
ABSTRACT
This study focuses on organically modifying a layered calcium silicate with two types of organic functional groups to control chemical stability. Solvothermal processing was used to prepare organically modified layered calcium silicates with different contents of aminopropyl and phenyl groups. By-products of polyhedral oligomeric silsesquioxanes derived from organosilanes (Rn-SiO1.5n) were also formed in the sample powders upon preparation with a high initial fraction of phenyltriethoxysilane. The formation of layered calcium silicates modified with both organic groups was confirmed by X-ray diffractometry, nuclear magnetic resonance spectroscopy, Fourier-transform infrared spectroscopy, and Raman spectroscopy. The contents of these organic groups in the samples reflected the molar fractions of silanes in the starting materials. Although all samples readily released calcium and silicate ions into a buffer solution of pH = 7.4 after soaking for 24 h, the amounts of released silicate and calcium ions tended to decrease as the initial molar fraction of phenyl groups was increased up to 75 mol%. The chemical stability of the layered calcium silicates increased with the increase of the hydrophobic functional groups incorporated into their structure. These results indicate the potential for applying these modifications in biomaterials with tunable solubility for the purpose of bone reconstruction.
1. Introduction
Calcium silicate-based materials have been important components of artificial bones since the invention of Bioglass 45S5 [Citation1]. A fundamental characteristic of calcium silicate-based materials is their capability of hydroxyapatite formation in the body [Citation2]. The formation of hydroxyapatite on calcium silicate-based materials leads to direct bonding to living bone (i.e., bone-bonding). This can be achieved by the dissolution of calcium ions into the surrounding bodily fluid, which increases the degree of supersaturation with respect to hydroxyapatite. The soluble products released from the calcium silicate-based materials have been known to genetically stimulate osteoblasts, leading to proliferation, differentiation, and calcification [Citation3,Citation4]. An inorganic layered compound calcium silicate hydrate (CSH, CaxSiyOx+2y·nH2O) is composed of sheet-like units (denoted as hetero-layer) consisting of calcium oxide polyhedral, crosslinked by chains of SiO4 tetrahedra or “dreierketten” [Citation5,Citation6]. We focused on the CSH as a candidate for drug delivery carriers for bone-reconstruction treatment because materials with binary CaO-SiO2 composition have bone-tissue-bonding abilities through the formation of hydroxyapatite within the body [Citation2]. Tuning the solubility of the CSH is the key to developing novel bone-bonding biomaterials as the solubility determines the releasing rate and the bone-bonding ability of the administered drug.
Organic functional groups can be incorporated between the layers through synthesis with selected starting materials thanks to their interlayer structural flexibility. The modified calcium silicate layer would then possess various chemical functionalities, implying that incorporated organic molecules or inorganic ions could act as biological factors as well as improve chemical stability. A precipitation process using various silicate and alkylsilane molecules was used in the fabrication of organically modified layered calcium silicates. Previous studies [Citation7,Citation8] reported that the interlayer distance of layered calcium silicates increases linearly with the alkyl chain length of the silane molecules. The dissolution behavior of layered calcium silicates is expected to become tunable by modifying the interlayer distance, charge density, and hydrophobicity. However, there have been no reports on layered calcium silicates modified using two different types of organic functional groups. We hypothesized that the modification of the silicate chain in calcium silicates with nonpolar organic functional groups could influence the hydrophobicity and hence enhance the chemical durability in an aqueous environment. On the other hand, modification with aminopropyl groups could afford sites for the incorporation of organic molecules or inorganic ions as biological factors. In this study, one-pot synthesis of layered calcium silicates modified with aminopropyl (C3H6NH2) and phenyl (C6H5) groups was attempted to investigate the effect of functional groups on the chemical stability of resultant compounds. Besides, a solvothermal process with preliminary conditions of 120°C for 48 h was selected instead of the conventional precipitation method. This selection was due to the assumption that the non-uniform reaction caused by the rapid hydrolysis and condensation reactions of aminopropylsilane [Citation9] could be suppressed.
