
ABSTRACT
Divalent ion-conducting solid electrolytes with NASICON-type three-dimensional network structures (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Ni, Mg, Ca, Sr) were successfully developed by introducing M2+ cations into HfNb(PO4)3 solids. The presence of high-valence cations such as Hf4+, Nb5+, and P5+ in the structures effectively reduced the electrostatic interaction between the conducting M2+ cations and the surrounding oxide anions, enabling the M2+ cations to migrate smoothly in the rigid crystal lattice. The relationship between the cation conductivity in NASICON-type solids and the ionic radius of the migrating divalent cation species was also clarified by taking into account the relative sizes of the conduction pathways in the structures.
1. Introduction
Solid electrolytes are considered promising candidates for use as key components of next-generation electrochemical devices such as all-solid-state batteries, fuel cells, and chemical sensors because of their high chemical/thermal stability and energy density [Citation1–Citation4]. In the field of solid electrolytes, it is generally well accepted that ion migration in a solid is strongly influenced by both the valence state and the ionic size of the conducting ion species. The higher the valence of the conducting ion species, the stronger the electrostatic interaction with the surrounding counterions. As the ionic size of the conducting ion species increases, moreover, their migration in rigid crystal structures becomes difficult, because most solids have a dense structure with insufficient open space to permit the free migration of bulky ions. As a result, the migration of high-valence and/or large ions is expected to be poor in crystalline solids [Citation5]. For this reason, many studies have focused on small Li+ or H+ ions with low valences, which are expected to migrate smoothly in solid lattices, and various kinds of Li+ or H+ ion conductors with high conductivity have been developed. With the gradual increase in demand for next-generation batteries with a high energy density that can be applied to electric vehicles and energy storage systems (ESS), however, divalent ion conductors that can theoretically carry twice as much electricity as monovalent ions are gaining increasing attention. Many studies have been focusing on small Mg cations as a conducting species in solid electrolytes [Citation6–Citation8].
To date, several divalent cations, including Ni2+, Mg2+, Ca2+, Sr2+, Pb2+, and Ba2+, have been reported to exhibit conductivity in solids with β″-alumina or β-Fe2(SO4)3-type structures [Citation9–Citation13]. However, these two-crystal compounds possess a layered or distorted three-dimensional network structure, respectively, that limits smooth ion migration in the solid because the corresponding conduction pathway is anisotropic or distorted; the divalent cation conductivities of these solids are therefore too low for application in practical electrochemical devices. Although some reports on divalent cation (Ca2+, Sr2+, Pb2+, and Ba2+) conduction in the well-ordered three-dimensional network structure of Na+ superionic conductor (NASICON)-type MZr4(PO4)6 (M = Ca2+, Sr2+, Pb2+, Ba2+) solids have been published [Citation12], the conductivities of these systems are significantly low. The development of highly conductive divalent cation materials requires a strict selection of both the crystal structure and the components. Incidentally, in a recent study we reported successful development of various kinds of trivalent and tetravalent cation conductors with NASICON-type structures [Citation14–Citation18]; we also showed that the presence of cations with a higher valence than that of the conducting cation is essential for achieving smooth ion migration, together with the selection of an appropriate crystal structure possessing large conduction pathways for the migration of bulky ions. Based on this strategy, it is expected that divalent cations incorporated in NASICON-type solids containing higher-valence cations may show high ion conductivity.
In this study, we developed high-conductivity divalent cation solid electrolytes with a NASICON-type structure based on the HfNb(PO4)3 material that was identified as a tetravalent Hf4+ cation conductor in our previous work [Citation19]. By introducing the lower-valence M2+ cation into the Hf4+ sites of the HfNb(PO4)3 solid, the conducting ion species should change from Hf4+ to the lower-valence M2+, because ion conduction is in general strongly influenced by the valence state, and the migration of lower-valence cations is easier due to their weaker electrostatic interaction with counterions. As conducting divalent cation species, we selected Ni2+ and Mg2+ as well as the Ca2+, Sr2+, and Ba2+ alkaline-earth metal ions, which maintain a stable divalent state.
