
ABSTRACT
Zika virus (ZIKV) can cause devastating congenital Zika syndromes in pregnant women and Guillain-Barre syndrome in adults. Understanding the molecular mechanism of ZIKV replication is essential for antiviral and vaccine development. Here we report the structural and functional characterization of ZIKV NS2A protein. Biochemical structural probing suggests that ZIKV NS2A has a single segment that traverses the ER membrane and six segments that peripherally associate with the ER membrane. Functional analysis has defined distinct NS2A residues essential for viral RNA synthesis or virion assembly. Only the virion assembly-defective mutants, but not the RNA synthesis-defective mutants, could be rescued through trans complementation with a wide-type NS2A protein. These results suggest that the NS2A molecules in virion assembly complex could be recruited in trans, whereas the NS2A molecules in viral replication complex must be recruited in cis. Together with previous results, we propose a flavivirus assembly model where NS2A plays a central role in modulating viral structural and nonstructural proteins as well as genomic RNA during virion assembly.
Introduction
Zika virus (ZIKV) is a recently emerged mosquito-borne flavivirus that is primarily transmitted by Aedes spp. mosquitoes. It can also be transmitted through sexual, maternal-to-fetal, and blood transfusion routes [Citation1–5]. ZIKV was first identified in 1947 from a sentinel rhesus monkey in Uganda [Citation6]. Since 2007, it emerged in the Pacific region, South America, Central America, and the Caribbean, posing a global public health threat [Citation7–11]. ZIKV infection causes mild illnesses, including fever, pruritis, arthritis, conjunctivitis, and asthenia [Citation12]. Importantly, it can cause devastating congenital Zika syndromes (CZS; including microcephaly, congenital malformation, and fetal demise) in pregnant women and Guillain-Barre syndrome (GBS) in adults [Citation13–16]. The explosive epidemics and its association with CZS had led the World Health Organization (WTO) to declare ZIKV as a Public Health Emergency of International Concern from February to November in 2016 [Citation17]. Since then, a number of vaccine candidates have been developed, some of which have already advanced to clinical trials [Citation18]. In addition, small molecule inhibitors and therapeutic antibodies have also been identified for antiviral development [Citation19–22].
ZIKV belongs to the Flavivirus genus within the Flaviviridae family. Besides ZIKV, many other flaviviruses are significant human pathogens, including dengue (DENV), yellow fever (YFV), West Nile (WNV), Japanese encephalitis (JEV), Saint Louis encephalitis (SLEV), and tick-borne encephalitis virus (TBEV) [Citation23]. Flavivirus genome is a positive-sense, single-stranded RNA of ∼11,000 nucleotides. It contains a 5’ untranslated region (UTR), a long open-reading-frame (ORF), and a 3’ UTR. The single ORF encodes a polyprotein that is processed into three structure proteins (capsid [C], pre-membrane [prM] and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The structural proteins, together with genomic RNA, are components of virions. The nonstructural proteins participate in viral RNA replication, virion assembly, and evasion of host immune response [Citation24–26].
Flavivirus NS2A is an ER-resident, transmembrane protein with a calculated molecular weight of ∼22 kDa. The N-terminus and C-terminus of NS2A are formed by cleavages with an unknown host protease and viral protease NS3 (in complex with NS2B), respectively [Citation27,Citation28]. Flavivirus NS2A has multiple functions: (i) Viral replication. Kunjin virus (KUNV) NS2A co-localizes with viral double-stranded RNA and interacts with the 3’UTR of genomic RNA, NS3, and NS5 protein [Citation29]. Mutagenesis studies confirmed the role of NS2A in viral RNA synthesis [Citation30,Citation31]. (ii) Virion assembly/secretion. Mutagenesis analyses of YFV, KUNV, and DENV have identified distinct NS2A amino acids that are essential for virion assembly/secretion [Citation30–34]. The virion assembly defects can be restored by second site mutations in NS2B or NS3 in YFV and DENV [Citation32,Citation35,Citation36]. (iii) Evasion of innate immunity. ZIKV NS2A inhibits type-I IFN induction at the step of TBK1 [Citation26]. DENV NS2A antagonizes type-I IFN signalling [Citation25]. JEV NS2A blocks dsRNA-activated protein kinase PKR [Citation37]. In WNV and KUNV, the NS2A-mediated inhibition of type-I IFN induction can be abrogated by specific NS2A mutations [Citation38,Citation39]. (iv) Disease pathogenesis. WNV NS2A modulates virus-induced cytopathic effect (CPE) and pathogenesis in mice [Citation40,Citation41]. ZIKV NS2A impairs mammalian cortical neurogenesis through depleting adherens junction proteins [Citation42].