2. Experimental method
Calcium nitrate tetrahydrate (5.3 g) (Ca(NO3)2 · 4H2O, Nacalai Tesque, Japan), sodium hydroxide (1.0 g) (NaOH, Nacalai Tesque, Japan), appropriate amounts of phenyltriethoxysilane (PhS, C6H5Si(OC2H5)3, Tokyo Chemical Industry, Japan), and aminopropyltriethoxysilane (APS, NH2C3H6Si(OC2H5)3, Sigma–Aldrich, USA) were added to a mixture of 50 cm3 of ethanol (C2H5OH, Kishida Chemical, Japan) and 25 cm3 of deionized water to obtain the initial composition with a Ca/Si molar ratio of 0.9. The formation of CSH that is free from the interlayer crosslinking has been reported at this Ca/Si molar ratio [Citation10]. A sample without organic modification was also prepared using tetraethylorthosilicate (TEOS, Si(OC2H5)4, Sigma–Aldrich, USA). The mixture was solvothermally treated in a Teflon-lined autoclave vessel at 120°C for 48 h. summarizes the sample codes and compositions of the starting materials. The precipitates were centrifuged, washed with ethanol, and dried at 40°C for 1 d to obtain the sample powders.
Table 1. Sample codes and composition of starting materials
The elemental analysis of the prepared samples was performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES, Optima 2000 DV, PerkinElmer, USA) and organic elemental analysis (OEA, 2400 Series II CHNS/O, PerkinElmer, USA). The samples for ICP-AES were prepared by sintering the sample powders in an electric furnace (SuperBurn, Motoyama, Japan) at 1200°C for 4 h with an increasing temperature rate of 5°Cmin−1, followed by dissolving the obtained powders in nitric acid (HNO3, Fujifilm Wako Pure Chemical, Japan). The powder X-ray diffraction (XRD) patterns of the sample powders were measured using CuKα radiation on an X-ray diffractometer (RINT-2100/PC, Rigaku, Japan). The structure of silicates in the organically modified samples (APxPh(100-x), x= 0 ~ 100) was analyzed using solid-state 29Si dipolar decoupling magic angle spinning nuclear magnetic resonance spectrometry (29Si DD-MAS NMR, Avance 300 wbs, Bruker, USA). The samples were also evaluated using Fourier-transform infrared spectroscopy (FT-IR, FT/IR-6100, Jasco, Japan) with the KBr pellet method, Raman spectroscopy (ApaRAMAN-Micro/CZ, Lucir, Japan) using a diode-pumped solid-state laser (532 nm, 150 mW), and differential thermal/thermogravimetric (TG) analysis (TG-DTA, DTG-60, Shimadzu, Japan). The onset and final temperatures of mass loss were estimated by the extrapolation of the TG curves (denoted hereafter as the extrapolation method) unless specifically mentioned otherwise. Briefly, the temperatures were estimated from the intersecting points between a tangential line drawn to the steepest point of the mass loss and the extrapolated baseline before and after the mass loss. The morphologies of the samples were observed using a field emission scanning electron microscope (FE-SEM, JSM-7500 F, JEOL, Japan).
The chemical stability of the samples was evaluated by immersion tests in a tris(hydroxymethyl)aminomethane buffer solution (Tris (HOCH2)3CNH2, Nacalai Tesque, Japan) and an aqueous solution of hydrochloric acid (HCl, Fujifilm Wako Pure Chemical, Japan). Tris (6.05 g) was dissolved in 1000 cm3 deionized water and maintained at 36.5°C using a water bath. The pH of the solution was adjusted to 7.4 by adding 1 mol·dm−3 aqueous HCl to obtain a Tris-HCl buffer solution with a Tris concentration of 50 mmol·dm−3. Each sample (100 mg) was dispersed in 10 cm3 of the Tris-HCl buffer solution by a vortex mixer (Deltamixer, Taitec, Japan), followed by 1 min of sonication. The resulting mixtures were maintained at 36.5°C for 24 h, after which the samples were filtered. Ca and Si concentrations of the resulting solutions were analyzed using the ICP-AES.
3. Results
The elemental compositions of the prepared samples are listed in . The molar ratio of Ca/Si was close to 1 in all prepared samples. It has been reported that the layered skeleton of pristine CSH was formed with the Ca/Si molar ratio ranging from 0.6 to 1.5 [Citation10]. The molar ratio of our sample was also within this range. However, the final molar ratios were greater than that of the starting material. This is due to APS or PhS in the starting material remaining in the liquid phase during the solvothermal processes. Among the samples prepared using APS and PhS, the mass fraction of N tended to increase and that of C tended to decrease as the APS/PhS molar ratio of the starting material increased.
