citation4]. Numerical methods and computational techniques employ computers to solve problems that are either iterative, repeating, or sequential in nature, and are frequently intractable or theoretically difficult to grasp [Citation5]. They are powerful problem-solving tools capable of dealing with enormous quantities of equations, complex geometries, and non-linearities. Computational methods simplify complex mathematics by reducing them to fundamental arithmetic processes. Among the many applications of computer modelling is the determination of cell survival/death [Citation6]. Computers are used in computational modelling to investigate and simulate the behaviour of complex systems in computer science, physics, and mathematics. This modelling is also an important component of big data analytics. To determine the major metabolic changes associated with cell division, signal processing approaches used for cell survival/death analysis employ a variety of computational methodologies [Citation7]. Extracellular signals such as cytokines, growth factors, and hormones are mediated by receptors that aid in the transduction of intracellular cues [Citation8]. Tumour necrosis factor- (TNF-) functions as a cue for programmed cell death [Citation9], whereas epidermal growth factor (EGF) [Citation10,Citation11] and insulin [Citation12,Citation13] function as survival cues. There are different types of proteins that help in the computer engineering hierarchy. In this paper, the Extracellular Signal-Regulated Kinase (ERK) protein that forms one of the pathways of Mitogenic (MAPK) proteins [Citation14] is investigated. MAPKs are threonine/serine protein kinases that are activated by MAP/ERK kinases (MEKs), which activates MEK kinase (MEKK). This pathway promotes cell survival by increasing transcription cell proliferation, Bcl2 family members, and inhibitors of cell death proteins (IAPs). The ERK MAP kinase cascade is one of the central pathways in growth factor signal transductions. ERK 1/2 are members of the MAPK superfamily that mediate apoptosis or cell proliferation. The RAS-Raf-MEK-ERK signalling cascade, which controls cell proliferation, has received a lot of attention in recent times. Because U0126 inhibits ERK1/2, which results in decreased branching, morphogenesis, increased mesenchymal apoptosis, and decreased epithetical proliferation in foetal lung explants, the ERK1/2 activation appears to be responsible for proper foetal lung development. ERK2 and MEK1 are more important than than ERK1 and MEK2 for embryonic development. ERK2 and MEK1 deficient mice develop a faulty placenta, whereas ERK1 or MEK2 deficient mice are viable, normal in size, and fertile. Defects in AKT signalling have been identified as the root cause of a wide range of human diseases, elucidating the downstream targets and functions that are critical to comprehend. Because the ability to knockout, knockdown, or pharmacologically inhibit specific AKT isoforms and related AGC kinases has greatly improved, previously identified substrates should be examined more rigorously. Cancer, insulin resistance, diabetes, cardiovascular disease, and autoimmune diseases are all caused by improper regulation of the AKT pathways/networks. It is regulated by growth signals/factors such as insulin. By increasing anabolism and decreasing catabolism, the AKT pathway promotes glucose uptake and utilisation, synthesis of glycogen, fatty acid, protein, and cell survival/proliferation. It is crucial for health practitioners and the academic community to develop an efficient model that will effectively regulate the signalling of the AKT pathways/networks. In this paper, the authors have worked on HT carcinoma cells.
The experimental data of the carcinoma cells was acquired for a period of 0–24 h for the three input proteins. The results were obtained based on different concentrations and different samples for ERK protein. The samples were preprocessed using visual tests (P-P plot, Q-Q plot, and box plot), Pearson Correlation Coefficients (PCC), and normality AD test. The AD value for different distribution functions explains how the sample data fits a particular distribution (Normal, Kaplan Meier (KM), and Herd Johnson (HJ) [Citation15]) which can be used for further analysis. The AD values for Maximum Likelihood (ML) and Least Square (LS) techniques for different distribution functions were calculated [Citation16]. A novel computational model is proposed using a machine learning technique and fuzzy system to help in the detection of cell survival/death. The model is validated by evaluating the predicted values and deterministic model and the predicted and observed values from the developed model were plotted. A deterministic model using difference equations was designed and the results agree with the results obtained from the computational model. The authors of this paper employed AD values for the evaluation of uniformity tests and a deterministic model for model validation. A combination of these strategies have not been used in any previous paper.