The structure and function of ZIKV NS2A have not been characterized. Although NS2A proteins from different flaviviruses perform similar functions (described above), each flavivirus may exert its NS2A functions in a virus-specific manner [Citation35]. The goals of this study are (i) to define the membrane topology of ZIKV NS2A and (ii) to characterize its roles in viral RNA synthesis and virion assembly.
Materials and methods
Cell lines
Vero cells, baby hamster kidney (BHK-21) cells and human embryo kidney 293T cells were cultured at 37°C with 5% CO2 in high-glucose Dulbecco’s modified Eagle medium (DMEM; Life Technologies) supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 g/ml streptomycin and 10% fetal bovine serum (FBS; HyClone Laboratories).
Bioinformatics
NS2A protein sequences from ZIKV, DENV1, DENV2, DENV3, DENV4, WNV, JEV, YFV, SLEV were aligned using CLC Main Workbench software (CLC Bio). Prediction of ZIKV NS2A was carried out using various web servers, which including TOPCONS, Philius, Polyphobius, SCAMPI, and TMHMM2. The amphipathic helices of ZIKV NS2A were predicted by HeliQuest.
Antibody
The following antibodies were used in this study: a mouse monoclonal antibody 4G2 cross-reactive with flavivirus E protein (American Type Culture Collection); Rabbit anti-HA monoclonal antibody (Cell Signaling Technology); Alexa Fluor®488 conjugated mouse anti-HA monoclonal antibody (Cell Signaling Technology); Rabbit anti-Calnexin polyclonal antibody(Abcam); Mouse anti-GFP monoclonal antibody (Sigma-Aldrich); Horseradish peroxidase(HRP) conjugated goat anti-mouse and anti-rabbit IgG polyclonal antibodies (Sigma-Aldrich); Alexa Fluor®488 conjugated goat anti-mouse IgG polyclonal antibody (Thermo Fisher Scientific); Alexa Fluor®568 conjugated goat anti-rabbit IgG polyclonal antibody (Thermo Fisher Scientific).
Plasmid construction
The infectious cDNA clone (pFLZIKV) bearing the full-length genome of ZIKV (strain FSS13025) or Renilla luciferase (Rluc) reporter ZIKV replicon (pZIKV Rep WT) were used as the backbone for introducing NS2A mutations [Citation43,Citation44]. XhoI and KpnI restriction sites were initially generated into pFLZIKV and pZIKV Rep WT replicon at positions 3018 and 5010 in ZIKV genome without changing amino acid sequences. The NS2A alanine-scanning mutagenesis was performed by using a QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) as described previously [Citation30].
A mammalian expression vector pXJ with a cytomegalovirus (CMV) promoter was used for constructing plasmids expressing NS2A and its derivatives. To construct the pXJ-E24-NS1-NS2A-HA plasmid, a gene cassette encoding the last 24 amino acids of E protein (E24), NS1 and NS2A with a C-terminal hemagglutinin (HA) tag was firstly amplified from the pFLZIKV and cloned into the pXJ vector. Next, NS2A mutations were individually introduced into the pXJ-E24-NS1-NS2A-HA by overlap PCR, resulting in plasmids expressing NS2A mutants.
To construct the pXJ-SPG-C16-NS2A-HA that was used for selecting cell line constitutively expressing NS2A protein, a DNA fragment containing the Gaussia luciferase signal peptide (SPG), the last 16 amino acids of NS1 protein (C16) and NS2A tagged with a C-terminal HA tag sequence were constructed by overlap PCR and cloned into the pXJ vector via NotI/XhoI restriction sites.
Plasmid pXJ-eGFP was constructed by inserting the enhanced green fluorescence protein (eGFP) sequence into the pXJ vector via BamHI/XhoI restriction sites. DNA fragments that encode different NS2A segments were individually inserted into above pXJ-eGFP vector, resulting various pXJ-smNS2A-eGFP plasmids (smNS2A represents NS2A segment). A DNA fragment that express NS2A or truncated NS2A and an eGFP with a C-terminal N-glycosylation acceptor site (Glyc) were amplified and cloned into the pXJ-SPG-C16-NS2A-HA vector, resulting in the plasmid pXJ-SPG-C16-NS2A-eGFP-Glyc and various pXJ-SPG-C16-ctNS2A-eGFP-Glyc plasmids (ctNS2A represents C-terminal truncated NS2A).
