
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The effect of oxygen vacancies on catalytic activity for direct N2O decomposition was demonstrated in Zr1−xYxO2−δ solid solutions with a fluorite-related structure. The fluorite-type structure was formed as samples with a small amount of Y3+ (0.05 ≤ x ≤ 0.19) and a large amount of Y3+ (x ≥ 0.80), leading to the generation of a C-type structure derived from the fluorite-type structure by removing a quarter of the oxide anions. The oxygen vacancies worked as N2O adsorption sites and catalytic active sites, with the highest activity realized by the YO1.5 catalyst with a C-type structure, which contained the largest number of oxygen vacancies in the ZrO2-Y2O3 system.
1. Introduction
Nitrous oxide (N2O) has a high global warming potential, 310 times higher than that of carbon dioxide, and it also contributes to the destruction of the ozone layer in the stratosphere [Citation1]. N2O is emitted anthropogenically from manufacturing plants such as nitric acid and adipic acid plants, and the atmospheric N2O concentration has increased continuously for several years [Citation2]. The removal of N2O from industrial exhaust gas to protect the environment is therefore an important issue.
Among the several N2O decomposition methods, direct catalytic decomposition of N2O into N2 and O2 is the ideal route, because this process is simple, clean, and sustainable. A large number of catalysts have been studied for N2O decomposition: these include noble-metal-based catalysts [Citation3–Citation7], spinels [Citation8,Citation9], perovskites [Citation10–Citation12], hexa-aluminates [Citation13,Citation14], and zeolites [Citation15,Citation16].
The reaction for direct N2O decomposition is thought to involve the following steps [Citation2]: adsorption of N2O on the active sites (Equation 1), decomposition to N2 gas and adsorbed oxygen (Equation 2), and, finally regeneration through recombination of adsorbed oxygen species (Equation 3) or direct reaction with N2O (Equation 4):
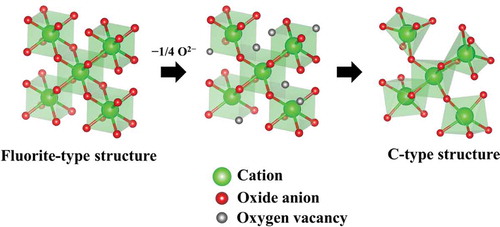
Since the reaction proceeds on the active sites in the lattice, the crystal structure is an important factor for decomposition. Recently, we have focused on the C-type rare-earth sesquioxide structure, which is related to the fluorite-type structure by removing one-quarter of the oxide anions (). The C-type structure consequently possesses large open spaces descended from the oxygen vacancies, and these spaces are expected to work as active sites. In fact, we have demonstrated that
and
with C-type structures effectively decomposed
[Citation17,Citation18]. However, the effect of open spaces on the activity remains unclear. In order to investigate the relationship between oxygen vacancies and the N2O decomposition ability of the fluorite-related structure, we selected
solid solutions. A small amount of the
in
solids leads to the formation of a defected fluorite-type structure, in which oxygen vacancies are disordered, i.e. converted to yttria-stabilized zirconia. In the case of large amounts of
, the structure is known to form the C-type structure [Citation19]. In addition, while it was reported that redox properties of the catalyst affected the
decomposition ability [Citation17,Citation18,Citation20,Citation21], the effect of the redox properties in the
solid is expected to be suppressed due to the single valence state of the constituent cations (
and
). Based on these concepts, we prepared
solids and investigated the effect of oxygen vacancies on direct N2O decomposition activity.
Figure 1. Crystal structure of the cubic C-type phase.
2. Experimental
catalysts were synthesized using the co-precipitation method. Stoichiometric amounts of aqueous solutions of 1.0 mol·L−1
and 0.1 mol·L−1
were mixed, and the resulting solution was added to a 1 mol·L−1 ammonium carbonate aqueous solution with stirring at room temperature. After the pH of the solution was controlled to 9 by dropwise addition of 4 mol·L−1 ammonia aqueous solution, the mixture was stirred for 6 h at room temperature. The precipitation product was collected by suction filtration, dried, and then calcined at 1600°C for 6 h in air.