In this article, we describe the divalent cation-conducting properties of NASICON-type (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Ni [Citation20], Mg [Citation21], Ca [Citation5], Sr, Ba) solids and discuss the relationship between conductivity and the ionic radius of the migrating M2+ species.
2. Experimental
(MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Ni, Mg, Ca, Sr, Ba) solids were prepared by a conventional co-precipitation method with the following high-purity starting materials: Ni(NO3)2 (99.99%, Sigma-Aldrich Chemistry), Mg(NO3)2 (99.9%, Kojundo Chemical Lab. Co., Ltd.), Ca(NO3)2 (99.9%, Wako Chemical Industries, Ltd.), Sr(NO3)2 (99.9%, Kojundo Chemical Lab. Co., Ltd.), Ba(NO3)2 (99.9%, Kishida Chemical Co., Ltd.), HfCl4 (99.9%, Sigma-Aldrich Chemistry), NbCl5 (99.9%, Mitsuwa Chemicals Co., Ltd.), and (NH4)2HPO4 (99.99%, Sigma-Aldrich Chemistry). (NH4)2HPO4 diluted in 3 N nitric acid was added dropwise into an ethanol solution in which stoichiometric amounts of M(NO3)2 (M = Ni, Mg, Ca, Sr, Ba), HfCl4, and NbCl5 had been dissolved in advance. The mixed solution was stirred at 130°C for 24 h to obtain solid powders, which were then dried at 130°C for several hours. After pre-calcination of the obtained powders at 600°C for 6 h in an air atmosphere, the sample powders were pelletized and calcined repeatedly at 1100–1350°C for 6 h in synthetic air (80% N2-20% O2) until a single-phase NASICON-type structure was formed. The calcined samples were identified by X-ray powder diffraction (XRD) analysis using the step scanning method in the 2ϴ range from 10° to 40°, with a step width of 0.04°, a scan speed of 8°min−1, and Cu Kα radiation (SmartLab, Rigaku Corp., 40 kV, 40 mA). The lattice volume of the calcined powder was calculated from the XRD peak positions with respect to α-Al2O3 powder as an internal standard. After identifying the crystal phase of the (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Ni, Mg, Ca, Sr, Ba) samples, the powders obtained as single-phase samples were pressed into a pellet shape and sintered at 1200–1350°C for 12 h in a synthetic air atmosphere.
The electrical conductivity of the sintered sample pellets was measured with platinum-sputtered layers formed at the center of both surfaces of the pellets in order to obtain good contact with the blocking Pt electrodes. The ac conductivity (σac) of the sintered sample pellets was measured using the complex impedance method in the frequency range of 5 Hz to 13 MHz (1260 impedance/gain-phase analyzer, Solartron) at temperatures between 300°C and 600°C in an air atmosphere. Both the ac and dc conductivities were measured under various oxygen partial pressures, ranging from 10−17 to 105 Pa (adjusted by mixing pure O2, synthetic air, 99.999% Ar, and 1% CO−99% CO2) to identify the conducting species in the (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3 solids. dc electrolysis was conducted on the sintered sample pellets (without Pt layers) by applying a dc voltage in an air atmosphere with platinum bulk electrodes. After the dc electrolysis, a cross-sectional line analysis was conducted using energy-dispersive X-ray spectroscopy (EDX; SSX-550, Shimadzu) to investigate the elemental distribution inside the electrolyzed sample pellets are a means of probing the migration of the cation species directly.