RNA in vitro transcription and electroporation
All full-length cDNA clone plasmids and subgenomic replicons of ZIKV were linearized with restriction enzyme ClaI, followed by phenol-chloroform extraction and ethanol precipitation. RNA transcripts were prepared by using the T7 mMessage mMachine kit (Ambion). For RNA transfection, 10 μg in vitro transcribed RNA was electroporated into 8 × 106 cells (BHK-21 or Vero) by using Gene Pulser Xcell™ Electroporation Systems (Bio-rad) as described previously [Citation30,Citation43].
Immunofluorescence assay
For indirect immunofluorescence assay (IFA), cells were seeded in an 8-well Nunc Lab-Tek II Chamber Slide (Thermo Fisher Scientific). At various time points, cells were washed three times with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS at room temperature for 30 min and permeabilized with 0.1% Triton X-100 in PBS at room temperature for 10 min. The permeabilized cells were then washed three times with PBS and incubated in a blocking buffer containing 1% FBS in PBS at room temperature for 1 h. E protein was stained with a 1:1000 dilution of mouse monoclonal antibody 4G2 in blocking buffer. After incubation for 1 h, the cells were washed three times with a washing buffer (1% FBS and 0.5% Tween 20 in PBS). After 1 h of incubation with a secondary antibody (Alexa Fluor®488 conjugated goat anti-mouse IgG), the cells were washed three times with washing buffer and counterstained with DAPI (4’,6-diamidino-2-phenylindole). Images were acquired using an Olympus fluorescence microscope. Images were processed in ImageJ software (National Institutes of Health, MD).
Plaque assay
2 × 105 Vero cells per well were grown in 24-well plates one day prior to viral infection. 10-fold serial dilutions of virus samples were prepared in DMEM containing 2% FBS and 1% penicillin/streptomycin and inoculated (100 µl) into each well of 24-well plates. After 1 h of incubation at 37°C with 5% CO2, the inoculum was replaced with 500 µl overlay medium (DMEM medium containing 0.8% methyl-cellulose, 2% FBS and 1% penicillin/streptomycin). Plaques formed after incubation at 37°C with 5% CO2 for 4–5 days. Cells were fixed with 4% paraformaldehyde for 30 min, followed by staining with 1% crystal violet. The numbers of plaque per millilitre (PFU/ml) of ZIKV were calculated.
Replicon transient-transfection assay
Briefly, 8 × 106 Vero cells were electroporated with 10 μg wild type or mutant Rluc-ZIKV replicon RNAs. After electroporation, 3 × 105 cells per well were seeded in 12-well plates. At given time points, the cells were washed twice with PBS and lysed in 200 μl of luciferase lysis buffer (Promega). The luciferase activity assay was performed according to the manufacturer’s protocol.
RNA extraction and cDNA sequencing
The culture fluids from infected cells were centrifuged at 1000 g for 10 min and then filtered through a 0.22 μm pore size polyethersulfone membrane (Millipore) to remove cellular debris. Viral RNAs in the supernatants were extracted using a QIAamp viral-RNA Minikit (Qiagen) by following the manufacturer’s protocol. Reverse transcription PCR (RT-PCR) was performed by using the SuperScript® III One-Step RT-PCR System (Thermo Fisher Scientific). The amplified cDNA fragments were purified and sent for Sanger sequencing at the GENEWIZ facilities.
Selection of NS2A-HA BHK-21 cell line
4 × 105 BHK-21 cells per well were seeded into a 6-well plate. The plasmid pXJ-SPG-C16-NS2A-HA that contains a neomycin resistance gene was transfected into BHK-21 cells (4 µg per well) using X-tremeGENE 9 DNA transfection reagent (Roche). At 24 h post-transfection, geneticin (G418) was added to the culture medium with a concentration of 1 mg/ml. The medium was changed every three days. After two weeks, several neomycin-resistant individual colonies were selected for propagation. The expression of NS2A-HA protein in stable cell lines was analyzed by immunofluorescence assay and Western blot.
In vitro proteinase K protection assay
Proteinase K protection assay was performed as previously described with minor modifications [Citation45,Citation46]. BHK-21 cells were transfected in 6-well plates with various pXJ-SPG-C16-ctNS2A-eGFP-Glyc plasmids. After incubated at 30°C for 24 h, cells were trypsinized, washed twice with PBS and centrifuged at 450 × g for 5 min. The cell pellets were resuspended in a hypotonic lysis buffer (10 mM Tris-HCl, pH 7.5, and 2 mM MgCl2) and kept on ice for 10 min. Subsequently, the samples were homogenized with 40 strokes and centrifuged at 1000 × g for 5 min. The postnuclear supernatants (PNS) were divided into three aliquots and centrifuged at 15,000 × rpm for 40 min. The pellets from three aliquots were then resuspended in PBS, 60 μg/ml proteinase K in PBS, or 60 μg/ml proteinase K in PBS in the presence of 0.5% Triton X-100, respectively. The samples were incubated on ice for 90 min. 20 mM phenylmethylsulfonyl fluoride (PMSF) was finally added to terminate the reaction. After further incubation on ice for 15 min, the samples were mixed with 4× Laemmli sample buffer, heated at 95°C for 15 min and analyzed by Western blotting.