The composition of the catalyst was measured by X-ray fluorescence spectrometry (XRF; Supermini 200, Rigaku). The Brunauer-Emmett-Teller (BET) surface area was measured at −196°C (Micromeritics TriStar 3000, Shimadzu) using nitrogen gas adsorption. The crystal structure was identified by X-ray powder diffraction (XRD; SmartLab, Rigaku) using radiation (40 kV and 30 mA). The lattice constants were estimated by refining the XRD peak angles using α-alumina as an internal standard.
The catalytic tests for the direct decomposition were performed using a conventional fixed-bed flow reactor with a 10 mm diameter quartz glass tube. Catalytic activities were measured in the temperature range from 400°C to 950°C using thermal conductivity detection gas chromatography (GC-8AIT, Shimadzu). A gas mixture of 0.5 vol% nitrous oxide and 99.5 vol% helium was fed at a rate of 60 mL·min−1 over 0.2 g of the catalyst, a catalyst weight/gas flow rate (W/F) ratio of 0.2 g·s·mL−1. Before the catalytic activity tests, each catalyst was preheated at 200°C for 1 h in a helium flow.
Diffusion reflectance infrared fourier transform spectroscopy (DRIFTS) analysis was performed with an in situ FT-IR spectrometer (FT/IR-6100, JASCO) under a 0.5 vol% nitrous oxide and 99.5 vol% helium flow (60 mL·min−1). An environmental DRIFTS chamber was equipped with calcium fluoride windows to allow thermal and water resistance. Each spectrum was recorded by averaging 100 scans with a resolution of 4 cm−1, where the spectrum was compensated by the helium underflow. Prior to measurement, the sample was preheated at 400°C under a helium flow.
The oxygen-species desorption characteristics of the catalysts were evaluated by oxygen temperature-programmed desorption (O2-TPD) measurement using oxygen gas as follows. After heating the catalyst in a flow of helium at 500°C for 30 min, the catalyst was exposed to oxygen gas at the same temperature for 1 h and then cooled to 50°C. The catalyst was heated again under a helium flow at a heating rate of 10°C·min−1, and the desorbed gas was monitored using a catalyst analyzer (BELCAT-B, MicrotracBEL).
The acidity of the catalysts was measured by ammonia temperature-programmed desorption () measurement. The sample was exposed to a mixture of 0.5 vol% ammonia and 99.5 vol% nitrogen at 50°C for 1 h after annealing at 500°C for 30 min under a helium flow. Subsequently, the sample was heated under a helium flow (30 mL·min−1) at a heating rate of 10°C·min−1 using the same analyzer (BELCAT-B, MicrotracBEL). The number of basic sites on the catalysts was estimated from the temperature-programmed desorption profile of carbon dioxide (
), assuming that one carbon dioxide molecule is adsorbed on one basic site. Much as in the previous study [Citation22], the sample was pretreated in a flow of hydrogen (30 mL·min−1) at 500°C for 30 min, and exposed to carbon dioxide (30 mL·min−1) at 50°C for 1 h. After evacuation at 50°C for 30 min, the catalyst was heated under a flow of helium at a rate of 10°C·min−1, and the desorbed gas was monitored using the same analyzer (BELCAT-B, MicrotracBEL).
3. Results and discussion
shows the compositions of measured by XRF and the number of oxygen vacancies (δ) simply estimated using the measured x: i.e.