This paper is organised as follows. Section 2 describes the materials and methods used in the development of the proposed computational model, while Section 3 presents the results and discussion of the model when the AD values for normal, KM and HJ were calculated and also presents the design of the computational model using neural network and fuzzy system. Finally, the paper is concluded in Section 4.
Materials and methods
Materials
The control of cell death/survival decision is based on understanding the information of the cell environment. The inputs are ten different combinations of TNF-EGF-Insulin for 0–24 h as shown in . In , 0-0-0 means, 0 ng/mL of TNF, 0 ng/mL of EGF, and 0 ng/mL of Insulin. Similarly, the rest of the values represent the concentrations of the three input proteins. The signal values were normalised (0: green; 0.5: black; 1: red) to the maximum. All the simulations were carried out in MINITAB, Statistica 11, and MATLAB 2018b software using an Intel (R) Core™ i3-2120 CPU @ 3.30 GHz processor.
Methods
Data was collected for ERK from the heat map in [Citation3,Citation16] for HT carcinoma cells. The computational model is developed using NN and a fuzzy system with the data that is pre-processed and various features were extracted. The various steps for the proposed computational model are shown in and the developed algorithm is presented in . The data was scrutinised, cleaned, and preprocessed. The visual tests, uniformity tests, and Pearson correlation coefficient tests were considered for pre-processing of data.
The visual tests include the quantile-quantile plot (Q-Q plot), frequency distribution (FD) (histogram), box-plot, and probability-probability plot (P-P plot) as shown in . Frequency distribution helps in plotting the frequency with the observed values. The cumulative probabilities of the variable v/s particular distribution are the P-P plots. A P-P plot is analogous to a Q-Q plot. The only change in a Q-Q plot is the quartiles of the dataset, large sample sizes, and its ease to understand.
In , the histogram represents the data and the solid line is the model of the normal distribution based on the mean and standard deviation of the corresponding sample plotted across a range of three standard deviations from the mean. Even though some of the samples significantly differ from the normal distribution (indicated with a star next to the p-value), histograms of some datasets visually resemble a normal distribution. The test performed rejected the null hypothesis of the sizes sampled from a normal distribution (Lilliefors test, p < .05). However, the histogram suggest that the discrepancies are driven by just a few outliers that skew the distribution, which are identical to the normal distribution. We, therefore, chose to assume the normal distribution for those which the discrepancies from the normal distribution are substantial.
In , the normal probability plot is a special case of the probability plot. The shape of the plot signifies the data.
fairly straight signifies symmetric and unimodal.
Inverted C shape signifies unimodal and skewed to the right.
S shape signifies multimodal and supposedly roughly symmetric.
Correlation tests
In this paper, authors have used the normal distribution function (ND), Weibull distribution function (WD), Lognormal 10 distribution function (LND), lognormal e-distribution function (LNe), logistic distribution function (LD) and exponential distribution function [Citation17]. presents the PCC values for different distribution functions for the least square method. Herd Johnson method also known as mean rank is calculated by using the expression i/(n + 1) and Kaplan Meier method calculated using i/n, where n is the number of observations and i is the rank of ith order observable x(i) data arranged from the smallest to largest. There are different types of Correlations tests, namely: Pearson Product-Moment Correlation or Pearson’s r, Point-Bi-serial r, Spearman’s r, and Phi (φ) Correlation. Pearson’s r is defined by EquationEquation (1)(1)
(1) .
(1)
(1)
where CCxy is the covariance of two variables x and y, CCxx and CCyy are the covariance of the single variables x and y respectively.
displays the results of the correlation coefficient, t-value, and p-value calculations for all concentrations used. Likewise, the PCC values are calculated for different distribution functions using the Normal, Kaplan, and HJ methods and are tabulated in . The PCC is only calculated for the least square- approach. PCC values lie in the range of 0 and 1. The higher the value of PCC, the better the distribution curve. From the table, it is seen that the best value of PCC is 0.959 which is obtained for 0 ng/mL of TNF, 100 ng/mL of EGF, and 0 ng/mL of insulin concentration for lognormal distribution function using the normal method.