Table 2. Elemental compositions of prepared samples. The mass fractions of aminopropyl (NH2(CH2)3) and phenyl (C6H5) groups were estimated from the mass fractions of N and C determined by organic elemental analysis
shows the powder XRD patterns of the prepared samples, which indicate that TE consisted of only CSH (PDF#34-0002) as a crystalline phase. The peak at 2θ = 6.9° (d= 1.3 nm) was assigned to the 002 diffraction originating from the interlayer distance (ID). The peaks at 2θ = 16.6°, 29.3°, and 50.5° originated from the in-plane diffractions of the hetero-layers consisting of calcium oxide polyhedra and silicate chains [Citation7]. Particularly, the peaks at 29.3º and 50.5º originated from the overlapped (020)+(20) reflections and (
40) diffraction, respectively [Citation11]. The samples prepared using APS and PhS also showed corresponding peaks of CSH, while a halo peak derived from the amorphous structure appeared at 2θ of around 15 ~ 25°. The AP25Ph75 and AP0Ph100 showed a more intense halo peak compared to AP100Ph0, AP75Ph25, and AP50Ph50. The 002 diffraction at 2θ of approximately 7° had shoulders in the patterns of AP25Ph75 and AP0Ph100. The shoulder peaks at 2θ = 7.2° (d= 1.2 nm) were analyzed by Voigt function fitting and are shown in . These shoulders are presumably due to the presence of polyhedral oligomeric silsesquioxane derived from organosilanes (Rn-SiO1.5n). 29Si DD-MAS NMR spectra of samples prepared using APS and PhS were measured to obtain an insight into the structure of silicates, as shown in . The connectivity of trifunctional silane is indicated by Tn (n= 0 ~ 3); T0 indicates a single silane molecule (R’-Si(-O-R)3), and Tn (n= 1, 2, or 3) represents the silane connected with -O-Si groups (R’-Si(-O-Si)n(-O-R)3−n). The AP100Ph0 showed the peaks of T1 and T2 units derived from the APS at the chemical shifts of −47~−51 ppm and −58~−67 ppm, respectively [Citation9,Citation12]. The AP0Ph100 showed the peaks of T1, T2, and T3 units derived from the PhS at −63 ppm, −69~−73 ppm, and −79 ppm chemical shifts, respectively [Citation13,Citation14]. The AP75Ph25 showed the T1and T2 units, while AP50Ph50 and AP25Ph75 showed T1, T2, and T3 units. Peaks of the APS-derived T2 unit and the PhS-derived T1 unit greatly overlapped in these three samples. T1 and T2 units of APxPh100−x samples reflected the presence of silicate chains, in which a pair of T1 units terminates a chain consisting of T2 units. This chain structure is a characteristic of the hetero-layer of the CSH. The T1APS and T2PhS units of APxPh100−x samples were observed as split peaks. The CSH crystal and its aluminum-doped derivatives have bridging and pairing positions (indicated as (i) and (ii) in ) for Si atoms, which caused the split of the Qn-derived peak in its 29Si NMR spectrum [Citation15,Citation16]. The split peaks of the T1APS and T2PhS units were believed to indicate the presence of the bridging and pairing sites. The relative intensity of the APS-derived T1 peak decreased, while that of the PhS-derived T2 peak increased with increasing the content of PhS in starting materials. These trends are in good agreement with the CHN data. The molar fractions of T3 units within the AP50Ph50, AP25Ph75, and AP0Ph100 were estimated to be 11, 36, and 35 mol%, respectively, from the integrated peak area ratio of the NMR spectra. The T3 units suggested the formation of amorphous components or the polyhedral oligomeric silsesquioxane, which is detected from the XRD patterns. The silsesquioxane consisting of APS and PhS was known to show excellent solubility in a mixed solvent of ethanol and water when the molar fraction of APS/PhS exceeds 0.8 [Citation17]. The T3 units are considered to be eliminated from AP100Ph0 and AP75Ph25 during their preparation procedures. Based on the analysis of 002 diffractions, the estimated IDs of the prepared samples are summarized in . While the estimated ID of the CSH sample without organic modification (TE) was 1.3 nm, and the calcium silicate prepared with APS and PhS had larger IDs of 1.5–1.9 nm. XRD pattern assignment of TE showed a formation of crystalline-phase CSH. The incorporation of APS and PhS increased the interlayer space of CSH, and the formation of silsesquioxanes as a by-product occurred with higher initial amounts of PhS (AP25Ph75 and AP0Ph100). On the other hand, samples with greater amounts of phenyl groups (AP25Ph75 and AP0Ph100) showed less expanded distances of approximately 1.5 nm than those of AP50Ph50, AP75Ph25, and AP100Ph0, which ranged from 1.7 to 1.9 nm.