. Here,
was regarded as a distorted fluorite-related structure with a large number of oxygen vacancies (δ = 0.5). The measured compositions of the catalysts were consistent with their stoichiometric values within the range of experimental error. also shows the BET surface area of the catalysts. The surface area decreased with increases in the Y content (x) due to the lower melting point of
(ca. 2200°C) [Citation23] compared to that of
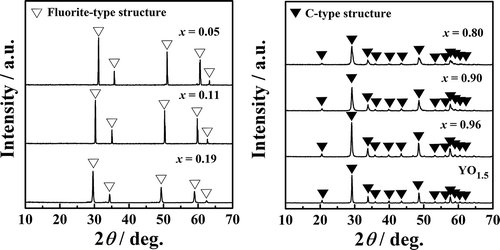
(ca. 2700°C) [Citation24]. As for the crystal structure (), the samples with x = 0.05, 0.11, and 0.19 exhibited a single-phase cubic fluorite-type structure of
, Fm
m. In the case of large amounts of x (x = 0.80, 0.90, 0.95, or 1), the patterns were assigned as a cubic C-type structure of YO1.5, Ia
. By contrast, the samples with x = 0.30, 0.50, 0.69 (Figure S1) were a mixture of a C-type structure and a monoclinic
phase. The crystal phase and the lattice constant are also summarized in . The lattice constant of the samples with fluorite-type structures increased with increases in x, because the Zr4+ ion sites (0.098 nm, coordination number [CN] = 8) [Citation25] were partially replaced by larger Y3+ ions (0.116 nm, CN = 8) [Citation25]. For catalysts with C-type structures, the lattice constant also increased with increases in x, indicating replacement of the Zr4+ sites (0.086 nm, CN = 6) [Citation25] with Y3+ (0.104 nm, CN = 6) [Citation25]. This structural change in the
system is consistent with that observed in the previous studies [Citation26,Citation27].
Table 1. Measured composition, number of oxygen vacancies (δ), BET surface area, crystal structure, and lattice constant of the Zr1−xYxO2−δ catalysts.
Figure 2. XRD patterns of Zr1−xYxO2−δ (x = 0.05, 0.11, 0.19, 0.80, 0.90, 0.96, and 1).
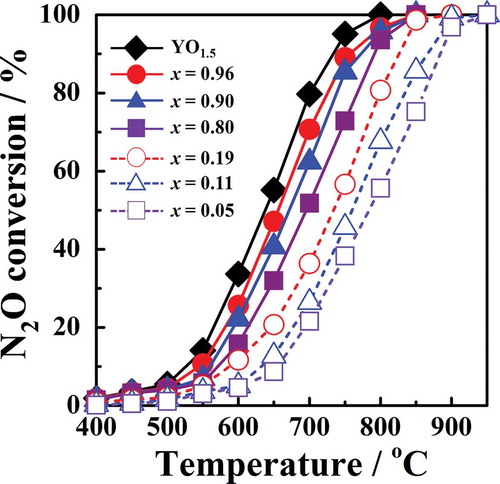
shows the temperature dependence of the conversion for the
catalysts with a single-phase fluorite-type or C-type structure. The catalytic activity increased continuously with increases in x, regardless of any decrease in the surface area. A high level of activity was obtained for
with a C-type structure. The apparent activation energy of the
decomposition was calculated using the following equations:
Figure 3. Temperature dependence of N2O conversion by Zr1−xYxO2−δ catalysts.
where r is the reaction rate, k is the apparent rate constant, C is the concentration, A is the pre-exponential factor, Ea is the apparent activation energy, R is the gas constant, and T is the absolute reaction temperature. The apparent activation energy of the
decomposition can be obtained from the slope of the Arrhenius plot, in which ln(k) is plotted against the reciprocal temperature. The results are listed in . The apparent activation energy decreased with increases in x, the same tendency as that of the N2O decomposition activity. Based on these results, it was determined that the oxygen vacancy (δ) was related to the catalytic activity and the apparent activation energy of the
decomposition.
Table 2. Apparent activation energy of Zr1−xYxO2−δ catalysts.