Uniformity tests
The uniformity of data was achieved by calculating the AD values for different distribution functions using maximum likelihood and least square tests for different concentrations and different samples. AD value is used for uniformity test when different distribution functions are used. An AD value for different distribution functions explains how the sample data fits a particular distribution and that distribution function is used for further analysis.
Assume that the random variables X1, · · ·, Xn form a random sample from an f (x|θ); if X is a continuous random variable, f (x|θ) is a Probability Density Function (pdf), and if X is a discrete random variable, f (x|θ) is a point mass function. The symbol is used to represent the distribution that is dependent on the parameter θ, where θ is a real-valued unknown parameter or a vector of parameters. All observed random sample, x1, · · ·, xn, are defined by EquationEquation (2)(2)
(2) .
(2)
(2)
If f (x|θ) is pdf, f (x1, · · ·, xn|θ) is the joint density function; if f (x|θ) is probability mass function (pmf), f (x1, · · ·, xn|θ) is the joint probability or f (x1, · · ·, xn|θ) as the likelihood function (LF). The main difference between pdf and pmf in terms of random variables is: pdf represents the likelihood of the random variable in the range of discrete values while pmf is the likelihood of the random variable in the range of continuous values.
Motivation for the introduction of LF
The LF depends on the unknown parameter θ, and it is denoted as L(θ). MLE requires the greatest possible LF L(θ) with respect to the unknown parameter θ. L(θ) is defined as a product of n terms that is difficult to maximise. Because log is a monotonic increasing function, maximising L(θ) is equivalent to maximising log L(θ). Log L(θ) is log LF, and is denoted as L(θ) as expressed in EquationEquation (3)(3)
(3) .
(3)
(3)
Maximising l(θ) with respect to θ results in MLE estimation.
The AD tests of all the distribution functions for normal, Kaplan Meier (KM), and Herd Johnson methods for maximum likelihood and least square approach were performed. AD test is a statistical test and an improvement of the CM test. This test gives more weight to the tails of the distribution. An AD value for different distribution functions explains how the sample data fits a particular distribution. This test is a test with the hypothesis that x1, x2…., xn a sample from a specified distribution K(x) is equivalent to a test that x1 = K(x1), x2 = K(x2)……. xn = K(xn) which is a sample from X (0, 1). The AD equation is given by EquationEquation (4)(4)
(4) .
(4)
(4)
Replacing the value of ϕ(z) in EquationEquation (4)(4)
(4) , we obtain EquationEquation (5)
(5)
(5) which is the AD statistic equation:
(5)
(5)
(6)
(6)
An AD test is a comparison of Kn(x) with K (x), where Kn(x) is an EDF or CDF. Considering the hypothesis
H0: F(x) = K(x), −∞ < x < ∞, is rejected(7)
The AD equation of EquationEquation (6)(6)
(6) can be written as EquationEquation (8)
(8)
(8) .
(8)
(8)
where n is the sample size and K(Xj) signifies the CDF for any distribution function.
A fuzzy system and machine learning technique (neural network) was used to develop the computational model which is used to predict cell death/survival for ERK protein. The predicted and observed values from the developed model were also plotted. A deterministic model using difference equations was designed and the results agree with the results obtained from the computational model.
Results and discussion
The experimental data of cell survival or cell death for ERK-MAPK proteins was taken from [Citation3,Citation16] for HT carcinoma cells and was treated with ten cytokine combinations of different input proteins. In every combination, there are 13 different platelets. From every platelet, 10 values were drawn randomly for further consideration. The authors have used perl software to write the code for selecting random values. For the ERK protein, the signal values were normalised (1: red; 0.5: black; 0: green) to the maximum value for ten combinations of three input proteins (ng/mL) for a period of 0–24 h. Since cell survival/death is highly context-dependent and changes with the stress faced by cells, it must be experimentally determined. The authors have performed similar experimental work for ERK protein in [Citation18,Citation19]. Preprocessing of data was done using visual tests, uniformity tests, and Pearson correlation coefficient tests. Normality AD test was used for two different approaches: based on different concentrations and the basis of samples.
