
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In this study, non-toxic mercury nanoparticle Prakasine (PRK-NP) was synthesized as per ‘Prakash theory of metal drugs’ and nanoparticle’s non toxicity has been demonstrated by employing in vitro MTT (dose = 320ug/ml), SBR (dose = 80ug/ml) and apoptosis assays (dose = 320ug/ml), and in vivo acute and chronic toxicity studies in mice (n = 12, dose = 900 mg/kg body weight oral), rat (n = 14, dose = 500 mg/kg body weight oral for 18 months), rabbit (n = 14, dose = 500 mg/kg body weight oral for 18 months) and dogs (n = 14, dose = 500 mg/kg body weight oral for 18 months). The MTT, SBR and apoptosis assays established no cytotoxicity, no genotoxicity and no cytolytic anticancer effects. The mice, rat, rabbit and dog studies also indicated nontoxicity. The PRK-NPs significantly reduced the breast cancer tumour in murine mammary tumour - C3H/HeJ model 35% and 43.7% in mice at doses of 200 mg/kg and 500 mg/kg respectively. Also, in xenograft mammary tumour mice model the tumour regressions are 25.7% and 83% in the doses of 500 mg/kg and 1000 mg/kg respectively, compared to standard positive control drugs without any adverse effects and toxicity. Thus, the current study beholds anticipation PRK-NPs may play a vital role in therapeutic.
Introduction
Nearly, 10 million deaths in 2020 due to cancer as it is a leading cause of death globally and the new cases reported were [Citation1]:
breast (2.26 million cases);
lung (2.21 million cases);
colon and rectum (1.93 million cases);
prostate (1.41 million cases);
skin (non-melanoma) (1.20 million cases); and
stomach (1.09 million cases).
The most common types of cancer deaths in 2020 were:
lung (1.80 million deaths);
colon and rectum (916 000 deaths);
liver (830 000 deaths);
stomach (769 000 deaths); and
breast (685 000 deaths).
Every year approximately 400000 children are being affected by the cancer. The most commonly affecting cancers differ from country to country, mainly, the cervical cancer is prevailing in almost 23 countries [Citation2]. The risk factors for cancer in low- and middle-income countries are tobacco use, alcohol consumption, unhealthy diet, physical inactivity, air pollution, noncommunicable diseases, and some chronic infections. The carcinogenic infections such as Helicobacter pylori, human papillomavirus (HPV), hepatitis B virus, hepatitis C virus, and Epstein-Barr virus have produced 13% of cancers in 2018 worldwide [Citation3]. The reason for more cancer deaths in the low-income countries is only 15% comprehensive treatment is reportedly available but more than 90% in high-income countries [Citation4].
Breast cancer (BC) is the most common cancer in women. The middle and older-aged women are mostly being affected by breast cancer globally. The median age at which breast cancer diagnosis is 62, a small number of women are being diagnosed with breast cancer are younger than 45. The rate of incidence have increased by 0.5% year by year and now, the breast cancer is the second leading killer disease among women next to lung cancer. The death rates in breast cancer have been steadily decreasing since 1989 to 2020 about 43% due to increased awareness, early diagnosis and better treatments but slowed down slightly in recent years. Presently, the breast cancer survivors in the United States including those who have completed treatment are more than 3.8 million [Citation5].
However, breast cancer is not limited to females. Carcinoma of the male breast accounts for 0.8%–1% of all breast cancers [Citation6]. When breast cancer is detected and treated early, the chances of survival are very high [Citation7]. Delays in breast cancer treatment >3 months have been associated with more advanced disease stage at diagnosis and poorer survival [Citation8,Citation9].
The factors contributing to the pathophysiology of breast cancer are heat shock protein 70 (HSP70), genes encoding cell signalling molecules (PR, oestrogen receptor (ER), TNF-alpha, glutathione S-transferase family (GSTM1, GSTP1), DNA repair genes (XRCC1, XRCC3, ERCC4/XPF), alcohol and one-carbon metabolism genes (ADH1C and MTHFR), high-penetrance genes such as (BRCA1, BRCA2, p53, PTEN, ATM, NBS1, or LKB1), and low-penetrance genes such as cytochrome P450 genes (CYP1A1, CYP2D6, CYP19) [Citation10]. Breast cancer is categorized as carcinoma and invasive (infiltrating) carcinoma in situ. It is further subclassified as ductal (ductal carcinoma in situ [DCIS]) and lobular (lobular cancer in situ [LCIS]) which are from lobular and mammary ducts origin respectively [Citation6,Citation11]. The diagnosis of breast cancer is being done with one or two or multiple diagnostic methods like Mammography, Magnetic resonance imaging (MRI), Molecular breast imaging (MBI), Breast biopsy, HER-2/neu detection assay and Blood-based assay [Citation6,Citation12–16]. The main types of conventional treatment for breast cancer are surgery, radiation therapy (RT), chemotherapy (CT), endocrine (hormone) therapy (ET), and targeted therapy [Citation17].
The recent trend and development in cancer treatment is immunotherapy. Cancer immunotherapy based on PD-1/PD-L1 immune checkpoint inhibitors and other immunotherapeutic strategies such as cancer vaccines, CAR-T cells, bispecific antibodies, and oncolytic viruses in all breast cancer subtypes are being employed in breast cancer treatment but every strategy and treatment have had certain limitations [Citation18]. Various nanocarriers have been introduced to improve the therapeutic efficacy of anticancer drugs, including liposomes, polymeric micelles, quantum dots, nanoparticles, and dendrimers, they also have the limitations [Citation6]. In spite of all the before said treatments for the breast cancer, targeting cancer cells while avoiding noncancerous cells is the Holy Grail of cancer therapy still [Citation6]. In my previous study, I have reported that the cancer cells could be destroyed by the Cancer specific Cytotoxic-T-Lymphocytes (CsCTL) by the elevated immune gene expression by the stimulation of Prakasine without any toxicity to other cells in zebrafish breast cancer model [Citation19] and in this present study, I have demonstrated that the Prakasine nanomedicine immunotherapy has regressed the tumour cells about 83% immunologically without disturbing the noncancerous cells in breast cancer mice model than the conventional drugs.
Material and methods
The Prakasine nanoparticle (PRK-NP) was synthesised as described in my previous publication [Citation19] and subjected for Ultraviolet-visible absorption spectra, in vitro and in vivo toxicological analysis and in vitro and in vivo immunological anti-tumour experiments. Animal experiments were performed as per guidelines to comply with ARRIVE guidelines. All procedures were performed according to protocols approved by the Institutional Animal Ethics Committee, ACTREC, Tata Memorial Centre, Navi Mumbai and all the research procedures have been carried out in accordance with applicable national or international guidelines.
Ultraviolet-visible absorption study
The step by step UV- visible absorption spectra of PRK-NP synthesis were recorded on VarianCary–500 spectrophotometer (Shimadzu UV 3600) [Citation20–23] to examine the complex formation.
Cytotoxicity study of Prakasine in HEK293 cell line
The cytotoxicity study of PRK-NP was performed by employing the protocols mentioned elsewhere [Citation24–30]. Briefly, the culture medium containing DMEM (Invitrogen), 10% foetal bovine serum (Invitrogen), penicillin (100 IU/ml), streptomycin (100 µg/ml) and 2 mM L-glutamine was seeded with HEK293 cell lines sourced from ATCC at the density of 50,000 cells /well in 96 well microtiter plates after cell dissociation with cell dissociating solution (0.2 % trypsin (Invitrogen), 0.02 % EDTA, 0.05 % glucose in PBS), incubated for 24 h at 37 °C, 5 % CO2 atmosphere and grown for the screening experiments. After trypsinization of the monolayer formed in the cell culture with the media containing 10% FBS the cell count was adjusted to 5.0 x 105 cells/ml and 100 µl of the diluted cell suspension (50,000cells/well) was added to each well of the 96 well microtiter plate. The microtiter plates were incubated at 37° C, 5 % CO2, 95 % air and 100 % relative humidity for 24 h prior to addition of experimental drug dosage concentrations after cell inoculation. The experimental drug Prakasine was dissolved in dimethyl sulfoxide (DMSO) at the concentration of 100 mg/ml and made the final concentrations as 10 μg/ml, 20 μg/ml, 40 μg/ml, 80 μg/ml, 160 μg/ml and 320 μg/ml by diluting with water and medium accordingly. These drug concentrations were added to the microtitre plates after partial monolayer was formed and incubated at 37 °C for 24 h in 5% CO2 atmosphere. The residual experimental drug was removed from the wells after incubation and 100 µl of MTT (5 mg/10 ml of MTT in PBS) was added to each well, again incubated for 4 h at 37 °C in 5% CO2 atmosphere, again the supernatant was discorded and 100 µl of DMSO was added. The formazan formed in this reaction was dissolved by gentle shaking of the plates. The absorbance was measured by a microplate reader at a wavelength of 590 nm. The percentage cell growth inhibition and concentration of test drug needed to inhibit cell growth by 50% (IC50) were calculated using the following formula
Acridine orange/ethidium Bromide (AO/EB) staining to detect apoptosis
Apoptosis study was performed by employing Acridine Orange/Ethidium Bromide (AO/EB) staining as per the protocols mentioned elsewhere [Citation31,Citation32]. Briefly, 25 µl (approx. 1x105 cells) of treated and untreated cells were taken separately in a micro centrifuge tubes and is stained with 5 µl of AO-EtBr (Acridine orange and Ethidium Bromide) for about 2 min followed by gentle mixing. 10 µl of cell suspension is placed onto a microscopic slide and covered with a glass coverslip and examined in a fluorescence microscope using a fluorescein filter. (Higher or lower magnification may be desired depending on cell type. Nuclear morphology should be discernible).