To investigate the role of oxygen vacancies in the decomposition process of
catalysts, in situ FT-IR measurements were carried out. shows the DRIFTS spectra of
catalysts under a 0.5 vol% nitrous oxide-99.5 vol% helium flow at 50°C. There are doublet peaks at ca. 2240 and 2210 cm−1, due to the asymmetric N-N stretching vibrations of
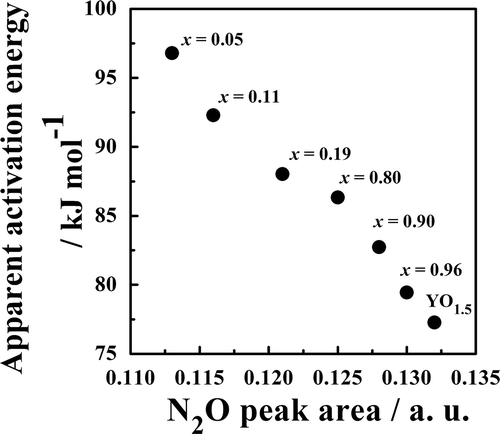
adsorbed on the catalysts [Citation28,Citation29]. With increases in the x value, the N2O adsorption peak intensity increased. presents the dependence of the N2O adsorption peak area on the number of oxygen vacancies (δ). For each fluorite-type (0.05 ≤ x ≤ 0.19) or C-type (x ≥ 0.80) structure, the adsorption area increased linearly with increases in the number of oxygen vacancies (δ). In addition, the fluorite-type structure has a high potential for N2O adsorption compared to the C-type structure when the same number of oxygen vacancies is assumed. Practically speaking, samples with a large number of oxygen vacancies (δ ≥ 0.40) exhibited the C-type phase, not the fluorite-type phase. shows the dependence of the apparent activation energy on the N2O adsorption peak area. The apparent activation energy was lowered monotonically by increasing the amount of N2O adsorption, irrespective of the crystal structure. YO1.5 solids with a large number of oxygen vacancies exhibited high catalytic activity. Oxygen vacancies are therefore considered to work as active sites for N2O decomposition.
Figure 4. (a) DRIFTS spectra of Zr1−xYxO2−δ and (b) the relationship between oxygen vacancies and the N2O peak area.
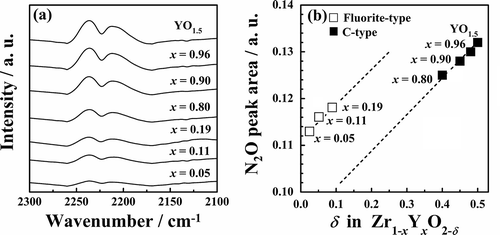
Figure 5. Relationship between the N2O peak area and apparent activation energy of Zr1−xYxO2−δ catalysts.
For further investigation of the catalysts, O2-TPD measurements were carried out (Figure S2). Two O2 desorption peaks were observed: O2 desorption peaks at lower and higher temperatures were assigned to desorption of physisorbed and chemisorbed oxygen, respectively [Citation8]. The amounts of physisorbed and chemisorbed oxygen species are listed in . Both the amounts of physisorbed and chemisorbed oxygen increased with increases in the number of oxygen vacancies (δ), a tendency similar to that reported in the previous paper [Citation21]. Thus, the O2-TPD results also support the proposition that the oxygen vacancies are gas adsorption sites.
Table 3. Volumes of oxygen physisorbed and chemisorbed by Zr1−xYxO2−δ catalysts.
Several researchers have reported that the gas adsorption on catalyst surfaces is related to the acidity and basicity of the catalyst surface [Citation30–Citation32]; the NH3-TPD and CO2-TPD levels were therefore measured to investigate the number of acidic and basic sites, respectively, on the catalysts. The NH3-TPD (Figure S3) and the CO2-TPD (Figure S4) profiles were used to calculate the numbers of acidic and basic sites, and the results are listed in . The number of acidic sites increased with increases in x, suggesting that oxygen vacancies with a positive charge served as acidic sites. Here, the O species in the N2O molecule possesses a stronger negative charge than the N species, since the electronegativity of oxygen is higher than that of nitrogen. O in N2O is therefore considered to be adsorbed on the acidic site preferentially, a conclusion supported by the DFT calculations reported previously [Citation33]. On the other hand, the number of basic sites decreased with increases in x, probably due to a decrease in the number of oxide anions in the lattice. This result suggests that basic sites have a negative effect on N2O adsorption. For these reasons, it is clear that the active sites for N2O decomposition are the oxygen vacancies in the Zr1−xYxO2−δ lattice.