Results of uniformity tests
Results based on ten different concentrations of input proteins
The AD statistics were used to measure the area between the fitted curve for the distribution and the non-parametric step function. shows the AD values for normal; KM and HJ technique for the least square (LS) and maximum likelihood (ML) approach for ten various values of three input proteins. A lower value of AD gives a better fitting distribution. In general, the KM method was used to get the AD values. The p-value for this statistic is not considered. shows the results of the different distribution functions (Weibull, Lognormal e, Lognormal 10, exponential, and logistic distribution). The best AD statistics was achieved for 0 ng/mL of TNF, 100 ng/mL of EGF, and 0 ng/mL of insulin concentration for ML and LS methods using normal, lognormal base 10, and Weibull distribution function, while 5 ng/mL of TNF, 0 ng/mL of EGF, and 5 ng/mL of insulin concentration was achieved for using Exponential function. When the statistic values are small, it means data was generated from the ND. In this case, the hypothesis of normality was not rejected. If the statistic values are large, then data can be generated using Cauchy and lognormal distribution. The hypothesis of normality was rejected at the 0.05 significance level.
Results based on samples of ERK
Based on the samples of ERK, the best AD values of Weibull, Lognormal e, exponential, and normal distribution plots using maximum likelihood and least-squares methods for normal, HJ, and KM were plotted. and show the AD (adj) value for ML and LS for four different distribution functions respectively. The AD values for maximum likelihood (ML) and least square (LS) technique using Kaplan–Meier (KM) and Herd Johnson don’ts how any significant changes. The results obtained are almost similar to that of the experimentations performed when the Normal method was used. shows the different values using KM and HJ methods. Comparative analysis is also performed for the two proposed approaches as tabulated in .
The computational analysis was done by calculating adjustment AD statistic values for different distribution functions using different samples of ERK. Using the KM method, 7.55 AD values for different concentrations and 18.4 AD values using different samples for the Weibull distribution function were obtained. Likewise for HJ, 7.6 AD values were achieved based on different concentrations of input proteins for ERK proteins. The results show that when the different distribution functions were used, the Weibull function yields remarkable performance.
After the pre-processing of data, different morphological features are detected and extracted. Some of the extracted features are tabulated in . signifies the minimum, maximum, median, mean, standard deviation, variance, and coefficient of variation value for one concentration of three input proteins. Likewise, for the rest of the nine concentrations of ERK protein, the authors have calculated the features.
Computational modeling using fuzzy system
Different rules for protein-protein interactions were introduced to represent the enzymatic activities and binding in cellular signalling. Encoding rules help in understanding how systems work in possible interactions, states, and bio-molecules [Citation11]. Rules can be processed for the automatic generation of computational or mathematical models which give predictive and explanatory descriptions of the system’s behaviour. Instead of changing a large number of lines of code or equations that require a conventional mathematical model, protein interactions can be modified by changing or adding a single rule which represents the interface. Structure identification is one of the major steps in fuzzy modelling for compound systems. represents the 3D model of ERK-RAS-EGF protein using the fuzzy system. Survival and death are unique options with 1 and 0 values. A fuzzy system includes values between 0 and 1 which is a research gap. To overcome this challenge, the authors have employed a machine-learning approach. In this paper, the authors have used a neural network for modelling.