3. Results and discussion
3.1. (NIxHf1−x)4/(4−2x)Nb(PO4)3 solids
) shows the XRD patterns of (NixHf1−x)4/(4−2x)Nb(PO4)3 (0.03 ≤ x ≤ 0.10) samples obtained by calcination at 1300°C. The diffraction pattern of the samples can be assigned to a trigonal NASICON-type structure with space group Rc, and a single phase of the NASICON-type solids was obtained in the x ≤ 0.06 compositional range. The samples with x > 0.06 consisted of a multiphase mixture of the NASICON-type solid with NbPO5 and HfP2O7 impurity phases. As depicted in ), moreover, the peaks assigned to the NASICON-type phase were shifted slightly to higher angles with increases in the Ni content (x) in the single-phase region (x ≤ 0.06). From all of the NASICON-type XRD peak positions at 10–40° in the 2ϴ range, we calculated the lattice volume of the NASICON-type phase, whose compositional dependence is illustrated in . The lattice volume of the NASICON-type phase for the single-phase samples (x ≤ 0.06) decreased with increases in x because of the smaller ionic radius of the Ni2+ cation (0.083 nm for a coordination number (CN) of 6) compared with that of the Hf4+ cation (0.085 nm, CN = 6) [Citation22]. On the other hand, no further lattice volume changes were observed in the range in which impurity phases appeared. This result strongly suggests that the smaller Ni2+ cations partially substitute the Hf4+ sites in the NASICON-type HfNb(PO4)3 samples.
Figure 1. (a) XRD patterns of the (NixHf1−x)4/(4−2x)Nb(PO4)3 (0.03 ≤ x ≤ 0.10) solids and (b) magnified XRD patterns.
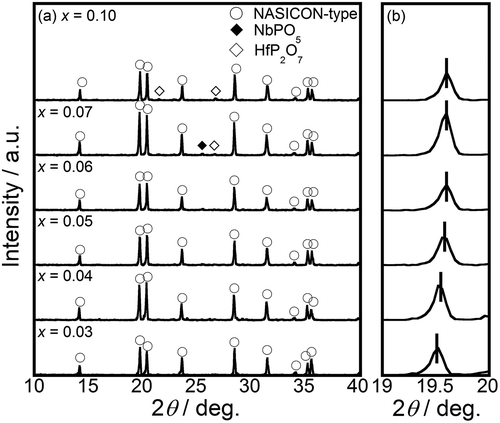
Figure 2. Compositional dependence of the lattice volume and ac conductivity of the (NixHf1−x)4/(4−2x)Nb(PO4)3 solids.
also shows the compositional dependence of the ac conductivity at 600°C for samples with x ≤ 0.10. Conductivity increased monotonically with the Ni2+ content up to x = 0.06, parallel to the changes in lattice volume, due to the increased amount of conducting Ni2+ in the solid. Conductivity decreased in the multiphase mixture region, however, due to the formation of insulating NbPO5 and HfP2O7 materials. This result also supports the idea that the solid solubility limit composition for NASICON-type (NixHf1−x)4/(4−2x)Nb(PO4)3 solids is around x= 0.06.
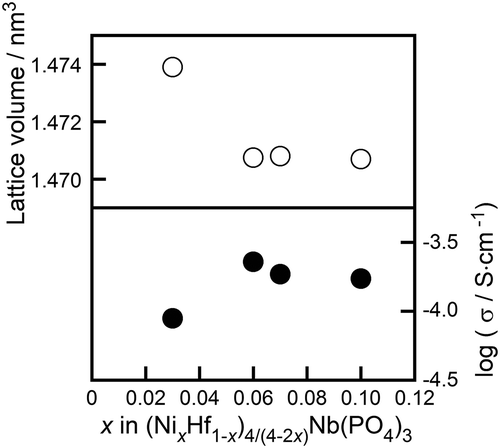
In order to identify the conducting species in the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid, the dc and ac conductivities were measured in atmospheres with an oxygen partial pressure ranging between 10−16 and 105 Pa, and the samples, cation transference number was then estimated. shows the dependence of ac conductivity on the oxygen partial pressure at 600°C. In the case of metal oxides, oxygen is generally released from solids at low oxygen partial pressures, resulting in n-type (electron-conducting) behavior of the material. On the other hand, p-type (hole-conducting) properties appear at high O2 partial pressures, due to the oxidation of metal cations. In the region of oxygen partial pressures where ion conduction is dominant in solids, moreover, conductivity is known to remain