Table 4. Number of acidic and basic sites on Zr1−xYxO2−δ catalysts.
4. Conclusion
In summary, Zr1−xYxO2−δ catalysts with fluorite-related structures were synthesized to investigate the relationship between oxygen vacancies and direct decomposition activity. The
decomposition activity was enhanced by increases in the Y3+ content (x), or the number of oxygen vacancies (δ). In situ FT-IR measurement showed that the amount of
adsorption increased with increases in the number of oxygen vacancies. Here, when the same number of oxygen vacancies is assumed, the fluorite-type structure possesses greater potential for
adsorption; however, catalysts with a large number of oxygen vacancies (δ ≥ 0.40) have C-type structures. The amounts of
adsorption and activation energy employed for
decomposition have a simple relationship, irrespective of the crystal structure. The highest activity was obtained for
solids with C-type structures, which possess the largest number of oxygen vacancies in the
system.
Supplemental Material
Download MS Word (6.7 MB)Acknowledgments
This study was supported in part by JSPS KAKENHI Grant No. JP19K15290.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Tricot C, Berger A. Modelling the equilibrium and transient responses of global temperature to past and future trace gas concentrations. Clim Dyn. 1987;2:39–61.
- Konsolakis M. Recent advances on nitrous oxide (N2O) decomposition over non-noble-metal oxide catalysts: catalytic performance, mechanistic considerations, and surface chemistry aspects. ACS Catal. 2015;5:6397–6421.
- Parres-Esclapez S, Illan-Gomez MJ, Salinas-Martinez de Lecea C, et al. On the importance of the catalyst redox properties in the N2O decomposition over alumina and ceria supported Rh, Pd and Pt. Appl Catal B: Environ. 2010;96:370–378.
- Yuzaki K, Yarimizu T, Ito SI, et al. Catalytic decomposition of N2O over supported rhodium catalysts: high activities of Rh/USY and Rh/Al2O3 and the effect of Rh precursors. Catal Lett. 1997;47:173–175.
- Hussain M, Fino D, Russo N. N2O decomposition by mesoporous silica supported Rh catalysts. J Hazard Mater. 2012;211–212:255–265.
- Rico-Pérez V, Salinas-Martínez de Lecea C, Bueno-López A. Preparation of RhOx/CeyPr1−yO2 N2O decomposition catalysts by rhodium nitrate impregnation with different solvents. Appl Catal A Gen. 2014;472:134–142.
- Konsolakis M, Yentekakis IV, Pekridis G, et al. Insights into the role of SO2 and H2O on the surface characteristics and de-N2O efficiency of Pd/Al2O3 catalysts during N2O decomposition in the presence of CH4 and O2 excess. Appl Catal B: Environ. 2013;138–139:191–198.
- Xue L, Zhang C, He H, et al. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl Catal B: Environ. 2007;75:167–174.
- Yan L, Ren T, Wang X, et al. Catalytic decomposition of N2O over MxCo1−xCo2O4 (M = Ni, Mg) spinel oxides. Appl Catal B: Environ. 2003;45:85–90.
- Kumar S, Vinu A, Subrt J, et al. Catalytic N2O decomposition on Pr0.8Ba0.2MnO3 type perovskite catalyst for industrial emission control. Catal Today. 2012;198:125–132.
- Ivanov DV, Sadovskaya EM, Pinaeva LG, et al. Influence of oxygen mobility on catalytic activity of La-Sr-Mn-O composites in the reaction of high temperature N2O decomposition. J Catal. 2009;267:5–13.
- Ivanov DV, Pinaeva LG, Isupova LA, et al. Insights into the reactivity of La1−xSrxMnO3 (x = 0 ÷ 0.7) in high temperature N2O decomposition. Catal Lett. 2011;141:322–331.
- Tian M, Wang A, Wang X, et al. Effect of large cations (La3+ and Ba2+) on the catalytic performance of Mn-substituted hexaaluminates for N2O decomposition. Appl Catal B: Environ. 2009;92:432–444.