SBR assay to detect cytotoxicity and anti-tumour activity
The SBR assay was performed by employing the protocols mentioned elsewhere [Citation1]. Briefly, the culture medium containing RPMI 1640, 10% foetal bovine serum and 2 mM L-glutamine was seeded with cells and grown for the screening experiments. The cells were inoculated into 96 well microtiter plates at the concentrations of 100 µL densities depending on the doubling time of individual cell lines. The micro titre plates were incubated at 37° C, 5 % CO2, 95 % air and 100 % relative humidity for 24 h prior to addition of experimental drugs after cell inoculation. The experimental drugs, Prakasine, Doxorubicin, Paclitaxel and Adriamycin were dissolved in dimethyl sulfoxide (DMSO) at the concentration of 100 mg/ml and made the final concentrations as 10 μg/ml, 20 μg/ml, 40 μg/ml, and 80 μg/ml by diluting with water and medium accordingly. These drugs concentrations were added to the micro titre plates which were already loaded with cells and incubated for 48 h. Finally, the biological reaction was stopped and cells were fixed in situ by the addition of 50 µl of cold 30 % (w/v) TCA (final concentration, 10 % TCA) and incubated for 60 min at 4 °C. The Sulforhodamine B (SRB) solution (50 µl) at 0.4 % (w/v) in 1 % acetic acid was added to each of the wells after discording the supernatant, washing the plates five times with tap water and air dried. The plates were incubated for 20 min at room temperature, staining, removed the unbound and residual dye with 1 % acetic acid by washing five times, air dried, the bound stain was once again removed by 10 mM trizma base. The absorbance was read by the plate reader at a wavelength of 540 nm with 690 nm reference wavelength. The percentage of cell growth was calculated for test wells relative to control wells and expressed as the ratio of average absorbance of the test well to the average absorbance of the control wells * 100. By employing the measurements of absorbance [time zero (Tz), control growth (C), and test growth in the presence of drug at the four concentration levels (Ti)], the percentage of cell growth was calculated for each drug concentration and the cell growth inhibition percentage was arrived by the formula [Ti/C] x 100 %.
The acute toxicity (maximum tolerated dose) experiment in mice
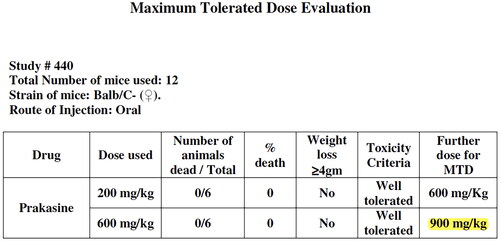
The maximum tolerated dose (MTD) determination study for PRK-NP was performed by studying acute toxicity. Acute toxicity for PRK-NP was determined by employing 12 Balb/C- female mice. Six animals were employed for each dose tested. Mortality and weight loss ≥ 4 g/ mouse were considered to indicate toxicity criteria. Two types of PRK-NP doses were employed for two groups for this MTD study. One is, 200 mg/kg body weight initial and 600 mg/kg body weight as final. The second dose is, 600 mg/kg body weight as initial and 900 mg/kg body weight as final were orally administered as one time dose into mice and mice were monitored for any physical signs of toxicity or mortality till day 5 post-dosing [Citation21,Citation23].
Toxicological study in rat, rabbit and canine in 1990's
The toxicological study in rat, rabbit and canine were performed in 1990s by following OECD guidelines in Kandasamy farms, Kerayur, Namakkal district, Tamil Nadu, India with approval by resolution of Indian Social Services, guided by Department of Laboratory Animal Sciences, Madras Veterinary College. The histopathological examinations were performed in Department of Pathology in Namakkal Veterinary College and Research Centre, Namakkal.
14 Wistar albino rats (7 males and 7 females), approximately 4–6 months of age, were obtained in good health from the breeder, Namakkal District, Tamil Nadu. The rats were acclimatized to the conditions of the testing facility for about 7 days before initiation of the experiment. Before initiation of Prakasine administration the body weights of rats were ranged from 90–100 grams. In a separate isolated clean room the temperature maintained at 18–25 °C, with 30–70% relative humidity, natural ventilation, and a 12-h light-dark cycle each group (4 control and 10 trial) of animals were housed in Torsons rat cages. Noise was controlled below 50 dB. The animals were fed 2 times daily with a medicated diet at 9:00 and 15:00. Ad libitum distilled water was provided to animals for the entire duration of 18 months study.
The Prakasine was administered at the dose rate of 500 mg/kg body weight daily along with feed. The blood picture, liver function tests, kidney function tests, neurological examination including cognisance, weight gain, morbidity and mortality data were generated before the study initiation, 6th, 12th months during the study and end of the study at 18th month [Citation1].
Fourteen New Zealand White (albino) breed rabbits (7 males and 7 females), approximately 4–6 months of age, were obtained in good health from the breeder, Namakkal District, Tamil Nadu. The rabbits were acclimatized to the conditions of the testing facility for about 7 days before initiation of the experiment and all rabbits were vaccinated as per the recommended vaccination schedule. Before the initiation of the Prakasine administration, the body weights of rabbits ranged from 1–1.1kgs. In a separate isolated clean room, the temperature was maintained at 18–25 °C, with 30–70% relative humidity, natural ventilation, and a 12-h light-dark cycle each group (4 control and 10 trial) of animals were housed in rabbit cages. Noise was controlled below 50 dB. The animals were fed 2 times daily with a medicated diet and fodder at 9:00 and 15:00. Ad libitum distilled water was provided to animals for the entire duration of 18 months study. The Prakasine was administered at the dose rate of 500 mg/kg body weight daily along with feed. The blood picture, liver function tests, kidney function tests, and neurological examination including cognisance, weight gain, morbidity and mortality data were generated before the study initiation, 6th, and 12th months during the study and end of the study 18th month [Citation1,Citation33].
14 Rajapalayam breed dogs (7 males and 7 females), approximately 4–6 months of age, were obtained in good health from the breeder, Namakkal District, Tamil Nadu. The dogs were acclimatised to the conditions of the testing facility for about 7 days before initiation of the experiment and all dogs were vaccinated as per recommended vaccination schedule. Before the initiation of Prakasine administration, body weights of the dogs ranged from 6.0–7.0 kgs. In a separate isolated clean room the temperature was maintained at 18–25 °C, with 30–70% relative humidity, natural ventilation, and a 12-h light-dark cycle the dogs were housed and were fed in individual stainless steel cages measuring 100 cm in height, 100 cm in length, and 90 cm in width. Noise was controlled below 50 dB. The animals were fed 2 times daily with a medicated diet at 9:00 and 15:00. Ad libitum distilled water was provided to animals for the entire duration of 18 months study. All the dogs were regularly subjected to exercise and social interaction. The Prakasine was administered at the dose rate of 500 mg/kg body weight daily along with food. The blood picture, liver function tests, kidney function tests, and neurological examination including cognisance, weight gain, morbidity and mortality data were generated before the study initiation, 6th, 12th months during the study and end of the study at the 18th month [Citation34,Citation35].
Breast cancer study with murine mammary tumour model
The donor and tumour-bearing mice were developed by passaging the cryopreserved murine mammary tumour cell lines—C3H/HeJ. The immunodeficient NOD-SCID mice were employed to develop the mammary tumour. The mice were randomized into five groups (n = 6/group) before starting the treatment such as tumour-bearing control group (Group A), Adriamycin- treated group as a positive control group (Group B), Paclitaxel - treated group as a second positive control group (Group C), Prakasine (200 mg/kg) as a first treatment group (Group D) and Prakasine (500 mg/kg) as a second treatment group (Group E). The Prakasine nanoparticle complex was dissolved in DMSO, two doses of PRK-NP complexes were administered at a dose of 200 mg/kg oral 5 days a week X 4 weeks (Group D) and 500 mg/kg oral 5 days a week X 4 weeks (Group E). The Adriamycin (ADR) in group B was administered at the dose rate of 2.5 mg/kg I.P weekly X 4 weeks and the Paclitaxel in group C was administered at the dose rate of 4 mg/kg I.P once a week X 4 weeks. The tumour growth measurements and tumour volume were measured by a digital Vernier calliper (Pro-Max, Electronic Digital Calliper, Fowler-NSK, USA). The body weight, tumour volume and mortality were observed in experimental mice at regular intervals for around 30 days. Three data were considered in this study for the assessment of the efficacy of PRK-NP such as Relative Tumour Volume (RTV in cc), T/C (ratio of test versus control) and survival. The tumour volume was arrived at by the formula [(w1 × w1 × w2) × (π/6)], where w1 is the smallest tumour in diameter (cm) and w2 is the largest tumour. RTV was calculated as tumour volume on the day of measurement/tumour volume on day 1. The antitumor efficacy is indicated by the T/C ratio and percent tumour regression values were calculated as follows:
Relative Tumour Volume (RTV)
T/C = RTV_Test/ RTV_Control
Tumour Regression % = 100-[T/C*100]
If the T/C values were ≤0.42 it is considered the biological activity is significant [Citation21,Citation23].