unaffected by changes in PO2. The (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid exhibited constant electrical conductivity in the O2 partial pressure range between 10−16 and 105 Pa, indicating that no electronic species (e− and h+) conduction can take place in a sample with such a wide PO2 range. This result shows that the conducting species in (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid are the constituent ions (Ni2+, Hf4+, Nb5+, P5+, or O2−). shows the dc to ac conductivity ratio (σdc/σac) for the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid in O2 (Po2 = 105 Pa) and Ar (Po2 = 103 Pa) atmospheres. When the oxide anion (O2−) is the conducting species, dc conductivity should be the same as the ac conductivity in an O2 atmosphere, because the O2− conducting species is supplied by the (O2) atmosphere. At a lower oxygen partial pressure, dc conductivity decreases because the Ar atmosphere cannot supply enough O2− species for continuous ion migration. As a result, in a low Po2 atmosphere the dc to ac conductivity ratio should decrease over time. In the case of cation conductors, no conducting species can be supplied from either the atmosphere or electrodes when Pt electrodes are used, suggesting that the dc conductivity should drop to almost zero in any atmosphere. According to , the σdc/σac ratio for the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid rapidly decreased with time, reaching a value of less than 0.01 after 30 min under both Ar and O2 atmospheres. This phenomenon indicates that cations are the conducting species in the sample. The cation transference number in the sample can be estimated as 1 − (σdc/σac), moreover, because the σdc/σac value represents the contribution of species other than cations to conductivity [Citation23]; the cation transference number of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 sample was estimated to be higher than 0.99.
Figure 3. Oxygen partial pressure dependence of the ac conductivity of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid at 600°C.
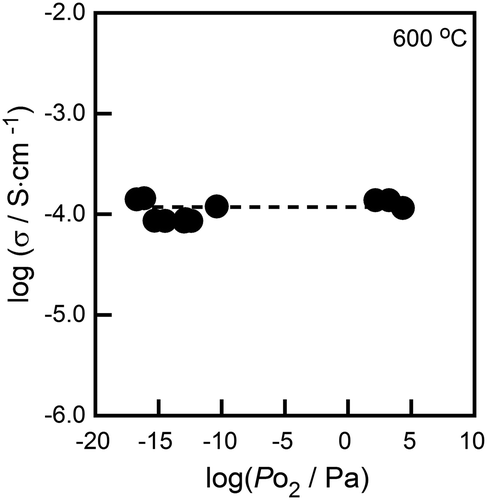
Figure 4. Time dependence of the σdc/σac ratio of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid in O2 and Ar atmospheres.
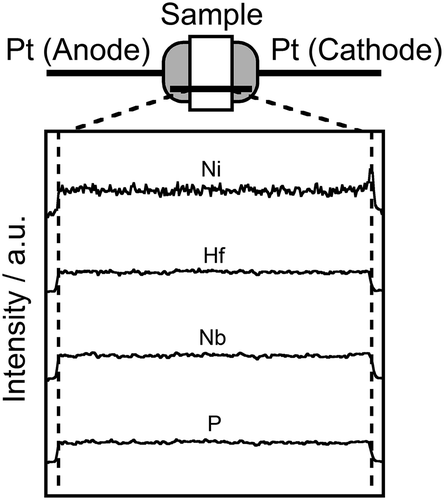
For purposes of direct analysis of Ni2+ cation migration in the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid, dc electrolysis was carried out by applying 4 V dc voltage to sintered pellets in the air at 650°C for 25 days. When dc voltage is applied to a sample, only the conducting cations migrate in the cathodic direction according to the potential gradient. illustrates the setup for dc electrolysis and theEDX line analysis results for the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid after dc electrolysis. A clear deposition of Ni was observed on the cathodic surface of the sample pellet, while no such deposition was observed on the cathodic surfaces or interior of sample pellets for other cation species. This result clearly demonstrates that Ni ions were the only conducting species in the solid.