Figure 1. (a) Powder XRD patterns of prepared samples. (b) Diffraction patterns (solid line) and Voigt function-fitted curves (chain, dotted, and dashed lines) of AP0Ph100 and AP25Ph75.
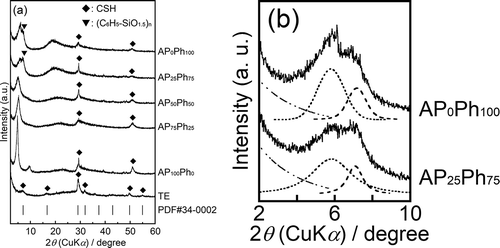
Figure 2. 29Si DD-MAS NMR spectra of the prepared samples
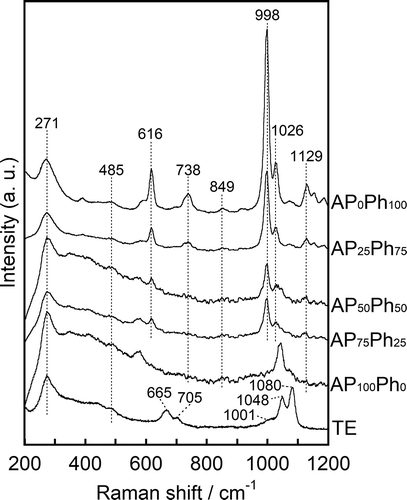
shows the FT-IR spectra of the prepared sample powders. TE showed a broad peak centered at 3400 cm−1, assigned to silanol groups and water molecules in the CSH interlayers [Citation7,Citation18]. AP100Ph0 showed two doublets centered at 3330 and 2900 cm−1 originating from N-H and C-H bonding in aminopropyl groups, respectively [Citation19]. The samples prepared using both APS and PhS showed a peak derived from the C-H bonding of phenyl groups in addition to the two doublets assigned to the aminopropyl groups [Citation20]. Moreover, the relative intensity of the phenyl- and aminopropyl-derived peaks (νphenyl/νaminopropyl) increased as the PhS/APS molar fraction was increased. These changes in the relative intensity support the mass fractions of phenyl- and aminopropyl groups estimated from OEA showed in . AP0Ph100 showed only the C-H phenyl group peak. From the detection of Tn units in the NMR spectra, the aminopropyl- and phenyl groups derived from APS and PhS were chemically bonded to the interlayer of the layered calcium silicates, and the contents of these groups were in agreement with the starting compositions. Raman spectra of the prepared sample powders were also measured, and the results are shown in . In the TE spectrum, the vibrational peak at 271 cm−1 is assigned to calcium oxide polyhedra in the layered calcium silicates [Citation21,Citation22]. The peaks at 665 and 1048 cm−1 are assigned to the symmetrical bending (SB) and symmetrical stretching (SS) modes of Si-O-Si in Q2 (Si(-O-Si)2(-O-X)2, X= Ca or H) group silicon, respectively [Citation23,Citation24], and the peaks at 705 and 1001 cm−1 are assigned to the SB and SS of Q1 (Si(-O-Si)(-O-X)3) group silicon, respectively [Citation25]. The peak at 1080 cm−1 is assigned to the vibration of carbonate (CO32−) ions [Citation22]. The samples prepared with APS and PhS also showed the calcium oxide polyhedra peak at 271 cm−1, and the vibrational peaks at 485 and 840 cm−1 are assigned to the SB and SS modes of Si-O-Si [Citation23], respectively. A peak originating from the stretching vibration of Si-C was observed at 738 cm−1 [Citation24]. The peaks at 616, 998, and 1026 cm−1 originated from the phenyl ring of PhS, the peak at 616 cm−1 was assigned to the deformation of C-C bonds in the phenyl ring, and the peaks at 998 and 1026 cm−1 were assigned to its stretching vibration [Citation20,Citation24]. These results revealed that both the amino and phenyl groups were incorporated into the organically modified layered calcium silicates, and their contents are dependent on the starting compositions.