Computational modeling using neural network
In recent years, innovations in molecular biology have experienced exciting developments. Most proteins communicate using primary structures of which many have a common evolutionary origin while some have unrelated families of proteins that have a common structure. These structures make protein classification difficult. To overcome this problem, a neural network (NN) was used in developing a computational model. Artificial Neural Network (ANN) has been used to solve composite problems in various areas like computer science, engineering, and bioinformatics. NNs are also appropriate for pattern recognition. In this work, the authors have designed an NN based protein classification system using Multilayer Perceptron (MLP) and Radial Basis Function (RBF). ANN is a computational model which functions by defining a set of processing units. A weight is assigned to each connection between neurons and it represents the influence of one neuron on the other. The evolving intelligent behaviour of an NN exists from the interactions between its processing units whose interactions are controlled by the input data. A NN is a set of topologies consisting of several layers, the number of processing units per layer, and connectivity among the layers using different transfer functions. NNs help us to classify and cluster a set of data. Normally to classify a dataset, the dataset must be trained. Training is a supervised procedure, in which a training set is accessible to the NN, and the results are compared with the real value. In each combination, there are 13 different platelets and each platelet comprises 10 values for a period of 0–24 h for every concentration. Out of the total of 130 values, 90 are utilised for training and 40 are used for testing. 10-fold cross-validation is ensured to prevent poor randomisation. In and , the chosen concentrations (TNF-EGF-Insulin) are shown as inputs, while PS exposure, Membrane permeability, DNA, and Caspase cleavage graphs are shown as the four outputs respectively. The chosen concentrations are shown as four inputs and one output (cell survival/death) using NN for MLP and RBF. Multilayer Perceptron results achieve 96.26% training perfection, 96.43% testing perfection, and 94.97% validation perfection using exponential function as hidden activation and Tanh function as output activation.
Validation of model
The developed computational model was validated using two ways. Firstly, values are predicted using the designed novel computational model. The model is validated by evaluating the predicted value which lies in the range of the observed values. The observed and predicted values are plotted and the results are shown in . The results validate the predicted data and indicate that it is up to the mark. The figure also shows the r2 and r2 (adj) values. The authors have also noted that in a biological system (in vivo, in vitro), the predictions are not acceptable. To overcome these authors have used mathematical validation. Secondly, a deterministic model using difference equations is designed.
The model by Bajzer et al. [Citation20] fitted experimental data well; we use it to describe the early events of TNF interactions with cells. Authors have modified the model in [Citation20] and [Citation21] as expressed in EquationEquation (9)(9)
(9) .
(9)
(9)
where square brackets denote molar concentrations of free receptors (R), NC is the complexes (like TNF/TNF-R) bound at the cell membrane, Nin is the internalised complexes, kon and koff are the association and dissociation rate constants for protein binding to its receptor, respectively, kin is the internalisation rate constant of 1 complex and kdeg is the rate constant of lysosomal degradation of the complexes. Our modelling results demonstrate that the pathway leads to a deterministic model for biochemical circuits utilising six molecular species, A, B, C, D, E, and F that collectively summarise the various reactions leading to cell survival and death respectively [Citation20,Citation21], with caspase-3, NF-κB/FLIP, JNK/FLIP, MEK/ERK, FKHR, and JAK/STAT, respectively as expressed by EquationEquation (10)
(10)
(10) . Authors have assumed that after the initial trigger pathways proceed irreversibly to their endpoint.
(10)
(10)
The apoptotic signal, which is modelled phenomenologically using the chemical species A that denotes the caspase 3 pathway, is proportional to the number of internalised ligand/receptor complexes (e.g. Nin) at a constant rate, α [Citation22]. The cell survival pathway inhibits the apoptotic one by destroying A with rates γ [B], φ [C], ε [D], ρ [E], and ζ [F].
The equations for B, C, D, E, and F are:
(11)
(11)
where the variables (NC and Nin) are the same as the differential equation in EquationEquation (9)
(9)
(9) , the differential equation in EquationEquation (11)
(11)
(11) shows that the cell survival signal, modelled phenomenologically with the chemical species B, C, D, E, and F, is proportional to the number of complexes at the cell surface (e.g. NC), with rate constant parameters, β, δ, η, μ and χ. Finally, F, E, D, C, and B can be degraded via ubiquitination and proteasome cleavage and/or irreversibly inhibited by other molecular species, as described by the rate constants kFdeg, kEdeg, kDdeg, kCdeg, and kBdeg, respectively. The differential equation that phenomenologically models the cell survival signal using the chemical species D depends on the number of EGF/EGFR complexes at the cell surface (e.g. NC), with rate constant parameter, η. EquationEquation (12)
(12)
(12) explains the function of ERK.