Breast cancer study with xenograft mammary tumour mice model
To obtain the donor mice the human breast cancer derived tumour (MCF-7) was passaged from the cryopreserved samples and small pieces of ∼4–5 mm of MCF-7 xenografts were subcutaneously implanted on to the flank region of male NOD-SCID mice for induction of tumour. The mice were randomized into five groups (n = 6/group) before starting the treatment once the tumour volume reached approximately 50–60 mm3 such as the tumour-bearing control group (Group A), Adriamycin- treated group as a positive control group (Group B), Paclitaxel - treated group as a second positive control group (Group C), Prakasine (500 mg/kg) as a first treatment group (Group D) and Prakasine (1000 mg/kg) as a second treatment group (Group E). The water was used as a vehicle for Adriamycin. The Prakasine nanoparticle complex was dissolved in DMSO, two doses of PRK-NP complexes were administered at a dose of 500 mg/kg oral 5 days a week X 4 weeks (Group D) and 1000 mg/kg oral once a week X 4 weeks (Group E). The Adriamycin (ADR) in group B was administered at the dose rate of 5 mg/kg I.P weekly X 4 weeks and the Paclitaxel in group C was administered at the dose rate of 4 mg/kg I.P once a week X 4 weeks. The tumour growth measurements and tumour volume were measured by digital Vernier calliper (Pro-Max, Electronic Digital Calliper, Fowler-NSK, USA). The body weight, tumour volume and mortality were observed in experimental mice at regular intervals for a period of around 30 days. Three data were considered in this study for the assessment of the efficacy of PRK-NP such as Relative Tumour Volume (RTV in cc), T/C (ratio of test versus control) and survival. The tumour volume was arrived at by the formula [(w1 × w1 × w2) × (π/6)], where w1 is the smallest tumour in diameter (cm) and w2 is the largest tumour. RTV was calculated as tumour volume on the day of measurement/tumour volume on day 1. The antitumor efficacy is indicated by T/C ratio and percent tumour regression values were calculated as follows:
Relative Tumour Volume (RTV)
T/C = RTV_Test/ RTV_Control
Tumour Regression % = 100-[T/C*100]
If the T/C values were ≤0.42 it is considered the biological activity is significant [Citation21, Citation23].
Statistical analysis
Group means were compared by Student’s t-test or analysis of variance as indicated.
Results
The non-toxicity of PRK-NP was demonstrated with in vitro and in vivo experiments. Subsequently, in-vitro and in-vivo studies performed to establish the immunological anticancer effect of PRK-NP in BC mice model.
Non-toxic PRK-NP complex formation confirmation by UV study
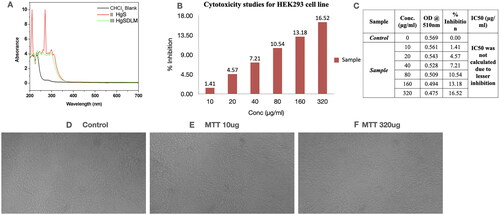
The nano chemical characters of PRK-NP has been reported in my previous publication [Citation19]. In this study, the UV Spectral Analysis of PRK-NP synthesis data has revealed complex formation. In , the ultraviolet-visible absorption spectra of CHCl3 in black line is the blank, HgS (Mercury Sulphide) in green line and HgSDLM (Mercury Sulphide DL-Methionine) in red line confirms the PRK-NP complex formation.
Figure 1. UV Characterization and MTT cytotoxicity assay of Prakasine. A UV Spectral Analysis of PRK-NP synthesis. Ultraviolet-visible absorption spectra of CHCl3 in black line is blank,HgS in green line and HgSDLM in red line reveal the PRK-NP complex formation. Citation1B, C, D, E, and F : MTT assay of Prakasine. The MTT assay reveals that there is no significant toxicity of PRK-NP in HEK293 cell line experiment even though the negligible toxicity in 320ug PRK-NP dose might be due to DMSO.
Abbreviations: PRK-NP-: Prakasine, CHCl3: chloroform, HgS: Mercury Sulphide, HgSDLM: Mercury Sulphide DL-Methionine.
Demonstration of non-toxicity of PRK-NP by in vitro MTT cytotoxicity, Acridine orange/ethidium Bromide (AO/EB) staining and SBR assays
The MTT assay () reveals that there is no significant toxicity of PRK-NP in HEK293 cell line experiment even though the negligible toxicity in 320 μg/ml PRK-NP dose might be due to DMSO.
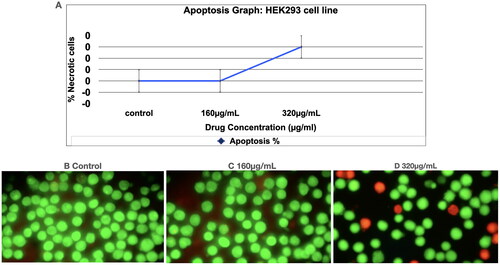
In Acridine Orange/Ethidium Bromide (AO/EB) staining, the dual staining was examined under a fluorescent microscope. Normal cells are seen with circular nucleus uniformly distributed in the centre of the cell which is seen in the control () and sample (, 160 μg/ml). At 320 μg/ml () some cells show orange fluorescence by EtBr staining. Cells that have taken up complete EtBr are the necrotic cells. The viable cells are in green fluorescence. The necrotic cells are with orange stain (short arrow) and late apoptotic cells with yellow green staining (long arrow). The apoptosis study () of Prakasine reveals that the Prakasine does not induce significant apoptosis in HEK293 cell line experiments with negligible necrotic cells in 320 µg/mL PRK-NP dosage might be due to DMSO. As there is no nuclear condensation in the treated cell except few necrotic cells in 320 μg/ml treated cells it is indicated that Prakasine does not have genotoxicity.
Figure 2. Acridine Orange/ethidium Bromide (AO/EB) staining assay to detect apoptosis.
: Apoptosis study of Prakasine. The apoptosis study of Prakasine reveals that the Prakasine does not induce significant apoptosis in HEK293 cell line experiments with negligible necrotic cells in 320µg/mL PRK-NP dosage might be due to DMSO. Viable cells _ Green fluorescence. Necrotic cells with orange stain (short arrow), Late apoptotic cells with yellow green staining (Long arrow)
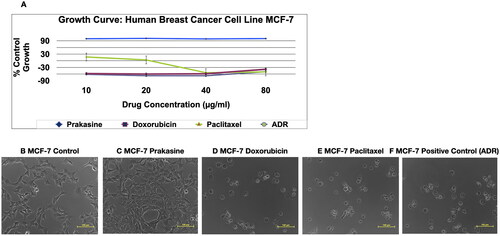
The SBR assay, , and C indicate that Prakasine does not have cytolytic anti-cancer activity and cytotoxicity in MCF-7 cell line culture as there is a 100% cell growth. The B is the control, the C, D, E and F are Prakasine, Doxorubicin, Paclitaxel and Positive control, ADR (Adriamycin). The ADR is the ACTREC’s positive control as per institute protocols. The percentages of cell growth are 100%, −38%, −50% and −49% in Prakasine, Doxorubicin, Paclitaxel and positive control, ADR respectively compared with control. have demonstrated the non-toxicity of the PRK-NP.
Figure 3. Non-cytolytic anti-Cancer activity and non-cytotoxicity of Prakasine by SBR assay. A, and C indicate that Prakasine does not have cytolytic anti-cancer activity and cytotoxicity in MCF-7 cell line culture as there is a 100% cell growth. The B is the control, The C, D, E and F are Prakasine, Doxorubicin, Paclitaxel and Positive control, ADR. The ADR is the ACTREC’s positive control as per institute protocols. The percentages of cell growth are 100%, −38%, −50% and −49% in Prakasine, Doxorubicin, Paclitaxel and positive control, ADR respectively compared with control. ADR: Adriamycin, ACTREC : Advanced Centre for Treatment, Research & Education in Cancer
Non-toxicity establishment of PRK-NP by Maximum Tolerated dose Evaluation study in mice
The maximum tolerated dose evaluation study in mice (), the gradual oral doses of PRK-NP 200 mg/kg body weight, 600 mg/kg body weight and 900 mg/kg body weight among 12 Balb/C- female mice for the duration of 5 days for each dose did not reveal any weight loss and toxicity. There was 0% death among 12 animals. This data demonstrates the non toxicity of PRK-NP in mice.