Figure 3. FT-IR spectra of the prepared samples
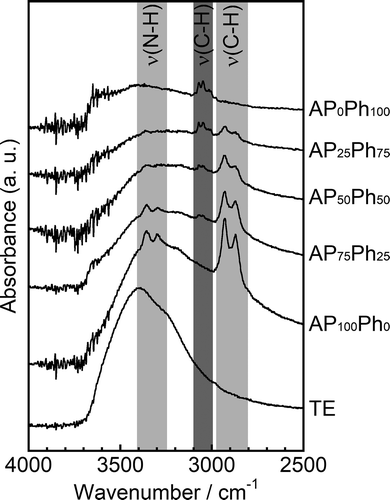
Figure 4. Raman spectra of the prepared samples
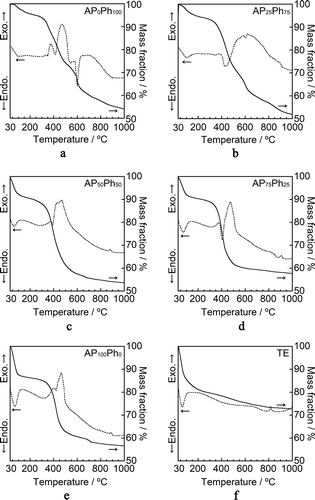
shows the TG curves (solid line) and DTA curves (dashed line) of the prepared sample powders. TE showed a significant mass loss below 200°C accompanied by an endothermic peak, which has been reported to come from the removal of interlayer water molecules of the CSH crystal [Citation25]. The mass loss with the broad endothermic peak around 450°C corresponds to the dehydration of silanol groups. The exothermic peak above 800°C indicates the exothermic transformation into β-wollastonite (β-CaSiO3), an anhydrous form of calcium silicate [Citation26,Citation27]. The mass losses and endothermic peaks of the samples prepared with APS and PhS below 160°C are due to the removal of water molecules. The TG curves of the samples prepared with APS and PhS also showed a mass loss between 300°C and 900°C. The DTA curves showed exothermic peaks due to the combustion of the organic components. Multi-stepped mass losses occurred in the temperature range 350–920°C in the TG curves of AP25Ph75 and AP0Ph100. Both samples showed broad exothermic peaks at this temperature range. Therefore, the mass losses may be attributable to the multi-step oxidation of the components of phenyl-modified silicates in ambient conditions [Citation28]. Using the TG results, the mass losses caused by the removal of water and combustion of organic components were estimated by the extrapolation method, and the results are summarized in . The initial temperature in the TG analysis (35°C) was used as the onset temperature for the removal of the water molecules due to the difficulty in determining the appropriate baseline for this method. The fraction of water mass lost decreased in the order of TE > AP100Ph0 > AP75Ph25 ≅ AP50Ph50 > AP25Ph75 > AP0Ph100, while the fraction of organic components lost decreased in the order of AP0Ph100 > AP25Ph75 > AP50Ph50 ≅ AP75Ph25 > AP100Ph0.
Figure 5. TG-DTA curves of the prepared samples
shows the FE-SEM images of the prepared samples. The images of TE showed non-modified CSH, consisting of 4–6 μm agglomerated particles that are seemingly composed of submicron-sized primary particles with flake morphology, as shown in . The samples prepared using APS and PhS were observed as irregularly shaped agglomerates consisting of 0.1–0.3 μm spherical and elliptical primary particles irrespective of the molar fraction of APS and PhS, as shown in .
Figure 6. FE-SEM images of the prepared samples. (a) TE, (b) AP100Ph0, (c) AP75Ph25, (d) AP50Ph50, (e) AP25Ph75, and (f) AP0Ph100.

shows the concentrations of calcium and silicon in the Tris-HCl solution after soaking the APxPh100−x (x = 100, 75, 50, 25, or 0) samples for 24 h. The concentrations of Ca and Si were the highest in the soaking of AP100Ph0 (indicated as 100 in ), suggesting that AP100Ph0 released the largest amounts of calcium and silicate ions among the examined organically modified samples. These concentrations decreased with increasing the fraction of PhS in the starting materials up to 75 mol%. AP25Ph75 (indicated as 25 in ) showed the lowest amounts of released calcium and silicate ions, with concentrations below AP0Ph100 (indicated as 0 in ) after soaking.