(12)
(12)
D can be degraded utilising proteasome cleavage and ubiquitination or irreversibly restrained by other molecular species, and these processes are described by the rate constants kEdeg. By solving the differential equation of EquationEquation (12)(12)
(12) (Solution in Appendix 1), EquationEquation (13)
(13)
(13) is obtained.
(13)
(13)
From [Citation20,Citation21] placing the values η = 0.33/min and kEdeg = 0.016/min, EquationEquation (13)(13)
(13) is obtained.
(14)
(14)
By varying the time value in EquationEquation (14)(14)
(14) , different values of E were calculated and plotted. The results are shown in . The coefficient of correlation (r2) gave 0.9569 which is a significant value. The results are shown in prove that the theoretical value and results from the deterministic model fit well. Generally, molecules undergo ballistic motion in a solution. But seeing that a big problem regime is reduced to a small problem with many approximations for different molecules; therefore the motion and dynamics of molecules were also considered.
Conclusion and future work
This paper presented a novel predictive model of cell survival/death-related effects of extracellular signal-regulated kinase protein using a neural network (NN) and fuzzy system. Experimentations were performed using ten different concentrations of 3 different proteins for 300 samples of ERK. The data is pre-processed such that Anderson darling values were evaluated considering different distribution function. Weibull distribution function achieved better results compared to all other functions. The results achieve indicate a 20.6% and 18.2% improvement for the Lognormal and Normal distribution respectively over Weibull based on samples using Kaplan Meier and a 3.2% and 6.9% improvement for the Lognormal and Normal distribution respectively over Weibull based on concentrations using Kaplan Meier. The values were predicted using the novel-designed model which yields remarkable results in comparison with the observed values. Furthermore, the results show that the deterministic model using the differential equations agrees with the designed model. Our designed model aids in the development of problem-solving tools capable of dealing with massive amounts of equations, complex geometries, and non-linearities. Among the many applications of computer modelling, this model helps in the determination of cell survival/death. In the future, authors will try to develop a model using other survival proteins.
Authors contributions
Both SJ and AOS contributed equally to this paper. SJ came up with the idea, ran simulations, and wrote some parts of the paper, while AOS came up with the approach, wrote the findings and discussion sections, and assisted in the writing of other sections. The paper has been read and approved by all authors.
Disclosure statement
No potential conflict of interest was reported by the author(s)
Data availability statement
The data analysed during the current study are not publicly available but is available from the corresponding author on reasonable request.
Additional information
Funding
References
- Keskin S. Comparison of several univariate normality tests regarding type I error rate and power of the test in simulation based on small samples. J Appl Sci Res. 2006;2(5):296–300.
- Jain S. Communication of signals and responses leading to cell survival/cell death using engineered regulatory networks [PhD dissertation]. Solan, Himachal Pradesh, India: Jaypee University of Information Technology; 2012.
- Weiss R. Cellular computation and communications using engineered genetic regulatory networks [PhD dissertation]. MIT; 2001.
- Farrell PJ, Rogers-Stewart K. Comprehensive study of tests for normality and symmetry: extending the Spiegelhalter test. J Stat Comput Simul. 2006;76(9):803–816.
- Salau AO, Jain S, Sood M. Computational intelligence and data sciences: paradigms in biomedical engineering. CRC Press; 2022. p. 272. https://www.routledge.com/Computational-Intelligence-and-Data-Sciences-Paradigms-in-Biomedical-Engineering/Salau-Jain-Sood/p/book/9781032123134
- Jain S, Salau AO. An image feature selection approach for dimensionality reduction based on kNN and SVM for AkT proteins. Cogent Eng. 2019;6(1):1–14.