Figure 5. Schematic illustration of the setup for the dc electrolysis experiment for the (Ni0.06Hf0.94)4/3.88Nb(PO4)3 solid, and the result of EDX line analysis of the electrolyzed pellets.
3.2. (MxHf1−x)4/(4−2x)Nb(PO4)3 solids (M = Mg, Ca, Sr, Ba)
The XRD patterns of the (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Mg, Ca, Sr) solid confirmed that these solids also form the NASICON-type phase (Figures S1–S3). Much as in the case of (NixHf1−x)4/(4−2x)Nb(PO4)3, single NASICON-type solid phases were successfully obtained for these samples, i.e., x ≤ 0.15 for Mg and x ≤ 0.05 for Ca and Sr. In the case of the Ba-doped samples, however, no single NASICON-type phase was obtained for any composition, and the NbPO5 impurity phase always appeared when NbCl5 was dissolved in nitric acid in a stoichiometric ratio during the sample preparation process (Figure S4). This result suggests that the Ba2+ ion (0.149 nm, CN = 6 [Citation22]) is too large to substitute the Hf4+ site (0.085 nm, CN = 6 [Citation22]) in NASICON-type HfNb(PO4)3 solids.
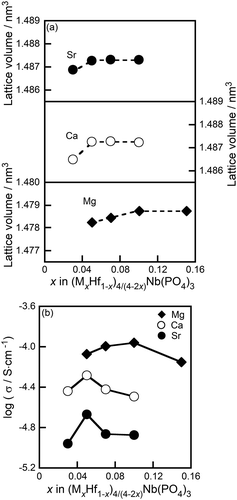
shows the compositional dependence of the lattice volume and the ac conductivity of (MxHf1−x)4/(4−2x)Nb(PO4)3 solids calcined at the optimal temperature, i.e., 1300°C for Mg and 1200°C for Ca and Sr. The lattice volume of the NASICON-type phase increased with increases in the M content (x) within the single-phase range because of the larger ionic radius of the M2+ species (0.086, 0.114, and 0.132 nm for Mg, Ca, and Sr, respectively, with CN = 6 [Citation22]) compared with that of the Hf4+ cation (0.085 nm, CN = 6 [Citation22]). No further lattice volume expansion was observed in the multiphase range, indicating that the M2+ cations were partially introduced into the Hf4+ sites of the (MxHf1−x)4/(4−2x)Nb(PO4)3 solids, up to the solubility limit composition (x = 0.10 for Mg, x = 0.05 for Ca and Sr) ()). The M2+ cation conductivity monotonically increased with increases in the M2+ cation content (x), moreover, up to the solubility limit composition ()).
Figure 6. Compositional dependence of the (a) lattice volume and (b) ac conductivity of the (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Mg, Ca, Sr) solids.
The σdc/σac ratios of the (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3 solids reached a value of less than 0.01 after 30 min in both O2 (105 Pa) and He (Po2 = 102 Pa) atmospheres and O2 (105 Pa) and Ar (103 Pa) atmospheres (Figures S5–S7), indicating that the estimated cation transference number of the (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3 samples was above 0.99. Much as in the case of the Ni2+ cation conductor, the EDX line analysis confirmed that M2+ cations were the only conducting species in (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3 solids (Figures S8–S10).