Figure 4. Maximum Tolerated Dose Evaluation of Prakasine.
Note: Toxicity Criteria: Mor and weight loss….
Non toxicity establishment in rat, rabbit and canine studies
In the rat toxicological study (), all the blood parameters (WBC − 3.9x 103/mm3 − 5.8x103/mm3, RBC − 7.9x106/mm3 − 8.2x106/mm3, Hb − 13 g/dL − 16 g/dL, Platelets - 450x103/mm3 - 490x103/mm3, Lymphocytes − 55% −65%, Monocytes − 7% − 8.2%, Neutrophils − 30% − 40%, Basophils − 0.3% − 0.5% and Eosinophils − 0.4% − 0.5%), liver function tests (SGOT − 60 IU/L − 65 IU/L and SGPT − 27 IU/L − 34 IU/L) and kidney functions tests (Blood urea nitrogen − 13 mg/dL − 15 mg/dL and creatinine − 0.31 mg/dL − 0.35 mg/dL) in all the animals are with in the normal range in before treatment, during treatment and after treatment have indicated that there is no toxicity and adverse effect of PRK-NP. The Neurological Examination including cognisance score range is between 8–10 indicates that there is no neurological and cognisance defect created by PRK-NP. The 0% morbidity, 0% mortality and some 8% increase in weight gain of the animal also confirm that there is no toxicity produced by the PRK-NP during the 18 months of treatment.
Table 1. Toxicological study in Wistar albino rats for the duration of 18 months (the readings are average values of triplicates of four animals in control and ten animals in trials from blood samples).
In the rabbit toxicological study (), all the blood parameters (WBC - 9x 109/L − 12 9x 109/L, RBC − 5.8 9x 1012/L − 6.8 9x 1012/L, Hb − 113 g/L − 139 g/L, Lymphocytes − 65% − 70%, Monocytes − 2% − 3%, Heterophils − 30% − 89%, Basophils − 0.5% − 1% and Eosinophils − 1% −2%), liver function tests (SGOT − 15 IU/L − 19 IU/L and SGPT − 7 IU/L − 8 IU/L) and kidney functions tests (Blood urea nitrogen − 13 mmol/L − 15 mmol/L and creatinine − 0.12 mmol/L − 0.14 mmol/L) in all the animals are within the normal range in before treatment, during treatment and after treatment have indicated that there is no toxicity and adverse effect of PRK-NP. The Neurological Examination including the cognisance score range is between 8–10 indicates that there is no neurological and cognisance defect created by PRK-NP. The 0% morbidity, 0% mortality and some 5% increase in weight gain of the animal also confirm that there is no toxicity produced by the PRK-NP during the 18 months treatment.
Table 2. Toxicological study in New Zealand White rabbits for the duration of 18 months (the readings are average values of triplicates of four animals in control and ten animals in trials from blood samples).
In the dog toxicological study (), all the blood parameters (WBC - 6x 103/mm3 − 8x103/mm3, RBC − 8.1x106/mm3 − 15x106/mm3, Hb − 15 g/dL − 18 g/dL, Platelets − 380x103/mm3 − 390x103/mm3, Lymphocytes − 24% − 37%, Monocytes − 7% − 8.2%, Neutrophils − 50% − 82%, Basophils − 0.4% − 1% and Eosinophils − 2% − 3%), liver function tests (SGOT − 50 IU/L − 55 IU/L and SGPT − 33 IU/L − 43 IU/L) and kidney functions tests (Blood urea nitrogen − 22 mg/dL − 25 mg/dL and creatinine − 1.2 mg/dL − 1.3 mg/dL) in all the animals are within the normal range in before treatment, during treatment and after treatment have indicated that there is no toxicity and adverse effect of PRK-NP. The Neurological Examination including cognisance score range is between 8–10 indicating that there is no neurological and cognisance defect created by PRK-NP. The 0% morbidity, 0% mortality and some 5% increase in weight gain of the animal also confirm that there is no toxicity produced by the PRK-NP during the 18 months treatment.
Table 3. Toxicological study in Rajapalayam dogs for the duration of 18 months (the readings are average values of triplicates of four animals in control and ten animals in trials from blood samples).
Immunological anti-tumour activity demonstration of PRK-NP with murine mammary tumour model
The in vitro anticancer activity cell-based assay results reveal that there is no direct cytolytic anti-cancer activity and no toxicity to the cells. This encouraging observation lead to evaluate the PRK-NP in vivo immunological anticancer activity study in animal models. The PRK-NP was evaluated in Balb/C mice to find out the Maximum Tolerated Dose (MTD) by oral administration route. Three concentrations of PRK–NP were orally administered to the Balb/C mice such as 200 mg/kg, 600 mg/kg and 900 mg/kg, and was observed that the animals well tolerated at the highest test dose of 900 mg/kg (), also observed that there was no infliction and harmful effects to the normal animals including body weight. Even though, there was a very higher dose (200 − 900 mg/kg) in acute toxicity study compared to actual dosing during efficacy study i.e. 200 mg/kg and 500 mg/kg the mice did not show any significant changes in locomotion, behaviour, neurological and regular activities. Based on these observations, for efficacy testing, the dosing protocols were designed as 200 mg/kg and 500 mg/kg oral 5 days a week X 4 weeks (mice per group n = 6).
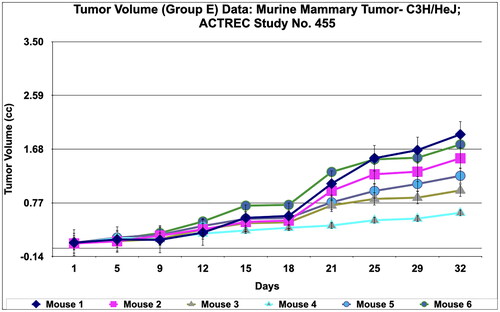
The immunological in vivo anticancer activity of PRK-NPs was tested against mammary tumour—C3H/HeJ in mice. The dosing protocol is, the two doses of PRK-NP complexes were administered at a dose of 200 mg/kg oral 5 days a week X 4 weeks (Group D) and 500 mg/kg oral 5 days a week X 4 weeks (Group E). The Adriamycin (ADR) in group B was administered at the dose rate of 2.5 mg/kg I.P weekly X 4 weeks and the Paclitaxel in group C was administered at the dose rate of 4 mg/kg I.P once a week X 4 weeks. Tumour volume, body weight, and signs of toxicity were monitored and recorded on respective days: 1, 5, 9, 12, 15, 18, 21, 25, 29 and 32. The results show that PRK- NPs exhibit potent anticancer activity against murine mammary tumour—C3H/HeJ. It was interesting to note that the PRK-NP demonstrated substantial tumour reduction with respect to tumour-bearing control in a time-dependent manner ().
Figure 5. Relative tumor volume and activity Criteria of Murine mammary tumor model treated with Prakasine for 32 days. A: The RTV of A, B, C, D, and E are 23.17, 9.87, 6.23, 16.26, and 14.86 respectively after 32 days of Prakasine treatment. The reduced values of 16.26 and 14.86 of D and E indicate that the Prakasine has reduced the tumour volume compare to control 23.17. (p < 0.05). B: The 32nd day T/C value of B, C, D, and E are 0.43, 0.27, 0.70 and 0.64 respectively indicate that the Prakasine is having lower tumour reduction effect compare to ADR and Paclitaxel, (p < 0.05) RTV: Relative Tumour Volume on day of measurement/Tumour Volume on day 1.*T/C: Tumour/ Control, * ADR - Adriamycin, ACTREC: Advanced Centre forTreatment,Research & Education in Cancer.
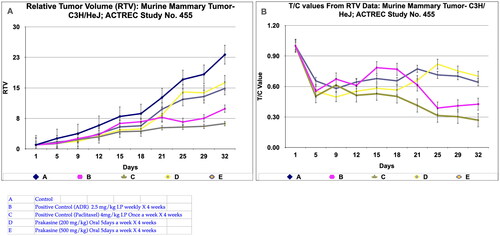
However, in Paclitaxel group C, the regression effect was observed as early as 5th day and got reduced to 80.5%, 95%, 35%, and 43.7% for the Adriamycin, Paclitaxel, PRK-NP 200 mg/kg and PRK-NP 500 mg/kg respectively at the end of the study. It was noticed that PRK-NPs have reduce the mammary tumour by 35% and 43.7% in D and E groups respectively (). As evident from relative tumour volume and T/ C ratio (Mean relative tumour volume for PRK-NP or standard drugs Adriamycin and Paclitaxel treated groups over that of tumour-bearing control group on individual day data), the PRK-NPs exhibited a significant reduction in tumour volume by day 5th in oral dosing protocol and continued till the end of the study (percent regression 35% and 43.7%; T/C = 0.7 and 0.64 (). The tumour size seen in the animal groups was found to be concordant with the tumour volume recorded.