In vitro deglycosylation assay
The samples were prepared by following a previously described protocol [Citation46,Citation47]. Briefly, the transfected BHK-21 cells in 6-well plates were incubated at 30°C for 24 h. Cells were trypsinized and suspended in DMEM medium containing 10%FBS. The cells were pelleted by centrifuging at 450 × g for 5 min. After two PBS washes, the cell pellets were resuspended in a solubilization buffer (20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM Mg(CH3COO)2, 1% Triton X-100, and EDTA-free protease inhibitors [Roche]) and incubated at 4°C for 2 h with gentle agitation. After centrifugation at 15,000 rpm 4°C for 20 min to remove cell debris, the supernatants were split into two aliquots. 50,000 units/ml N-glycosidase F (PNGase F) or PBS (no-treatment control) were added to those two aliquots. After incubation at 37°C for 2 h, the samples were mixed with 4× Laemmli sample buffer, heated at 70°C for 15 min and analyzed by Western blot.
SDS-PAGE and Western blot
Protein samples were separated by 12% SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane using Trans-Blot Turbo transfer system (Bio-Rad), after which the membrane was blocked in TBST (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 0.1% Tween 20) supplemented with 5% nonfat milk for 1 h at room temperature. The blots were probed with primary antibodies (1:1,000 dilution) overnight at 4°C, washed three times with TBST and incubated with horseradish peroxidase (HRP) conjugated goat anti-mouse and anti-rabbit IgG polyclonal antibodies (1:10,000 dilution) for 1 h. After three thorough TBST buffer washes, the blots were incubated with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Chemiluminescence signals were detected in ChemiDoc System (Bio-Rad).
Quantification and statistical analysis
IFA images processing and cell counting were performed in software ImageJ (NIH). Densitometry analysis was performed using Image lab version 6.0 (Bio-Rad Laboratories, Hercules, CA). All numerical data are presented as the mean ± SEM (standard error of mean). The size of each study or number of replicates, along with the statistical tests performed can be found in corresponding Figure Legends.
Results
The rationale for ZIKV NS2A mutagenesis
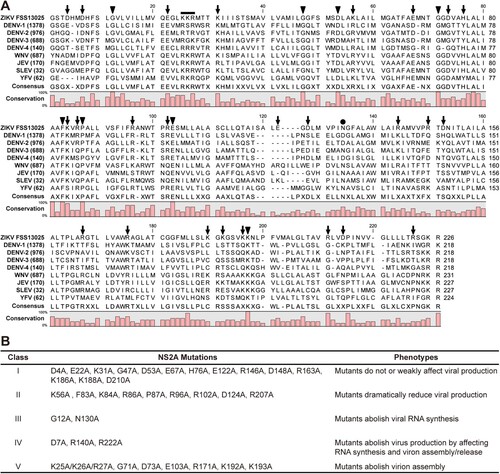
To identify critical residues in ZIKV NS2A, we performed a systematic alanine-scanning mutagenesis using an infectious cDNA clone of ZIKV strain FSS13025 [Citation43]. Sequence alignment of NS2A proteins from ZIKV, DENV, WNV, JEV, SLEV, and YFV revealed a high diversity: only 31 out of 218–227 amino acids with a consensus of >80% among different flaviviruses (A). A total of 35 residues were selected for mutagenesis: (i) All 8 identical non-alanine residues, including G12, G47, D53, G71, F83, P87, E103, and K193. The amino acid numbering is based on the ZIKV sequence. (ii) All charged residues, including D4, D7, E22, K31, K56, E67, D73, H76, K84, R86, R96, R102, E122, D124, R140, R146, D148, R163, R171, K186, K188, K192, R207, D210, and R222. Charged residues were selected because they likely participate in inter- or intra-molecular interactions; alternatively, they may alter the locations where NS2A would traverse the ER membrane since charged residues are hydrophilic. In addition, NS2A D73H mutation was previously shown to attenuate WNV replication [Citation48]. (iii) A K25/K26/R27 motif that was previously reported to be critical for YFV and DENV virion assembly [Citation35,Citation40]. (iv) Residue N130 that was previously shown to be required for NS1-NS2A cleavage and viral replication in DENV [Citation27,Citation30]. The selected residues were mutated to alanine in the context of ZIKV genomic RNA. Equal amounts of wild-type (WT) and mutant RNAs were electroporated into Vero cells. After electroporation, RNA replication and virus spread were monitored by immunofluorescence assay (IFA); the production of infectious viruses was quantified by plaque assay and RT-PCR. The 35 mutants exhibited distinct phenotypes that could be categorized into five groups (B), as detailed in the following sections.