3.3. Relationship between M2+ conductivity and ionic size in (MxHf1−x)4/(4−2x)Nb(PO4)3 solids
shows the temperature dependence of the ac conductivity of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, (Sr0.05Hf0.95)4/3.9Nb(PO4)3, and MZr4(PO4)6 (M = Ni, Mg, Ca, Sr) solids [Citation12]. The M2+ conductivity at 600°C and the activation energy for ion migration in these solids are listed in . Although the MZr4(PO4)6 (M = Ni, Mg, Ca, Sr) solids were reported to exhibit different structures depending on their ionic species (e.g., a distorted β-Fe2(SO4)3-type structure for M = Ni and Mg and a well-ordered NASICON-type structure for M = Ca and Sr), the activation energies for M2+ migration in MZr4(PO4)6 (M = Ni, Mg, Ca, Sr) solids were similar. It can therefore be assumed that the structures of NASICON-type MZr4(PO4)6 (M = Ca, Sr) solids might also be distorted to narrow their ion conduction pathways, resulting in both low ion conductivity and high activation energy. The solids prepared in this study, (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3, exhibited much higher conductivity and lower activation energy compared with those of the corresponding MZr4(PO4)6 (M = Ni, Mg, Ca, Sr) solids in all temperature ranges, even though the M2+ amounts in the present solids were lower than those of MZr4(PO4)6 (M = Ni, Mg, Ca, Sr). These high M2+ conductivities are thought to have originated from the well-ordered three-dimensional ion conduction pathway by maintaining a NASICON-type structure whose structural distortion was smaller than that of the β-Fe2(SO4)3-type structure. In addition, the electrostatic interaction between M2+ and the surrounding O2− ions was also effectively reduced by the presence in the structure of various kinds of high-valence cations (Hf4+, Nb5+, P5+).
Table 1. Ionic conductivity at 600°C and activation energy of (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3 solids.
Figure 7. Temperature dependence of the ac conductivity of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, (Sr0.05Hf0.95)4/3.9Nb(PO4)3, and MZr4(PO4)6 (M = Ni (––), Mg (––), Ca (––), Sr (···)) solids [Citation9].
![Figure 7. Temperature dependence of the ac conductivity of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, (Sr0.05Hf0.95)4/3.9Nb(PO4)3, and MZr4(PO4)6 (M = Ni (––), Mg (––), Ca (––), Sr (···)) solids [Citation9].](/cms/asset/bee9871a-7c5d-4e68-bbe2-85a30011a073/tace_a_1606141_f0007_b.gif)
The ion conduction phenomenon in solids is influenced by the crystallinity of the material, which is an especially important factor for the well-ordered three-dimensional network structures examined in this study. Distortions of the crystal structure lead to deterioration of the conduction pathways and, as a result, ion migration becomes difficult. Because the crystallinity of the samples was strongly dependent on the sintering temperature, we should take the sintering conditions into account when discussing the relationship between the ion conduction and ionic size of the conducting species. The M2+ (M = Ni, Mg, Ca, Sr) ion conductivities of the (Ni0.06Hf0.94)4/3.88Nb(PO4)3, (Mg0.1Hf0.9)4/3.8Nb(PO4)3, (Ca0.05Hf0.95)4/3.9Nb(PO4)3, and (Sr0.05Hf0.95)4/3.9Nb(PO4)3 solids discussed above applied to samples with solid solubility limit compositions that were sintered at optimum temperatures, i.e., 1300°C for Ni or Mg and 1200°C for Ca or Sr. In the case of Ca and Sr samples, when the calcination temperature was raised above 1200°C, an additional NbPO5 impurity phase appeared together with the NASICON-type solid due to sample decomposition. When discussing the effect of the ionic size of the M2+ ions on the conduction properties of NASICON-type solids, it is essential to compare the conductivity of solids with the same M2+ concentration and subjected to the same calcination temperature. We therefore compared the conduction properties of the (M0.05Hf0.95)4/3.9Nb(PO4)3 solids (M = Ni, Mg, Ca, Sr) sintered at 1200°C, whose compositions were within the solid solubility range for all systems. We introduced a parameter A corresponding to the ratio of the divalent cation and lattice volumes (A = Vion/Vlattice) [Citation24–Citation26], moreover, which should reflect the size of the conducting ion species relative to that of the conduction pathway.