Figure 7. Elemental concentrations of silicon and calcium in Tris-HCl buffer solution after soaking of APxPh100-x (x = 100, 75, 50, 25, or 0) samples for 24 h
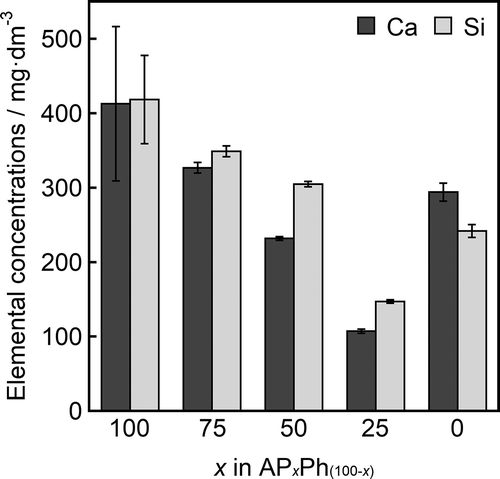
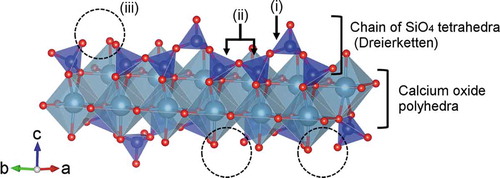
Figure 8. Structural model of single hetero-layer of calcium silicate hydrate: (i) bridging SiO4 tetrahedron, (ii) pairing SiO4 tetrahedra, and (iii) defect site of bridging tetrahedra
4. Discussion
Calcium silicate crystals were prepared by precipitation using TEOS and alkylsilane molecules [Citation7,Citation8]. The ID of the layered calcium silicates increased linearly with increasing the silane alkyl chain length. Here, calcium silicate crystals were prepared using APS and PhS molecules through the one-pot solvothermal process. The XRD patterns of the prepared samples confirmed the successful synthesis of layered calcium silicates with organic interlayer modification by solvothermal process with two types of alkoxysilanes. The XRD patterns showed peaks corresponding to the in-plane diffraction of hetero-layers in the layered calcium silicates corresponding to CSH. By estimating the 002 reflections (), organically modified samples showed an increase of 0.2–0.6 nm in the IDs compared with non-modified CSH. The 29Si DD-MAS NMR spectra showed the presence of T1, T2, and T3 units originating from the APS and PhS. Especially T1 and T2 units indicate the presence of silicate chain (dreierketten) peculiar to the hetero-layer of the CSH, confirming the formation of a layered skeleton. NMR, FT-IR, and Raman spectra also suggested that the interlayers were organically modified with both aminopropyl and phenyl groups in the samples prepared with functionalized alkoxysilane (APS and PhS) molecules. The amounts of amino and phenyl groups determined by OEA and the absorbance ratios in FT-IR are dependent on the molar ratio of APS/PhS of the starting materials. The presence of hetero-layers in the prepared samples was further proven by the calcium oxide polyhedra peak in the Raman spectra. These results indicated the successful preparation of calcium silicate modified by aminopropyl and phenyl groups.
Table 3. Estimated interlayer distances of the calcium silicate hydrate
Table 4. Estimated mass losses corresponding to interlayer water molecules and organic functional groups in the samples. The estimated temperature ranges were determined by the extrapolation method
The TG–DTA curves indicated that the organically modified samples contained organic components 31–40% in mass corresponding to the existence of aminopropyl and phenyl groups attached to alkoxysilane molecules. These organic mass fractions are in good agreement with the values calculated from OEA. The removal of interlayer water from CSH occurred below 150°C. All organically modified calcium silicate samples in this research showed smaller mass losses than the non-modified CSH (TE) below 160°C. The mass loss was minimized to 2% when only PhS was used as the silane source (AP0Ph100). These results revealed that the presence of organic groups affects the extent of interlayer dehydration. Amino groups are polar and are thus expected to hold the interlayer water molecules, while phenyl groups exclude water molecules due to their nonpolarity. Despite the similarity in the molecular length of C6H5-Si unit (0.57 nm) and H2NC3H6-Si unit (0.61 nm), the samples with greater amounts of phenyl groups (AP25Ph75 and AP0Ph100) tend to have smaller IDs, namely 1.5 nm, than those of AP50Ph50, AP75Ph25, and AP100Ph0, which were 1.7–1.9 nm. The presence of interlayer water molecules in the vicinity of aminopropyl groups is expected to contribute to the expansion of the IDs.