Figure 1. ZIKV NS2A mutagenesis. (A) Sequence alignment of NS2A proteins among ZIKV (strain FSS13025), DENV-1-4 serotypes, WNV, JEV, SLEV, and YFV using CLC Main Workbench software (CLC Bio). Thirty-five amino acids were selected for mutagenesis: (i) All 8 identical non-alanine residues (indicated by triangles), (ii) 25 charged residues (arrows), (iii) a K25/K26/R27 motif (line), (iv) residue N130 (circle). The consensus and conservation percentage are indicated below each amino acids. (B) A summary of ZIKV NS2A mutagenesis results. An infectious cDNA clone and a luciferase replicon of ZIKV strain FSS13025 were used to analyze the function of each amino acid. Based on the replication phenotypes, the 35 mutants could be categorized into five classes as indicated.
Class I mutations have no or weak effects on viral replication
Fourteen of the 35 NS2A mutant RNAs, including D4A, E22A, K31A, G47A, D53A, E67A, H76A, E122A, R146A, D148A, R163A, K186A, K188A, and D210A, replicated at levels comparable to the WT RNA. These mutants were categorized as class I (B). The IFA images and plaque morphologies of three representatives (D53A, D148A, and R163A), as well as the WT, are shown in (A and B), respectively. Most class I mutants (e.g. D148A and R163A) yielded comparable E-expressing cells as the WT from 24 to 48 h post-transfection (p.t.). At 72 h p.t., severe CPE occurred among D148A, R163A, and WT RNA-transfected cells. Some class I mutants (e.g. D53A) produced fewer E-positive cells than the WT at 48 h p.t., and spread to infect more cells at 72 h p.t (A). All class I mutants produced viruses with plaques similar to the WT virus (B). At 72 h p.t., the class I mutants produced infectious titers <4-fold lower than the WT did (C). Sequencing of the mutant viruses (collected at 72 h p.t.) confirmed the retaining of the engineered mutations without other changes (data not shown).
Figure 2. Characterization of class I, II, and III NS2A mutants. The results of representative mutants from class I (A–C), II (D–F), and III (G–K) are shown. (A) Vero cells were electroporated with equal amounts of ZIKV wild-type (WT) or class I mutant genomic RNAs. At indicated time points, the cells were fixed and subjected to IFA. Viral E protein and nuclei were stained with antibody 4G2 (green) and DAPI (blue), respectively. Cytopathic effect (CPE) is indicated at 72 h post-transfection. (B) Plaque morphologies. Recombinant viruses were subjected to plaque assay. Plaques were developed at day 4.5 post-infection. (C) Viral titers of class I mutants at 72 h post-transfection. The mean values from two independent experiments are shown. Error bars indicate the standard deviations. The limit of detection (LOD) of plaque assay is 10 PFU/ml. (D) IFA of class II mutant RNA-transfected Vero cells. (E) Plaque morphology of class II mutant viruses. (F) Viral titers of class II mutant viruses at 72 h post-transfection. (G) IFA of class III mutant RNA-transfected Vero cells. (H) Plaque morphologies of class III mutant viruses. Plaque assay was performed using culture supernatants collected at day 3 (for WT) and day 5 (for G12A and N130A) post-transfection. (I) RT-PCR analysis. At 5 day p.t., the mutant RNA-transfected cells were washed twice with PBS to remove residual input RNAs. At day 8 p.t., the supernatants of G12A and N130A RNA-transfected cells were subjected to RT-PCR. The WT virus (collected at 72 h p.t.) was used as a positive control. (J) A ZIKV luciferase replicon. C38 and E30 represent the first 38 amino acids of C protein and the last 30 amino acids of E protein, respectively. Renilla luciferase: Rluc. Foot-and-mouse-disease virus 2A sequence: 2A. (K) Transient replicon assay. WT, G12A, N130A, and NS5ΔGDD replicon RNAs were electroporated into Vero cells. Luciferase activities were measured at indicated time points. An average of 3 experiments are presented. Bars represent standard deviations.