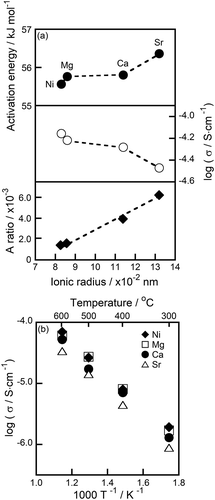
shows the M2+ ionic radius dependencies of the A ratio, the ion conductivity at 600°C, and the activation energy required for M2+ migration ()) as well as the temperature dependence of the M2+ conductivity for (M0.05Hf0.95)4/3.9Nb(PO4)3 solids sintered at 1200°C ()). The A ratio linearly increased with increases in the M2+ ionic radius, denoting the lack of a proportional relationship between the expansion degree of the lattice volume and the conducting ion volume; the structures of the samples containing smaller divalent M2+ cations exhibit relatively large conduction pathways. Because large conduction pathways favor smooth ion migration, a monotonous decrease in conductivity was observed with increases in the ionic radius of the M2+ ion, which is the opposite trend to that observed for the A ratio. Although the activation energies of the samples with M = Mg and Ca showed only small differences, moreover, their values increased in the following order: Ni2+ (55.56 kJ·mol−1) < Mg2+ (55.76 kJ·mol−1) < Ca2+ (55.80 kJ·mol−1) < Sr2+ (56.36 kJ·mol−1). If the ion conduction in a crystal lattice is influenced only by the relationship between the size of the conducting ion species and the lattice volume (A ratio), the ion conductivity and activation energy for the Ca2+-based sample are expected to be lower and higher, respectively, than those of the samples containing other ions. The reason for the improved Ca2+ conduction performance compared with the expected performance is the weak interaction between Ca2+ and O2− ions in the structure. In general, ionic bonds are weaker than covalent bonds in a solid material. Because the electronegativity of Ca (1.00) is smaller than that of Mg (1.31), the covalency of Ca-O bonds is smaller than that of Mg-O bonds, which indicates that the Ca-O bonding interaction in solids is weaker than that of Mg-O. This makes the migration of Ca2+ ions in a (Ca0.05Hf0.95)4/3.9Nb(PO4)3 solid relatively easy.
Figure 8. (a) Relationship of the M2+ ionic radius with the A ratio, ion conductivity at 600°C, and activation energy; and (b) temperature dependence of the M2+ conductivity of the (M0.05Hf0.95)4/3.9Nb(PO4)3 (M = Ni, Mg, Ca, Sr) solids.
4. Conclusions
We have synthesized divalent cation-conducting (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Ni, Mg, Ca, Sr, Ba) solid electrolytes with well-ordered three-dimensional NASICON-type structures and investigated their M2+ ion-conducting properties. Among the samples prepared, only the solids with M = Ba were unobtainable as single-phase samples, because the ionic size of Ba is too large to substitute Hf sites. In the case of (MxHf1−x)4/(4−2x)Nb(PO4)3 (M = Ni, Mg, Ca, Sr) solids obtained as single-phase samples, the conduction properties (conductivity and activation energy) were mainly affected by the relative size of the conduction pathway. Thus, the solid whose conducting species was Ni2+ (M = Ni) showed the highest conductivity and lowest activation energy, because its structure exhibited the largest conduction pathway among the investigated samples. A comparison of (Mg0.05Hf0.95)4/3.9Nb(PO4)3 and (Ca0.05Hf0.95)4/3.9Nb(PO4)3 samples revealed, moreover, that the divalent ion conduction in NASICON-type solids was also influenced by the electronegativity of the conducting species.
Supplemental Material
Download MS Word (1.2 MB)Acknowledgments
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) through KAKENHI Grant No. JP16K05936 (Grant-in-Aid for Science Research (C)), and by a research grant from the Murata Science Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material can be accessed here.
Related Research Data
References
- Takada K. Progress and prospective of solid-state lithium batteries. Acta Mater. 2013;61:759–770.
- Tatsumisago M, Nagao M, Hayashi A. Recent development of sulfide solid electrolytes and interfacial modification for all-solid-state rechargeable lithium batteries. J Asian Ceram Soc. 2013;1:17–25.
- Teng S, Tan J, Tiwari A. Recent developments in garnet based solid state electrolytes for thin film batteries. Curr Opin Solid State Mater Sci. 2014;18:29–38.
- Xu X, Yang T, Shui M, et al. The preparation and lithium mobility of zinc based NASICON-type solid electrolyte Li1+2x+2yAlxZnyTi2−x−ySixP3−xO12. Ceram Int. 2014;40:3819–3822.