- Kamal MS, Chowdhury L, Khan MI, et al. Hidden Markov model and Chapman Kolmogrov for protein structures prediction from images. Comput Biol Chem. 2017;68:231–244.
- Jain S. Parametric and non-parametric distribution analysis of AkT for cell survival/death. Int J Artif Intell Soft Comput. 2017;6(1):43–55.
- Jain S, Salau AO. Detection of glaucoma using two dimensional tensor empirical wavelet transform. SN Appl Sci. 2019;1(11):1417.
- Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling. Cancer Gene. 2006;366:2–16.
- Jain S, Naik PK, Bhooshan SV. Mathematical modeling deciphering balance between cell death and cell survival using insulin. Netw Biol. 2011;1(1):46–58.
- Jain S, Chauhan DS. Mathematical analysis of receptors for survival proteins. Int J Pharma and Bio Sci. 2015;6(3):164–176.
- Kevin JA, John AG, Suzanne G, et al. A systems model of signaling identifies a molecular basis set for cytokine-Induced apoptosis. Science. 2005;310(5754):1646–1653.
- Salau AO, Jain S. Feature extraction: a survey of the types, techniques, and applications. Proceeding of the 5th IEEE International Conference on Signal Processing and Communication (ICSC); Noida, India; 2019. p. 158–164.
- Jain S. Design of survival-hazard and mathematical model for high osmolarity glycerol protein using parametric and nonparametric methods. Int J Emerg Technol. 2019;10(3):1–9.
- Gaudet S, Janes KA, Albeck JG, et al. A compendium of signals and responses triggered by prodeath and prosurvival cytokines. Mol Cell Proteomics. 2005;4(10):1569–1590.
- Jain S. Compendium model using frequency/cumulative distribution function for receptors of survival proteins: epidermal growth factor and insulin. Netw Biol. 2016;6(4):101–110.
- Salau AO, Jain S. Adaptive diagnostic machine learning technique for classification of cell decisions for AKT protein. Inform Med Unlocked. 2021;23(1):1–9.
- Salau AO, Jain S. Computational modeling and experimental analysis for the diagnosis of cell survival/death for AKT protein. J Genet Eng Biotechnol. 2020;18(1):11.
- Bajzer Z, Myers AC, Vuk-Pavlović S. Binding, internalization and intracellular processing of proteins interacting with recycling receptors. J Biol Chem. 1989;264(23):13623–13631.
- Roberto C, Marcello F, Diego L, et al. Balance between cell survival and death: a minimal quantitative model of tumor necrosis factor alpha cytotoxicity 2009 biological sciences. Biophys Comput Biol. 2009:1–31. https://arxiv.org/abs/0905.4396
- Salau AO, Demilie WB, Akindadelo AT, et al. Artificial intelligence technologies: applications, threats, and future opportunities. Proceedings of the Advances in Computational Intelligence, its Concepts and Applications (ACI 2022); 2022. p. 265–273.
Appendix 1
Let us consider the differential equation in EquationEquation (1)(1)
(1) .
(1)
(1)
Replacing it in EquationEquation (1)
(1)
(1) , we get
(2)
(2)
Now, we know the
(3)
(3)
Integrating Both Sides, we get
Finally the solution of equation is
(4)
(4)
Now our main equation is (5)
If we compare the equations, EquationEquation (5)(5)
(5) with EquationEquation (2)
(2)
(2) we get
Putting all these values in EquationEquation (4)(4)
(4) , we get
(6)
(6)
(7)
(7)
After solving the EquationEquation (7)(7)
(7) , we get
(8)
(8)
Applying the initial condition, i.e. t = 0 and D = 0 in the EquationEquation (8)(8)
(8) , we get
Now putting the value of c in EquationEquation (8)(8)
(8) , we get
(9)
(9)
Figure 1. Proposed Novel Computational Model for Extracellular Signal Regulated Kinase protein.