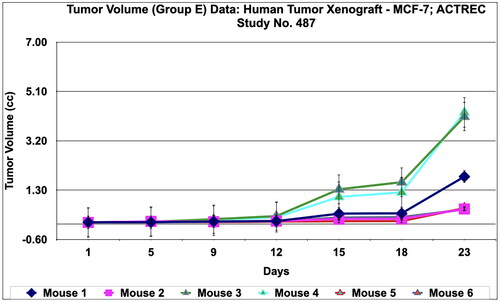
During in vivo anticancer study, the activity of PRK-NPs were compared with standard drugs Paclitaxel and ADR. The two positive control standard drugs have more tumour reduction activity than PRK-NPs. The present study showed that the mice survival rate was 100 % in PRK-NPs (). All mice displayed normal activities, and no significant bodyweight loss was noted (). In all the five groups, the tumour volume has increased gradually in all the six animals from 1st day to 32nd day ().
Figure 6. Survival and animal body weight (grams) data of Murine mammary Tumour- C3H/HeJ model treated with Prakasine for 32 days. A: All the groups from A-E, the animals survival rate is 100% in the study duration of 32 days. B: The Prakasine treated groups D and E is slightly having more weight gain compared to A, B and C groups indicates the efficacy of the Prakasine in the study duration of 32 days.
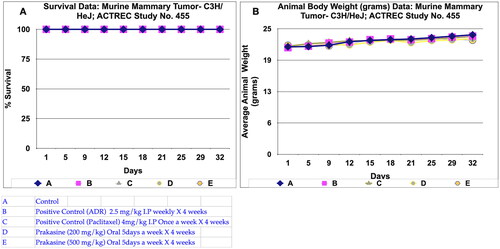
Figure 7. Tumour volume of group a and tumor volume of group B data of Murine mammary Tumour- C3H/HeJ model treated with Prakasine for 32 days. A and B: In both the group A and B the tumour volume has increased gradually in all the six animals from 1st day to 32nd day.
Figure 8. Tumour volume of group C and tumor volume of group D data of Murine mammary Tumour- C3H/HeJ model treated with Prakasine for 32 days. A and B: In both the group C and D the tumour volume has increased gradually in all the six animals from 1st day to 32nd day.
Figure 9. Tumour volume of group E data of Murine mammary Tumour- C3H/HeJ model treated with Prakasine for 32 days. In the group E the tumour volume has increased gradually in all the six animals from 1st day to 32nd day.
Immunological anti-tumour activity demonstration of PRK-NP with xenograft mammary tumour mice model
The in vivo immunological antitumor property of PRK-NP was further evaluated in the human xenograft mammary tumour model by employing Adriamycin and Paclitaxel as the reference antitumor agents.The dosing protocol is, the two doses of PRK-NPs complexes were administered at a dose of 500 mg/kg oral 5 days a week X 4 weeks (Group D) and 1000 mg/kg oral once a week X 4 weeks (Group E). The Adriamycin (ADR) in group B was administered at the dose rate of 5 mg/kg I.P weekly X 4 weeks and the Paclitaxel in group C was administered at the dose rate of 4 mg/kg I.P once a week X 4 weeks. Tumour volume, body weight, and signs of toxicity were monitored and recorded on respective days: 1, 5, 9, 12, 15, 18, and 23. The results show that PRK- NPs exhibit potent anticancer activity against xenograft mammary tumour. It was interesting to note that the PRK-NP demonstrated substantial tumour reduction with respect to tumour bearing control in a time- dependent manner ().
Figure 10. Relative tumor volume and activity Criteria of human tumor Xenograft - MCF-7 mice model treated with Prakasine for 23 days. A: The RTV of A, B, C, D, and E are 116.8, 42.81, 96.94, 90.24, and 33.44 respectively after 23 days of treatment with Prakasine. The reduced values of 90.24 and 33.44 of D and E indicate that the Prakasine has reduced the tumour volume compare to control 116.8 (p < 0.05). B: The 23nd day T/C value of B, C, D, and E are 0.37,0.83, 0.77 and 0.29 respectively indicate that the Prakasine is having better tumour reduction effect compare to ADR and Paclitaxel in group E, (p < 0.05). RTV = Relative Tumour Volume = Tumour Volume on day of easurement/Tumour Volume on day 1.*T/C: Tumour/ Control, * ADR: Adriamycin, ACTREC : Advanced Centre for Treatment, Research & Education in Cancer.
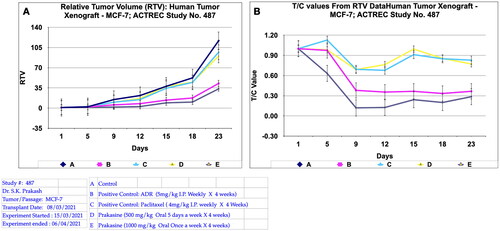
However, in PRK-NP group E , the regression effect was observed as early as 5th day and got reduced to 89%, 18%, 25.7% and 83% for the Adriamycin, Paclitaxel, PRK-NP 500 mg/kg and PRK-NP 1000 mg/kg respectively at the end of the study. It was noticed that PRK-NPs have reduced the xenograft mammary tumour by 25.7% and 83% in D and E groups respectively (). As evident from relative tumour volume and T/ C ratio (Mean relative tumour volume for PRK-NP or standard drugs Adriamycin and Paclitaxel treated groups over that of tumour-bearing control group on individual day data), the PRK-NPs exhibited a significant reduction in tumour volume by day 5th in oral dosing protocol and continued till the end of the study (percent regression 25.7% and 83%; T/C = 0.77 and 0.29 (). The tumour size seen in the animal groups was found to be concordant with the tumour volume recorded.
During in vivo anticancer study, the activity of PRK-NPs were compared with standard drugs Paclitaxel and ADR. The two positive control standard drugs have less tumour reduction activity than PRK-NPs. The present study showed that the mice survival rate was 100 % in PRK-NPs (). All mice displayed normal activities, and no significant bodyweight loss was noted (). In all the five groups, the tumour volume has increased gradually in all the six animals from 1st day to 23rd day ().
Figure 11. Survival and animal body weight (grams) data of human tumor Xenograft - MCF-7 mice model treated with Prakasine for 23 days. A : All the groups from A -E except B , the animals survival rate is 100% in the study duration of 23days. B : The Prakasine treated groups D and E is slightly having more weight gain compared to A, B and C groups indicates the efficacy of the Prakasine in the study duration of 23 days.
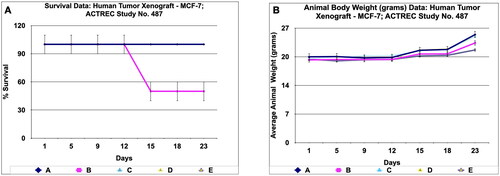
Figure 12. Tumour volume of group a and tumor volume of group B data of human tumor Xenograft - MCF-7 mice model treated with Prakasine for 23 days. A and B : In both the group A and B the tumour volume has increased gradually in all the six animals from 1st day to 23rd day.
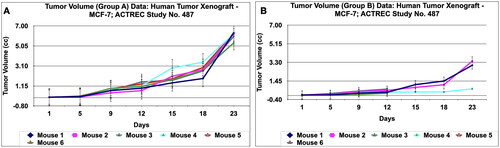
Figure 13. Tumour volume of group C and tumor volume of group D data of human tumor Xenograft - MCF-7 mice model treated with Prakasine for 23 days. A and B : In both the group C and D the tumour volume has increased gradually in all the six animals from 1st day to 23 rd day.
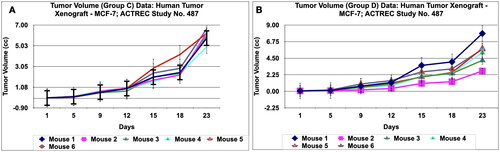
Figure 14. Tumour volume of group E data of human tumor Xenograft - MCF-7 mice model treated with Prakasine for 23 days. In the group E the tumour volume has increased gradually in all the six animals from 1st day to 23 nd day.
Discussion
Unless the breast cancer is detected in early stage it is impossible to increase the survival rate of the patients with the conventional treatment for breast cancer such as surgery, radiation therapy (RT), chemotherapy (CT), endocrine (hormone) therapy (ET), and targeted therapy inspite of good diagnostic methods employed such as Mammography, Magnetic resonance imaging (MRI), Molecular breast imaging (MBI), Breast biopsy, HER-2/neu detection assay and Blood-based assays.
Cancer immunotherapy, which helps the host’s immune system for producing necessary immune components to recognize the tumour antigen and finally attacks the tumour harbouring cells by the Cancer Specific Cytotoxic T-Lymphocytes (CsCTL), is a promising strategy for cancer treatment. The limitation of the cancer immunotherapy is the restriction by the immunosuppressive (i.e. immunologically cold) tumour microenvironment (TME) [Citation36]. Employing the immune system to fight against cancer by immunotherapy is the recent, emerging, possible, powerful and potentially revolutionising treatment strategy and approach to treat cancer including the secondary metastasis.