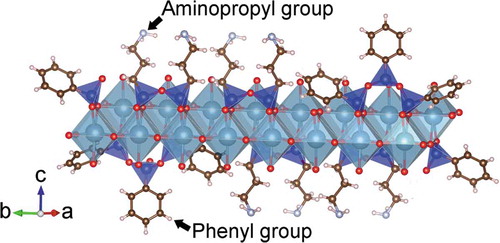
Several models are proposed for the nanostructure of CSH crystals, and the most common model is based on the crystalline structures of 1.4 nm tobermorite (Ca5Si6O16(OH)2 · 7H2O) and jennite (Ca9Si6O18(OH)6 · 8H2O) proposed by Cong and Kirkpatrick [Citation6]. The schematic model is depicted in . A single layer of CSH is composed of sheet-like units consisting of calcium oxide polyhedra crosslinked by chains of SiO4 tetrahedra and is referred to as “dreierketten”. The SiO4 tetrahedra have positions termed as either pairing or bridging. A bridging tetrahedron shares an oxygen with a calcium oxide polyhedron and possesses a single hydroxy (OH) group. In this research, the in-plane diffraction peaks were observed in all the samples. Moreover, the presence of a silicate chain and CaO polyhedra, the building units of the hetero-layer in CSH, were detected from the NMR and Raman spectrometry. Therefore, non-modified and organically modified calcium silicate showed resemblance in the nanostructure of the single hetero-layer. The expected structure of the organically modified CSH prepared with both APS and PhS is schematically shown in . A similar structural model of organically modified calcium silicate has been proposed by Minet et al. [Citation8].
Figure 9. Schematic representation of expected structure of organically modified calcium silicate hydrate prepared in the present study
The soaking test in a Tris-HCl buffer solution revealed a decrease in the release of calcium and silicate ions from the samples with higher contents of phenyl groups. However, AP0Ph100 showed a slight increase in the ion release compared with AP25Ph75. Modification of the silicate chain in calcium silicate with a nonpolar organic functional group may reduce its hydrophobicity and hence enhance its chemical durability in an aqueous environment. Although it was difficult to accurately evaluate the release of calcium and silicate ions due to the co-existence of the amorphous phase and silsesquioxanes, phenyl group incorporation provides hydrophobicity and is presumed to chemically stabilize calcium silicates against an aqueous environment. Tracking analyses of structural changes during the soaking in the buffer solution will be required for further understanding of the correlation between the chemical durability and the nanostructure of the layered calcium silicates with organic modification.
5. Conclusions
Layered calcium silicates modified with aminopropyl and phenyl groups were prepared by solvothermal process using APS and PhS. The fractions of aminopropyl and phenyl groups were varied via the molar fraction of silanes in the starting materials. The samples showed a release of calcium and silicate ions in the Tris-HCl buffer solution, and the amounts of released ions gradually decreased when the initial molar fraction of the PhS was increased. These results indicate their potential application as calcium silicate-based biomaterials with tunable solubility for bone reconstruction with further functionalities in biological systems.
Acknowledgments
This research was supported in part by Leading Initiative for Excellent Young Researchers (LEADER), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan and by the Kazuchika Okura Memorial Foundation. Some figures were prepared using VESTA software [Citation29].
Disclosure statement
The authors declare no conflicts of interest associated with this work.
References
- Hench L, Wilson J. An introduction to bioceramics. 1993. Singapour: World Scientific; 1971. p. 200.
- Ohtsuki C, Kokubo T, Yamamuro T. Mechanism of apatite formation on CaOSiO2P2O5 glasses in a simulated body fluid. J Non·Cryst Solids. 1992;143:84–92.
- Xynos I, Hukkanen M, Batten J, et al. Bioglass® 45S5 stimulates osteoblast turnover and enhances bone formation in vitro: implications and applications for bone tissue engineering. Calcif Tissue Int. 2000;67(4):321–329.
- Xynos ID, Edgar AJ, Buttery LD, et al. Ionic products of bioactive glass dissolution increase proliferation of human osteoblasts and induce insulin-like growth factor II mRNA expression and protein synthesis. Biochem Biophys Res Commun. 2000;276(2):461–465.
- Gard J, Taylor H. Calcium silicate hydrate (II)(“CSH (II)”. Cem Concr Res. 1976;6(5):667–677.
- Cong X, Kirkpatrick RJ. 29Si and 17O NMR investigation of the structure of some crystalline calcium silicate hydrates. Adv Cem Based Mater. 1996;3(3):133–143.
- Minet J, Abramson S, Bresson B, et al. New layered calcium organosilicate hybrids with covalently linked organic functionalities. Chem Mater. 2004;16(20):3955–3962.
- Minet J, Abramson S, Bresson B, et al. Organic calcium silicate hydrate hybrids: a new approach to cement based nanocomposites. J Mater Chem. 2006;16(14):1379–1383.