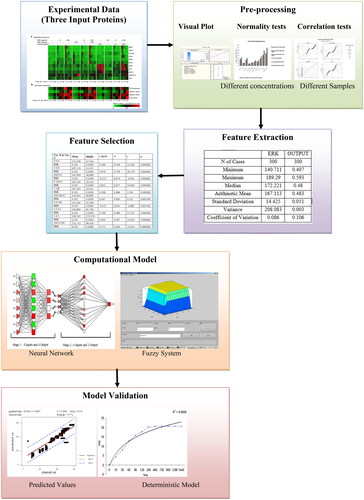
Figure 2. Statistical summary of Extracellular Signal Regulated Kinase protein (a) Kolmogorov-Smirnov plot (b) P-P plot, (c) various statistics values and (d) box plot.
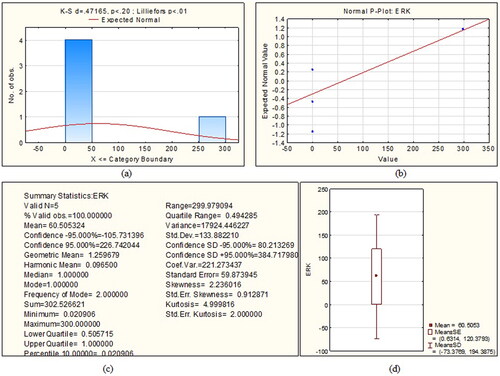
Figure 3. The normal, Kaplan Meier (KM) and Herd Johnson (HJ) method values for the Least Square (LS), and Maximum Likelihood (ML) technique.
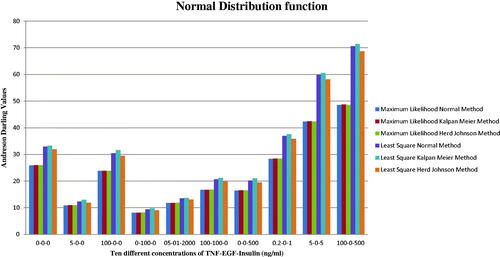
Figure 4. Plots for the Maximum Likelihood technique using normal distribution function.
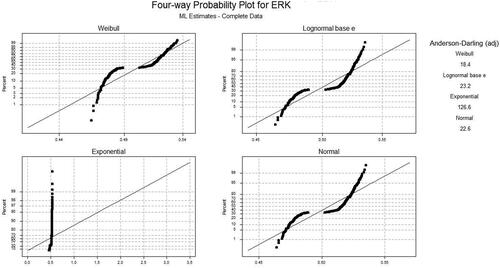
Figure 5. Different plots for the Least Square technique using normal distribution function.
Figure 6. Computational Fuzzy model for predicting cell death /survival.
Figure 7. Computational model using Neural Network.
Figure 8. Graph of observed and predicted value.
Figure 9. Results of deterministic model using difference equations.
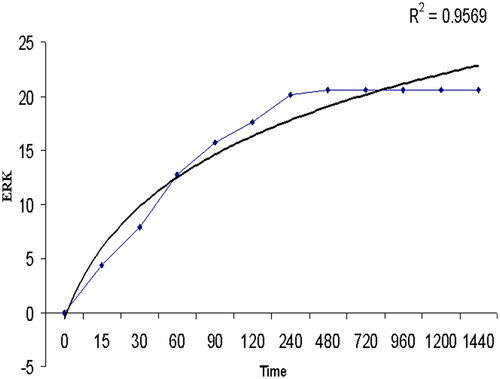
Table 1. Different concentrations of tumour necrosis factor (TNF)-epidermal growth factor (EGF)-insulin.
Table 2. Algorithm for the detection of cell survival/ death.
Table 3. Pearson correlation coefficient (PCC) for least square method.
Table 4. Correlation values for extracellular signal regulated kinase protein.
Table 5. Kaplan Meier, Normal, and Herd Johnson values using maximum likelihood of the different distribution functions for Extracellular Signal Regulated kinase protein.
Table 6. Anderson Darling values for different distribution functions based on different samples.
Table 7. Comparison of Anderson Darling values for the two different computational models.
Table 8. Different features of Extracellular Signal Regulated Kinase (ERK).