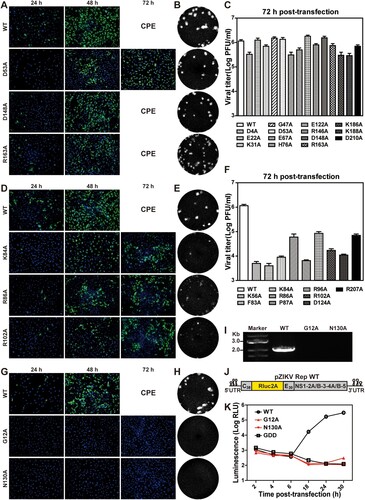
Class II mutations significantly attenuate viral replication
Nine NS2A mutants, including K56A, F83A, K84A, R86A, P87A, R96A, R102A, D124A, and R207A, significantly attenuated viral replication. These mutants were categorized into class II (B). Class II RNA-transfected cells produced fewer E-positive cells than the WT did (D). At 72 h p.t., only 30–50% of E-positive cells were detected in the class II RNA-transfected cells, whereas the WT RNA-transfected cells exhibited CPE. Class II mutant viruses formed smaller, opaque plaques (E), and the transfected cells produced >10-fold lower infectious titers than the WT RNA-transfected cells at 72 h p.t. (F). Complete genome sequencing of the class II mutant viruses (collected at 96 h p.t.) did not reveal reversions or any other mutations (data not shown). Because of the weak phenotypes of class I and II mutants, we did not further characterize them.
Class III mutations are lethal for viral RNA synthesis
Class III includes two mutants, G12A and N130A, which were completely defective in viral RNA synthesis (B). The G12A and N130A genomic RNA-transfected cells did not produce any E-positive cells at 24 and 48 h p.t. (G). On day 5–8 p.t., no infectious viruses were detected from culture medium by plaque assay (H) and RT-PCR (I). To confirm the above results, we introduced G12A or N130A mutation into a ZIKV luciferase replicon (J). A replication-defective replicon, containing an NS5 polymerase active site ΔGDD mutation, was included as a negative control [Citation44]. Vero cells were electroporated with equal amounts of WT, G12A, N130A, or NS5 ΔGDD replicon RNAs. At 2–6 h p.t., all replicons generated similar levels of luciferase signals, suggesting comparable levels of viral RNA translation and transfection efficiencies (K). However, at 18–30 h p.t., G12A and N130A replicons generated low luciferase signals as the non-replicative NS5 ΔGDD replicon did, whereas the WT replicon produced increasing luciferase signals (K). These results demonstrate that class III mutations completely abolish viral RNA synthesis.
Class IV mutations impair both viral RNA synthesis and virion production
Class IV includes three mutants, including D7A, R140A, and R222A (B). These mutant genomic RNAs produced fewer E-positive cells in the transfected cells than the WT RNA (A). For D7A or R140A mutants, the RNA-transfected cells did not produce infectious viruses, as suggested by plaque assay (B) and RT-PCR (C). Incubation of naïve Vero cells with culture supernatants from the D7A or R140A transfected cells did not generate any viral E-positive cells (D). Replicon analysis showed that D7A or R140A significantly compromised viral RNA synthesis (E). Since neither mutants could produce extracellular viruses (B and C), and could replicate at attenuated levels inside cells (E), D7A and R140A mutations may impair both viral RNA synthesis and virion production.
Figure 3. Characterization of class IV NS2A mutants. (A) IFA of transfected Vero cells. (B) Plaque morphologies of WT, D7A, R140A, and R222A viruses. (C) Extracellular viral titers were measured at 24, 48, and 72 h p.t. using plaque assay. At 72 h p.t., extracellular viral RNAs were subjected RT-PCR analysis. The RT-PCR products were sequenced to confirm the engineered mutations and to detect potential second site mutation(s) in the NS2A gene. N.A., not applicable. (D) The supernatants of transfected cells were collected on day 8 p.t. to infect naïve Vero cells. At 24 h p.t., the cells were analyzed for viral E expression by IFA. (E) Transient luciferase replicon analysis. Vero cells were electroporated with ZIKV replicon RNAs and measured for luciferase activities at indicated time points. An average of 3 experiments are presented. Bars represent standard deviations. (F) Analysis of NS2A proteolytic processes. NS2A D7A, G12A, N130A, R140A, and R222A mutations were individually engineered into expression plasmid pXJ-E24-NS1-NS2A-HA. After transfecting the plasmids into 293 T cells, total cellular protein samples were harvested at 24 h p.t. and analyzed for NS2A process using Western blot. An HA-tag antibody was used to detect both unprocessed NS1-NS2A-HA and processed NS2A-HA proteins.