- Lee W, Tamura S, Imanaka N. New calcium ion conducting solid electrolyte with NASICON-type structure. Chem Lett. 2017;46:1486–1489.
- Aubrey ML, Ameloot R, Wiers BM, et al. Metal–organic frameworks as solid magnesium electrolytes. Energy Environ Sci. 2014;7:667–671.
- Shao Y, Rajput NN, Hu J, et al. Nanocomposite polymer electrolyte for rechargeable magnesium batteries. Nano Energy. 2015;12:750–759.
- Zhao-Karger Z, Zhao X, Wang D, et al. Performance improvement of magnesium sulfur batteries with modified non-nucleophilic electrolytes. Adv Energy Mater. 2015;5:1401155.
- Farrington GC, Dunn B. Divalent beta″-aluminas: high conductivity solid electrolytes for divalent cations. Solid State Ion. 1982;7:276–281.
- Stricker DW, Carlson WG. Ionic conductivity of cubic solid solutions in the system CaO—Y2O3—zrO2. J Am Ceram Soc. 1964;47:122–127.
- Dunn B, Ostrom RM, Seevers R, et al. Divalent cation conductivity in beta″ alumina. Solid State Ion. 1981;5:203–204.
- Nomura K, Ikeda S, Ito K, et al. Framework structure, phase transition, and transport properties in MIIZr4(PO4)6 compounds (MII = Mg, Ca, Sr, Ba, Mn, Co, Ni, Zn, Cd, and Pb). Bull Chem Soc Jpn. 1992;65:3221–3227.
- Ikeda S, Takahashi M, Ishikawa J, et al. Solid electrolytes with multivalent cation conduction. 1. Conducting species in Mg-Zr-PO4 system. Solid State Ionics. 1987;23:125–129.
- Tamura S, Yamane M, Imanaka N. Trivalent gallium ion conduction in NASICON-type solid. J Asian Ceram Soc. 2016;4:390–393.
- Imanaka N, Tamura S. Development of multivalent ion conducting solid electrolytes. Chem Soc Jpn. 2011;84:353–362.
- Nunotani N, Tamura S, Imanaka N. Highly tetravalent hafnium ion conducting solids with a NASICON-type structure. Electrochemistry. 2012;80:743–745.
- Nunotani N, Ohsaka T, Tamura S, et al. Tetravalent Sn4+ ion conductor based on NASICON-type phosphate. ECS Electrochem Lett. 2012;1:A66–A69.
- Nunotani N, Sawada M, Tamura S, et al. Enhancement of Hf4+ ion conductivity in a NASICON-type solid. Bull Chem Soc Jpn. 2010;83:415–418.
- Imanaka N, Itaya M, Adachi G. First identification of tetravalent Hf4+ ion-conducting solid. Mater Lett. 2002;53:1–5.
- Lee W, Tamauchi S, Tamura S, et al. Divalent Ni2+ cation conduction in NASICON-type solid. Mater Lett. 2019;234:261–263.
- Tamura S, Yamane M, Hoshino Y, et al. Highly conducting divalent Mg2+ cation solid electrolytes with well-ordered three-dimensional network structure. J Solid State Chem. 2016;235:7–11.
- Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A. 1976;32:751–767.
- Kobayashi Y, Egawa T, Tamura S, et al. Trivalent Al3+ ion conduction in aluminum tungstate solid. Chem Mater. 1997;9:1649–1654.
- Aono H, Imanaka N, Adachi G. High Li+ conducting ceramics. Acc Chem Res. 1994;27:265–270.
- Tamura S, Imanaka N, Adachi G. Trivalent cation conduction in R1/3Zr2(PO4)3 (R: rare earths) with the NASICON-type structure. J Alloy Compd. 2001;323-324:540–544.
- Imanaka N, Adachi G. Rare earth ion conduction in tungstate and phosphate solids. J Alloy Compd. 2002;344:137–140.