The solid tumours destroying efficacy of the therapeutic cancer vaccines, chimeric antigen receptor (CAR) T cells, and other cancer immunotherapy strategies have been still challenging even though these kinds of immunotherapy strategies have shown promising success in both pre-clinical and clinical studies of hematological malignancies [Citation32,Citation37,Citation38,Citation39]. The key factors that contribute to the poor clinical outcomes of immunotherapy of solid tumours are immunosuppressive (i.e. immunologically cold) tumour microenvironment (TME) including malignant cells, deactivated/compromised immune cells, and soluble factors [Citation36,Citation40–45]. In the immune induction study in metastatic lobular breast cancer with the combination of carboplatin to the PD-L1 blockade, the carboplatin alone did not produce any significant role, especially, neither in immune cell composition nor in MHC class I expression and increase in cGAS–STING signalling but with the addition of anti-PD-L1, there is increasing in CD8+ T cell infiltration and higher expression of immune-related gene signatures particularly in triple-negative ILC (TN-ILC) patients but these responses were not longstanding [Citation46].
Immunotherapy plays an important role by enhancing immune network cascading mechanism of the host (the patient) to recognize cancer as a foreign antigen and destroy the cancer cells by the CTL lysis from antigen presentation to T-cells (primarily by dendritic cells) for anti-tumour immunity leading to trafficking and infiltration of the effector T cells to the tumour bed. The infiltrating effector/cytotoxic T cells sense and recognize tumour cells and eliminate them as in the case of infection or in other foreign antigens such as an allograft. Even though, William Cooley reported the immunotherapy phenomenon 100 years back, the field of immunotherapy yet to reach the milestone to produce clinically meaningful interventions outcome despite the contemporary successes with checkpoint inhibitors [Citation47]. The Nobel Prize was awarded in 2018 to James Allison at MD Anderson Cancer Centre who discovered CTLA-4, and Tasuku Honjo at Kyoto University who discovered PD-1. Their discovery has been recognized by the scientific community and these checkpoints has been a important stepping stone in cancer immunotherapy research. In the treatment of melanoma, lung cancer, and microsatellite unstable colon cancer the Immune checkpoint inhibitors (ICIs) have been successfully employed and have produced some clinical outcomes in the treatment of cancer patients but the same degree of success has not been obtained in breast cancer as in the case of melanoma and lung cancer by the ICIs [Citation46].
Nano-immunotherapy has not cured the cancer so far but has given some beneficial outcome in breast cancer patients.
To improve efficacy, therapeutic delivery and better immunostimulation in cancer treatment, importance is being given to the drugs which can reprogram cancer-associated fibroblasts (CAFs) [Citation48]. Additionally, the research is also being focused on nano-immunotherapy (combination of nano-drugs and immunotherapy) to increase cancer patient outcomes with high potential as it has been already demonstrated in triple-negative breast cancer patients with the combination of nanoparticle albumin-bound paclitaxel and the immune checkpoint blocker (ICB) antibody, atezolizumab [Citation48–51]. However, the CAF reprogramming drugs and nano-immunotherapy have the modest effect with median survival for patients having the cancer history of less than two years as the bottleneck tumour microenvironment (TME) inhibits the delivery of nanoparticles, low antibodies, induced hypoxia. The abnormalities of TME can also causes immunosuppression which leads to tumour progression, metastasis, drug resistance and compromised therapeutic outcomes eventually [Citation52–54]. The stiffening of the tumour and accumulation of mechanical forces at the tumour site have been developed due to dense tumour interstitial space, abundant cancer-associated fibroblasts (CAFs), collagen and hyaluronan in triple-negative breast cancers by the TME abnormalities [Citation55–59]. As the tumour vessel compression due to mechanical forces within the TME there would be low drug delivery, limitation of tissue oxygenation and vascular dysfunction [Citation60,Citation61]. The ‘normalization’ of the TME in desmoplastic tumour types is being aimed by an established approach to improve the efficacy of drug regimens [Citation53]. Therefore, the repairing of tumour blood vessel abnormalities, improving perfusion and simultaneously promoting the delivery of nanomedicines in the TME could be achieved by normalizing the structural components of the tumour stroma [Citation48,Citation62–65]. Nanomedicines, due to the small size in nanometres (nm), specific site targeting, increased bioavailability, fewer toxic side effects, they are being applied as alternative treatments as anticancer agents for the cancer patients [Citation66,71]. In this present study, the in vitro assays, the MTT assay result () reveals that the PRK-NP has not produced any cytotoxicity and cytolytic anti-cancer activity. The apoptosis assay, Acridine Orange/Ethidium Bromide (AO/EB) Staining Assay result () reveals that PRK-NP has not produced any significant apoptosis which indicates it does not cause the genotoxicity. The SBR assay result also reveals () that the PRK-NP has not produced any cytotoxicity and cytolytic anti-cancer activity as well when compared to standard drugs. In the in vivo toxicity studies, the MTD study (acute toxicity) result () indicates that PRK-NP does not have toxicity even beyond 900 mg/kg body weight dose in mice. In the chronic toxicity studies in rat, rabbit and dog, the results () indicate that the PRK-NP has not produced any toxicity and adverse reaction in the study duration of 18 months at the dose rate of 500 mg/kg body weight. All these results confirm the non-toxicity of the Prakasine. In the immunological anti-tumour breast cancer efficacy studies, the results () of the Murine Mammary Tumour- C3H/HeJ model reveals that the PRK-NP has reduced the tumour growth by 35% in 200 mg/kg body weight and 43.7% in 500 mg/kg body weight oral doses compared to standard drugs. The results () of the Human Tumour Xenograft - MCF-7 mice model, PRK-NP has reduced the tumour growth by 25.7% in 500 mg/kg body weight and 83% in 1000 mg/kg body weight oral doses compared to standard drugs. It has been observed from this study, the PRK-NP possesses the non-toxic, non-cytolytic anti-cancer activity without damaging the normal cells unlike chemotherapy. So, the PRK-NP has the potential breast cancer treatment application with further studies.
The science behind the Prakasine is, I had observed some type of special molecular and atomic articulations and its principles while learning the Traditional Tamil Medicine alchemy. I explored them, made a research and I understood few formulatory rules are equivalent to present scientific principles as science have born just 500 years back. Based on my understanding, I posit a theory and named it as ‘Prakash theory of metal drugs’ (patent registration Ref. No. 5776/CHE/2014). My theory is as follows:
When metal atoms are attached to sulphur atoms with a coordinated covalent bond gives rise to a metallic sulphur complex, if this metallic sulphur complex is attached further to the organic compound (ligand), the organometallic complex will be formed, which will not produce any toxicity or toxicity in less quantity.
This metallic sulphur complex will form the disulphide bond in vivo and evince therapeutic action with less adverse effects.
Some metals particularly mercury attaches with sulphur and form a covalent bond even though other elements present in a biochemical reaction. The bond between Hg and S cannot be broken by biochemical reactions both in vivo and in vitro.
The theory posits that the attachment of metal atoms to sulphur atoms and subsequent attachment of ligands yields complexes without metal toxicity due to the stable disulphide bonds. The stable disulphide bonds (coordinated covalent bond) between metal-sulphur-ligands (Hg-S-X) atoms could not be breakable by any of the biochemical reactions inside the human body and avoid the free metal atoms (Hg++) getting attached with sulphur of the sulphur-containing amino acids. Thus, the adverse effects, accumulations, and toxicity are being avoided as well.
Elaborately, Prakasine is engineered as nontoxic based on my Prakash theory of metal drugs by locking either side of the Hg as S-Hg-S with coordinated covalent bond, then ligands are attached in this molecule, finally it becomes as ligand-S-Hg-S-ligand complex. So that the ligand only attaches with the biomolecules, evince the therapeutic effects and excreted in biliary and urinary routes without accumulating in the biological system unlike regular Hg and its compounds [Citation67–69,Citation19]. The coordinated covalently bound mercury in the Prakasine will only serve as metal battery atom to pass the electronic resonance to the ligand-S- and -S-ligand compounds to pass the electrons signals to these molecules to enhance the biochemical reactions after attaching to the receptor in the unique domain in the cell membrane. Also, this Prakasine is very easily passes through the glomerular filtration as it is less than 100 nm which is less than the nephron lumen diameter to pass easily in the urine. The Hg in the Prakasine will not gets attached with the sulphur atoms of the sulphur containing amino acids cysteine, cystine and methionine to affect the biological function in the way Hg is engineered and the S-Hg-S bond could not be detached by any kind of physical, chemical and biochemical reactions. This non-toxic concept is confirmed in cell lines, zebra fish, birds, mice and human. The data were published in scientific journals [Citation67–70,Citation19,72,73,74,75]. In addition to these publications, I have been treating more than 1000 HIV patients at the dose rate of 2000 mg TID last 30 years with this Prakasine as it is a non-conventional traditional medicine and every 90 days once I use to collect the virology, immunology and toxicological data. The virology and immunology data reveal that Praksine is having immunotherapy character and the toxicology data reveal non-toxicity of Prakasine [Citation68–70]. The regular Hg and the liberated mercury ions/atoms from the regular mercury compounds HgCl, HgCl2,HgCH3 etc gets attached with the sulphur atoms of the sulphur containing amino acids cysteine, cystine and methionine and affects the biological function and will keep on accumulating in the body.