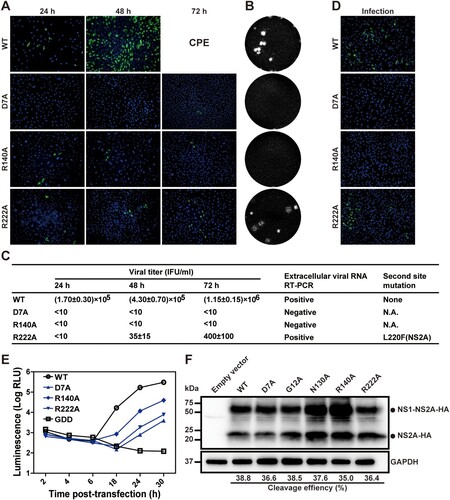
For R222A, the number of E-positive cells slightly increased in the genomic RNA-transfected cells from 24 to 72 h p.t. (A). A low level of infectious virus was detected from the culture medium by plaque assay (B) and RT-PCR (C). These data were supported by the positive-IFA results after incubating naive Vero cells with the supernatant from the R222A RNA-transfected cells (D). Sequencing the virus (collected at 72 h p.t.) revealed that, besides the engineered R222A mutation, the virus had acquired a second site mutation of L220F in NS2A (C). Replicon analysis showed that R222A significantly lowered viral RNA synthesis (E); no secondary mutation (i.e. L220F) was detected in the replicon RNA at 30 h p.t. (data not shown). These results suggest that (i) R222A mutant is defective in viral RNA synthesis and/or virion assembly, and (ii) a second site mutation L220F in NS2A could rescue the defects.
Class III and IV mutations do not affect NS2A maturation
Since cleavage between NS1 and NS2A is essential for flavivirus RNA synthesis [Citation30], we examined whether the replication defects of class III and IV mutants were caused by defective maturation of NS2A. To test this hypothesis, we introduced individual mutations into an expression plasmid pXJ-E24-NS1-NS2A-HA that encodes a polyprotein containing the C-terminal 24 amino acids of the E protein (serving as a signal peptide to translocate NS1 to ER lumen), NS1, and NS2A with a C-terminal HA tag. After transfecting the plasmids into 293 T cells, the expression of un-cleaved NS1-NS2A-HA intermediate and cleaved NS2A-HA product were analyzed using Western blot (F). None of the mutations significantly affected the cleavage efficiencies of NS1-NS2A-HA to NS2A-HA, indicating that the replication defects of class III and IV mutants are not caused by impaired NS2A cleavage.
Class V mutations block virion assembly
Class V includes G71A, D73A, E103A, R171A, K192A, K193A, and a triple K25A/K26A/R27A. Cells transfected with class V mutant genomic RNAs yielded E-positive cells at 24 h p.t., but the number of E-positive cells did not increase afterward (A), suggesting that the mutants are defective in virus spread. No infectious mutant viruses, except E103A, were detected in the supernatants of transfected cells by plaque assay (B) or RT-PCR (C). No IFA-positive cells were detected after naïve cells were incubated with the supernatants from transfected cells (D). Sequencing of the recovered E103A mutant virus revealed an A103D change in the NS2A gene (C), suggesting that an E103A-to-A103D substitution is responsible for the rescue of viral replication.
Figure 4. Characterization of class V NS2A mutants. (A) IFA of Vero cells that were transfected with WT or class V genomic RNAs. Viral E-positive cells are indicated in green. (B) Plaque morphologies of class V mutant viruses. Only the WT and E103A mutant generated plaques. (C) Supernatants from the RNA-transfected cells were quantified for viral titers by plaque assay. RT-PCR was also performed on the supernatants at day 3 p.t. for the WT virus and at day 8 p.t. for the mutants. Positive RT-PCR products were sequenced for potential second site mutation(s). N.A., not applicable. (D) Naïve Vero cells were infected with supernatants from the RNA-transfected cells and subjected to IFA for viral E protein expression. (E) Luciferase replicon analysis. Equal amounts of WT and mutant replicon RNAs were electroporated into Vero cells. Luciferase activities were measured at the indicated time points. (F) Analysis of intracellular infectious viruses. Intracellular viruses from ZIKV RNA-transfected cells were harvested at 24 h p.t. as previously described [Citation46]. The harvested samples were used to infect naïve Vero cells. IFA was performed to examine viral E protein expression at 24 h post-infection.