- M-C BS, Bayle P-A, Abdelmouleh M, et al. Kinetics of hydrolysis and self condensation reactions of silanes by NMR spectroscopy. Colloids SurfPhysicochem Eng Asp. 2008;312(2):83–91.
- Grangeon S, Claret F, Roosz C, et al. Structure of nanocrystalline calcium silicate hydrates: insights from X-ray diffraction, synchrotron X-ray absorption and nuclear magnetic resonance. J Appl Crystallogr. 2016;49(3):771–783.
- Grangeon S, Claret F, Lerouge C, et al. On the nature of structural disorder in calcium silicate hydrates with a calcium/silicon ratio similar to tobermorite. Cem Concr Res. 2013;52:31–37.
- Yoshino A, Okabayashi H, Shimizu I, et al. Kinetics of interaction of 3-aminopropyltriethoxysilane with silica gel using elemental analysis and 29Si NMR spectra. Colloid Polym Sci. 1997;275(7):672–680.
- Sun X, Xu Y, Jiang D, et al. Study on the ammonia-catalyzed hydrolysis kinetics of single phenyltriethoxysilane and mixed phenyltriethoxysilane/tetraethoxysilane systems by liquid-state 29Si NMR. Colloids SurfPhysicochem Eng Asp. 2006;289(1):149–157.
- Yoshinaga I, Yamada N, Katayama S. Effect of metal alkoxide complexes on condensation reactions of hydrolyzed phenyltriethoxysilane. J Sol-Gel Sci Technol. 2003;28(1):65–70.
- Pardal X, Brunet F, Charpentier T, et al. 27Al and 29Si solid-state NMR characterization of calcium-aluminosilicate-hydrate. lnorg Chem. 2012;51(3):1827–1836.
- L’Hôpital E, Lothenbach B, Le Saout G, et al. Incorporation of aluminium in calcium-silicate-hydrates. Cem Concr Res. 2015;75:91–103.
- Liu S, Lang X, Ye H, et al. Preparation and characterization of copolymerized aminopropyl/phenylsilsesquioxane microparticles. Eur Polym J. 2005;41(5):996–1001.
- Yu P, Kirkpatrick RJ, Poe B, et al. Structure of calcium silicate hydrate (C‐S‐H): near‐, mid‐, and far‐infrared spectroscopy. J Am Ceram Soc. 1999;82(3):742–748.
- Okabayashi H, Shimizu I, Nishio E, et al. Diffuse reflectance infrared fourier transform spectral study of the interaction of 3-aminopropyltriethoxysilane on silica gel. Behavior of amino groups on the surface. Colloid Polym Sci. 1997;275(8):744–753.
- Li Y-S, Wang Y, Ceesay S. Vibrational spectra of phenyltriethoxysilane, phenyltrimethoxysilane and their sol–gels. Spectrochim Acta A Mol Biomol Spectrosc. 2009;71(5):1819–1824.
- Kirkpatrick RJ, Yarger JL, McMillan PF, et al. Raman spectroscopy of C-S-H, tobermorite, and jennite. Adv Cem Based Mater. 1997;5(3):93–99.
- Garbev K, Stemmermann P, Black L, et al. Structural features of C–S–H(I) and its carbonation in air—a Raman spectroscopic study. part I: fresh phases. J Am Ceram Soc. 2007;90(3):900–907.
- de Ferri L, Lorenzi A, Lottici PP. OctTES/TEOS system for hybrid coatings: real-time monitoring of the hydrolysis and condensation by Raman spectroscopy. J Raman Spectrosc. 2016;47(6):699–705.
- Panitz J-C, Wokaun A. Characterization of the sol-gel process using Raman spectroscopy organically modified silica gels prepared via the formic acid-alkoxide route. J Sol-Gel Sci Technol. 1997;9(3):251–263.
- Fordham CJ, Smalley IJ. A simple thermogravimetric study of hydrated cement. Cem Concr Res. 1985;15(1):141–144.
- Taylor HFW. 33. Hydrated calcium silicates. part V. the water content of calcium silicate hydrate (I). J Chem Soc. 1953;163–171. DOI:10.1039/jr9530000163
- Alizadeh R, Raki L, Makar JM, et al. Hydration of tricalcium silicate in the presence of synthetic calcium–silicate–hydrate. J Mater Chem. 2009;19(42):7937–7946.
- Fina A, Tabuani D, Carniato F, et al. Polyhedral oligomeric silsesquioxanes (POSS) thermal degradation. Thermochim Acta. 2006;440(1):36–42.
- Momma K, Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr. 2011;44(6):1272–1276.