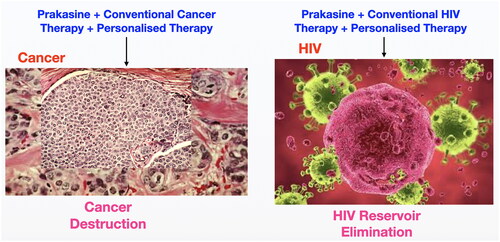
As the Prkasine has the potency to increase the humoral immunity [Citation67] and cell mediated immunity to reduce cancer [Citation19] and eliminate HIV [68–Citation70] without any toxicity (the non-toxicity has been demonstrated in 7 species including human) this could be a potential drug for HIV and cancer. I suggest the possible strategies () to cure HIV and Cancer with Prakasine from the out comes of my present and previous works with further studies.
Figure 15. Proposed mechanism of Prakasine in HIV and cancer cell destruction.
The increased CD8+ T cells by the stimulation of Prakasine gets sensitised with cancer and HIV antigens and become Cancer Specific Cytotoxic T-Lymphocytes (CS-CTL) and HIV Specific Cytotoxic T-Lymphocytes (HS-CTL). These CS-CTL and HS-CTL kills cancer and HIV.
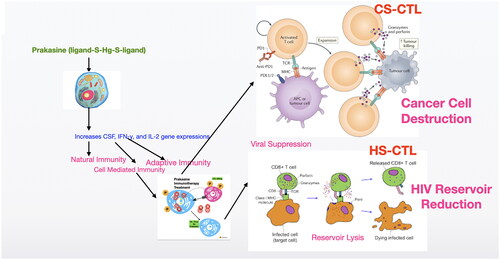
Figure 16. Possible strategies to cure HIV and cancer with Prakasine.
Conclusion
Prakasine, a non-toxic mercury nanoimmunotherapy has the potential to reduce the breast cancer by the elevated immune gene expression stimulation. Thus this non-toxic mercury nanoparticle and its immunotherapy potential are the novelty to combine with the conventional drugs. Prakasine has potential therapeutic effects and can be applied to treat cancer and HIV. The study concludes the significant effectiveness of PRK-NP in breast cancer reduction in mice better than existing standard anti-cancerous drugs. Future clinical investigations should be performed to assure the therapeutic value of PRK-NP in humans.
Author’s contribution
Dr SK Prakash was involved in the conception and design, or analysis and interpretation of the data; the drafting of the paper, revising it critically for intellectual content; and the final approval of the version to be published
Acknowledgments
1. Dr Jyoti Kode, Anti-Cancer Drug Screening Facility (ACDSF), Advanced Centre for Treatment, Research & Education in Cancer, Tata Memorial Centre, Kharghar, Navi Mumbai-410210
2. Skanda’s laboratory, Bangalore
Disclosure statement
The author reports no conflicts of interest in this work. This work was funded by the Naval AIDS Research Centre-Indian Social Services, Namakkal. No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author, [Prakash SK], upon reasonable request.
Additional information
Funding
References
- Ferlay J, Ervik M, Lam F, et al. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer; 2020 (https://gco.iarc.fr/today., accessed February 2021).
- World Health Organization. Cancer; 2020. https://www.who.int/news-room/fact-sheets/detail/cancer#:∼:text.
- de Martel C, Georges D, Bray F, et al. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020;8(2):e180–e190. doi: 10.1016/S2214-109X(19)30488-7.
- Ren Minghui. Assessing national capacity for the prevention and control of noncommunicable diseases: report of the 2019 global survey. Geneva: World Health Organization; 2020. https://iris.who.int/bitstream/handle/10665/331452/9789240002319-eng.pdf?sequence=1
- Key Statistics for Breast Cancer. American Cancer Society; 2023. https://www.cancer.org/cancer/types/breast-cancer/about/how-common-is-breast-cancer.html.
- Nounou MI, ElAmrawy F, Ahmed N, et al. Breast cancer: conventional diagnosis and treatment modalities and recent patents and technologies. Breast Cancer . 2015; 9(Suppl 2):17–34. PMID: 26462242; PMCID: PMC4589089 doi: 10.4137/BCBCR.S29420.
- Ophira Ginsburg MD, Cheng-Har Yip MD, Ari Brooks MM, et al. Breast cancer early detection: a phased approach to implementation. Cancer. 2020;126 Suppl 10(Suppl 10):2379–2393. doi: 10.1002/cncr.32887.
- Unger-Saldana K, Miranda A, Zarco-Espinosa G, et al. Health system delay and its effect on clinical stage of breast cancer: multicenter study. Cancer. 2015;121(13):2198–2206. doi: 10.1002/cncr.29331.
- Richards MA, Westcombe AM, Love SB, et al. Influence of delay on survival in patients with breast cancer: a systematic review. Lancet. 1999;353(9159):1119–1126. doi: 10.1016/s0140-6736(99)02143-1.
- Dumitrescu RG, Cotarla I. Understanding breast cancer risk—where do we stand in 2005? J Cell Mol Med. 2005;9(1):208–221. doi: 10.1111/j.1582-4934.2005.tb00350.x.
- Logan GJ, Dabbs DJ, Lucas PC, et al. Molecular drivers of lobular carcinoma in situ. Breast Cancer Res. 2015;17(1):76. doi: 10.1186/s13058-015-0580-5.
- Kerlikowske K, Hubbard RA, Miglioretti DL, et al. Comparative effectiveness of digital versus film-screen mammography in community practice in the United States: a cohort study. Ann Intern Med. 2011;155(8):493–502. doi: 10.7326/0003-4819-155-8-201110180-00005.
- O'Connor M, Rhodes D, Hruska C. Molecular breast imaging. Expert Rev Anticancer Ther. 2009;9(8):1073–1080. doi: 10.1586/era.09.75.
- Palmer ML, Tsangaris TN. Breast biopsy in women 30 years old or less. Am J Surg. 1993;165(6):708–712. doi: 10.1016/s0002-9610(05)80793-7.
- Kroese M, Zimmern RL, Pinder SE. HER2 status in breast cancer—an example of pharmacogenetic testing. J R Soc Med. 2007;100(7):326–329.
- Brooks M. Breast cancer screening and biomarkers. Methods Mol Biol. 2009;472:307–321. doi: 10.1007/978-1-60327-492-0_13.
- Dhankhar R, Vyas SP, Jain AK, et al. Advances in novel drug delivery strategies for breast cancer therapy. Artif Cells Blood Substit Immobil Biotechnol. 2010;38(5):230–249. doi: 10.3109/10731199.2010.494578.
- Debien V, De Caluwé A, Wang X, et al. Immunotherapy in breast cancer: an overview of current strategies and perspectives. Npj Breast Cancer. 2023;9(1):7. doi: 10.1038/s41523-023-00508-3.
- Prakash SK. Immunogenic antitumor potential of prakasine nanoparticles in zebrafish by gene expression stimulation. Artif Cells Nanomed Biotechnol. 2023;51(1):41–56. doi: 10.1080/21691401.2023.2173217.
- Ramyadevi J, Jeyasubramanian K, Marikani A, et al. Copper nanoparticles synthesized by polyol process used to control hematophagous parasites. Parasitol Res. 2011;109(5):1403–1415. doi: 10.1007/s00436-011-2387-3.
- Kholiya F, Chatterjee S, Bhojani G, et al. Seaweed polysaccharide derived bioaldehyde nanocomposite: Potential application in anticancer therapeutics. Carbohydr Polym. 2020; 240:116282. (https://www.sciencedirect.com/science/article/pii/S0144861720304562). doi: 10.1016/j.carbpol.2020.116282.
- Tokala R, Sana S, Lakshmi UJ, et al. Design and synthesis of thiadiazolo-carboxamide bridged β-carboline-indole hybrids: DNA intercalative topo-IIα inhibition with promising antiproliferative activity. Bioorg Chem. 2020;105:104357. (https://www.sciencedirect.com/science/article/pii/S0045206820316552). doi: 10.1016/j.bioorg.2020.104357.
- Kode J, Kovvuri J, Nagaraju B, et al. Synthesis, biological evaluation, and molecular docking analysis of phenstatin based indole linked chalcones as anticancer agents and tubulin polymerization inhibitors. Bioorg Chem. 2020; 105:104447–102068. (https://www.sciencedirect.com/science/article/pii/S0045206820317454) (For MTD assay) doi: 10.1016/j.bioorg.2020.104447.