- Zhang Y, Wang X, Zhu Y, et al. Stabilization mechanism and crystallographic sites of Ru in Fe-promoted barium hexaaluminate under high-temperature condition for N2O decomposition. Appl Catal B: Environ. 2013;129:382–393.
- Benjamin RW, Reimer JA, Alexis TB. Studies of N2O adsorption and decomposition on Fe-ZSM-5. J Catal. 2002;209:151–158.
- RSda C, Mascarenhas AJS, Andrade HMC. Co-ZSM-5 catalyst for N2O decomposition. Appl Catal B: Environ. 1998;18:223–231.
- Cho CM, Watanabe Y, Nunotani N, et al. Direct decomposition of nitrous oxide using Yb2O3-Pr6O11 with C-type cubic structure. Chem Lett. 2018;47:996–999.
- Cho CM, Nunotani N, Imanaka N, et al. Direct decomposition of N2O over C-type cubic Yb2O3-Co3O4 catalysts. Bull Chem Soc Jpn. 2019;92:1148–1153.
- Masaki H, Masui T, Imanaka N. Direct decomposition of nitric oxide into nitrogen and oxygen over C-type cubic Y2O3-ZrO2 solid solutions. J Alloy Compd. 2008;451:406–409.
- Pan KL, Yu SJ, Yan SY, et al. Direct N2O decomposition over La2NiO4-based perovskite-type oxides. J Air Waste Manage Assoc. 2014;64:1260–1269.
- Zhu J, Xiao D, Li J, et al. Effect of Ce on NO direct decomposition in the absence/presence of O2 over La1−xCexSrNiO4 (0 ≤ x ≤ 0.3). J Mol Catal A: Chem. 2005;234:99–105.
- Tsujimoto S, Masui T, Imanaka N. Direct decomposition of nitrogen monoxide over C-type cubic Y2O3-Pr6O11 solid solutions. RSC Adv. 2014;4:1146–1149.
- Reddy RG, Kumar SG. Solubility and thermodynamic properties of Y2O3 in LiF-YF3 melts. Metall Mater Trans B. 1994;25B:91–96.
- Duran P, Gonzalez M, Moure C, et al. A new tentative phase equilibrium diagram for the ZrO2-CeO2 system in air. J Mater Sci. 1990;25:5001–5006.
- Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A. 1976;32:751–767.
- Feighery AJ, Irvine JT, Fagg DP, et al. Phase relations at 1500°C in the Ternary system ZrO2-Y2O3-TiO2. J Solid State Chem. 1999;143:273–279.
- Scott HG. Phase relationships in the zirconia-yttria system. J Mat Sci. 1975;10:1527–1535.
- Chen L, Chen HY, Lin J. FTIR, XPS and TPR studies of N2O decomposition over Cu-ZSM-5. Surf Interface Anal. 1999;28:115–118.
- Kim SS, Lee SJ, Hong SC. Effect of CeO2 addition to Rh/Al2O3 catalyst on N2O decomposition. Chem Eng J. 2011;167:173–179.
- Bhattacharyya K, Danon A, Vijayan BK, et al. Role of the surface lewis acid and base sites in the adsorption of CO2 on titania nanotubes and platinized titania nanotubes: an in Situ FT-IR study. J Phys Chem C. 2013;117:12661–12678.
- Denysenko D, Jelic J, Magdysyuk OV, et al. Elucidating lewis acidity of metal sites in MFU-4lmetal-organic frameworks: N2O and CO2 adsorption in MFU-4l, CuI-MFU-4l and Li-MFU-4l. Microporous Mesoporous Mater. 2015;216:146–150.
- Kustov LM. New trends in IR-spectroscopic characterization of acid and basic sites in zeolites and oxide catalysts. Top Catal. 1997;4:131–144.
- Zhang B, He G, Shan Y, et al. Experimental and DFT study of the adsorption of N2O on transition ion-exchanged ZSM-5. Catal Today. 2019;327:177–181.