- Crouch SP, Kozlowski R, Slater KJ, et al. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxcity. J Immunol Methods. 1993;160(1):81–88. doi: 10.1016/0022-1759(93)90011-u.
- Gonzalez RJ, Tarloff JB. Evaluation of hepatic sub cellular fractions for alamar blue and MTT reductase activity. Toxicol in Vitro. 2001;15(3):257–259. doi: 10.1016/S0887-2333(01)00014-5.
- Hattori N, Sakakibara T, Kajiyama N, et al. Enhanced microbial biomass assay using mutant luciferase resistant to benzalkonium chloride. Anal Biochem. 2003;319(2):287–295. doi: 10.1016/s0003-2697(03)00322-1.
- Kangas L, Grönroos M, Nieminen AL, et al. Bioluminescence of cellular ATP: a new method for evaluating cytotoxic agents in vitro. Med Biol. 1984;62(6):338–343.
- Lundin A, Hasenson M, Persson J, Pousette A. Estimation of biomass in growing cell lines by adenosine triphosphate assay. Methods Enzymol. 1986;133:27–42. doi: 10.1016/0076-6879(86)33053-2. PMID: 3821540.
- Cell viability and proliferation. Mark Frei, BioFiles. 2011; 6(5):17–21.
- Denizot FF, Lang RR. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. Immunol Meth. 1986;89(2):271–277. doi: 10.1016/0022-1759(86)90368-6.
- Kasibhatla S, Amarante-Mendes GP, Finucane D, et al. Acridine orange/ethidium bromide (AO/EB) staining to detect apoptosis. Cold Spring Harb Protoc. 2006;2006(3):pdb.prot4493. doi: 10.1101/pdb.prot4493.
- Liu K, Liu P-C, Liu R, et al. Dual AO/EB staining to detect apoptosis in osteosarcoma cells compared with flow cytometry. Med Sci Monit Basic Res. 2015;21:15–20. doi: 10.12659/MSMBR.893327.
- Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. doi: 10.1038/nprot.2006.179.
- Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer drug screening J. J Natl Cancer Inst. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107.
- Vigneshwar R, Arivuchelvan A, Mekala P, et al. Sex-specific reference intervals for wistar albino rats: hematology and clinical biochemistry. ijah. 2021;60(1):58–65. doi: 10.36062/ijah.60.1.2021.58-65.
- Simona P, Ana-Maria Teodoru NL, Coman C. Hematological and biochemical dynamics of rabbits and guinea pigs used for scientific purposes at Cantacuzino Institute, Bucharest. Rev RomMed Vet. 2021;31, 2:69–80. ISSN:1220-3173; E-ISSN:2457–7618.
- Poljičak-Milas N, Kardum-Skelin I, Vuđan M, et al. Blood cell count analyses and erythrocyte morphometry in New Zealand white rabbits. Vet. arhiv. 2009;79(6):561–571.
- Khan SA, Epstein JH, Olival KJ, et al. Hematology and serum chemistry reference values of stray dogs in Bangladesh. Open Vet J. 2011;5(2):13–20. doi: 10.5455/OVJ.2011.v1.i0.p13.
- Lumsden JH, Mullen K, McSherry BJ. Canine hematology and biochemistry reference values. Can J Comp Med. 1979;43(2):125–131. PMID: 497885; PMCID: PMC1319907.
- Ou W, Stewart S, White A, et al. In-situ cryo-immune engineering of tumor microenvironment with cold-responsive nanotechnology for cancer immunotherapy. Nat Commun. 2023;14(1):392. doi: 10.1038/s41467-023-36045-7.
- Sharma P, Wagner K, Wolchok JD, et al. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11(11):805–812. doi: 10.1038/nrc3153.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5.
- Wculek SK, Cueto FJ, Mujal AM, et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24. doi: 10.1038/s41577-019-0210-z.
- Foy, S.P., Jacoby, K., Bota, D.A. et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 615, 687–696 (2023). doi: 10.1038/s41586-022-05531-1
- Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52:17–35. doi: 10.1038/s41586-022-05531-1.
- Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x.
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586.
- Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16(6):356–371. doi: 10.1038/s41571-019-0175-7.
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022.
- Xiao Y, Chen J, Zhou H, et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat Commun. 2022;13(1):758. doi: 10.1038/s41467-022-28279-8.
- Voorwerk L, Isaeva OI, Horlings HM, et al. PD-L1 blockade in combination with carboplatin as immune induction in metastatic lobular breast cancer: the GELATO trial. Nat Cancer. 2023;4(4):535–549. doi: 10.1038/s43018-023-00542-x.
- Tokumaru Y, Joyce D, Takabe K. Current status and limitations of immunotherapy for breast cancer. Surgery. 2020; ; 167(3):628–630. doi: 10.1016/j.surg.2019.09.018.
- Panagi M, Mpekris F, Chen P, et al. Polymeric micelles effectively reprogram the tumor microenvironment to potentiate nano-immunotherapy in mouse breast cancer models. Nat Commun. 2022;13(1):7165. doi: 10.1038/s41467-022-34744-1.
- Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. Article CAS PubMed Google Scholar doi: 10.1056/NEJMoa1809615.
- Adams S, Diamond JR, Hamilton E, et al. Atezolizumab plus nab-paclitaxel in the treatment of metastatic triple-negative breast cancer with 2-year survival follow-up: a phase 1b clinical trial. JAMA Oncol. 2019;5(3):334–342. Article PubMed Google Scholar doi: 10.1001/jamaoncol.2018.5152.
- Martin JD, Cabral H, Stylianopoulos T, et al. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat Rev Clin Oncol. 2020;17(4):251–266. doi: 10.1038/s41571-019-0308-z.
- Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi: 10.1038/nrclinonc.2010.139.
- Stylianopoulos T, Munn LL, Jain RK. Reengineering the physical microenvironment of tumors to improve drug delivery and efficacy: from mathematical modeling to bench to bedside. Trends Cancer. 2018;4(4):292–319. doi: 10.1016/j.trecan.2018.02.005.
- Martin JD, Seano G, Jain RK. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu Rev Physiol. 2019;81(1):505–534. doi: 10.1146/annurev-physiol-020518-114700.
- Stylianopoulos T, Martin JD, Chauhan VP, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. 2012;109(38):15101–15108. doi: 10.1073/pnas.1213353109.
- Nia HT, Munn LL, Jain RK. Physical traits of cancer. Science. 2020;370(6516):eaaz0868. doi: 10.1126/science.aaz0868.
- Voutouri C, Stylianopoulos T. Evolution of osmotic pressure in solid tumors. J Biomech. 2014;47(14):3441–3447. doi: 10.1016/j.jbiomech.2014.09.019.
- Kalli M, Stylianopoulos T. Defining the role of solid stress and matrix stiffness in cancer cell proliferation and metastasis. Front Oncol. 2018;8:55. doi: 10.3389/fonc.2018.00055.
- Voutouri C, Stylianopoulos T. Accumulation of mechanical forces in tumors is related to hyaluronan content and tissue stiffness. PLOS One. 2018;13(3):e0193801. doi: 10.1371/journal.pone.0193801.
- Stylianopoulos T, Martin JD, Snuderl M, et al. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res. 2013;73(13):3833–3841. doi: 10.1158/0008-5472.CAN-12-4521.
- Chauhan VP, Boucher Y, Ferrone CR, et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell. 2014;26(1):14–15. doi: 10.1016/j.ccr.2014.06.003.
- Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362.
- Chauhan VP, Martin JD, Liu H, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4(1):2516. doi: 10.1038/ncomms.3516.
- Sheridan C. Pancreatic cancer provides testbed for first mechanotherapeutics. Nat Biotechnol. 2019;37(8):829–831. Article CAS PubMed Google Scholar doi: 10.1038/d41587-019-00019-2.
- Martin JD, Miyazaki T, Cabral H. Remodeling tumor microenvironment with nanomedicines. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2021;13(6):e1730. doi: 10.1002/wnan.1730.
- Rasool M, Malik A, Waquar S, et al. New challenges in the use of nanomedicine in cancer therapy. Bioengineered. 2022; 13(1):759–773. PMID: 34856849; PMCID: PMC8805951. doi: 10.1080/21655979.2021.2012907.
- Prakash SK. Effect of feed supplementation of mercury nanoparticles on immunostimulation of live lentogenic Newcastle disease vaccine in layer birds. Indian Vet J. 2017;94:11–13.
- Prakash SK. Immunological and virological effects of novel prakasine nanomedicine in HIV-infected patients in South India: a preliminary study. Virol Antivir Res. 2020;9(2):195.
- Prakash SK. Phytochemical therapy as a possible cure for asymptomatic AIDS patients [dissertation]. India: Bharathidasan University; 2010.
- Prakash SK. Immunological and virological effects of prakasine nanomedicine in HIV eradication: a preliminary study. Fifth Eastern Europe and Central Asia AIDS conference. ; 2016 23–25. Moscow