![Figure 4. Characterization of class V NS2A mutants. (A) IFA of Vero cells that were transfected with WT or class V genomic RNAs. Viral E-positive cells are indicated in green. (B) Plaque morphologies of class V mutant viruses. Only the WT and E103A mutant generated plaques. (C) Supernatants from the RNA-transfected cells were quantified for viral titers by plaque assay. RT-PCR was also performed on the supernatants at day 3 p.t. for the WT virus and at day 8 p.t. for the mutants. Positive RT-PCR products were sequenced for potential second site mutation(s). N.A., not applicable. (D) Naïve Vero cells were infected with supernatants from the RNA-transfected cells and subjected to IFA for viral E protein expression. (E) Luciferase replicon analysis. Equal amounts of WT and mutant replicon RNAs were electroporated into Vero cells. Luciferase activities were measured at the indicated time points. (F) Analysis of intracellular infectious viruses. Intracellular viruses from ZIKV RNA-transfected cells were harvested at 24 h p.t. as previously described [Citation46]. The harvested samples were used to infect naïve Vero cells. IFA was performed to examine viral E protein expression at 24 h post-infection.](/cms/asset/150d1890-7fff-41c9-9cb1-62d85a2851a4/temi_a_1598291_f0004_oc.jpg)
To determine the stage of viral replication that was disrupted by the class V mutations, we analyzed the mutational effect on viral replication in ZIKV replicon (E). Compared with the WT replicon, all class V mutants exhibited equivalent levels of viral translation and RNA synthesis (<4-fold difference), suggesting that the mutations only marginally affect viral RNA synthesis. Next, we tested if the class V genomic RNAs could generate intracellular infectious viruses. Incubating naïve Vero cells with WT RNA-transfected cell extracts yielded IFA-positive cells, whereas no IFA-positive cells were detected after incubating naive cells with the mutant RNA-transfected cell extracts (F). Altogether, the results suggest that class V mutants are competent in viral RNA replication, but defective in assembly of infectious viruses.
WT NS2A rescues virion assembly for class V mutants
To test trans complementation of NS2A mutants, we constructed a BHK-21 cell line that constitutively expressed WT ZIKV NS2A protein with a C-terminal HA-tagged (NS2A-HA). The cell line was constructed by transfecting plasmid pXJ-SPG-C16-NS2A-HA encoding a Gaussia luciferase signal peptide (SPG), the C-terminal 16 amino acids of NS1 protein (C16), and NS2A-HA, as well as a neomycin resistance gene (Neo). The SPG and C16 sequences were engineered to enable the correct formation of NS2A membrane topology when expressed in cells. Individual BHK-NS2A-HA cell lines were generated through colony purification under G418 (1 mg/ml) selection. For each BHK-NS2A-HA cell line, all cells were IFA-positive for HA-NS2A expression (A). Western blot revealed an expected band of ∼22-kDa band (B). The expression level of NS2A-HA did not decrease in the BHK-NS2A-HA cells after 20 passages in culture (4 days per passage; B), indicating the stability of the cell line.
Figure 5. Trans complementation of class III and V mutants. (A) IFA of BHK-NS2A-HA cells. The BHK-NS2A-HA cells constitutively express WT ZIKV NS2A fused with a C-terminal HA-tag. The cells were fixed with paraformaldehyde and stained with an HA-tag antibody. (B) Western blot of BHK-NS2A-HA cells. The expression of WT NS2A-HA from BHK-NS2A-HA cells was detected using an HA-tag antibody. Both passage one (P1) and 20 (P20) BHK-NS2A-HA cells were analyzed. Naïve BHK-21 cells were included as a negative control. (C) WT and class III and V genomic RNAs were electroporated into BHK-NS2A-HA cells. IFA was performed on the transfected cells to detect viral E protein expression. (D) Viruses rescued from the BHK-NS2A-HA cells (harvested at 96 h p.t.) were used to infect naïve Vero cells. The infected Vero cells were subjected to IFA for E protein expression. (E) Extracellular viruses were harvested from the supernatants of RNA-transfected BHK-NS2A-HA cells. The harvested viruses were serially diluted to infect naïve Vero cells in 96-well plates. At 24 h p.i., IFA was performed to quantify the viral titers by counting the number of E-positive cells. The results show averages and standard deviations from triplicate samples. Viral RNAs in the supernatants of transfected BHK-NS2A-HA cells were isolated at 96 h p.t. and subjected to RT-PCR. Positive RT-PCR products were sequenced to confirm the engineered mutations and to identify potential second site change(s). (F) The supernatants of infected Vero cells from (D) were collected at 72 h p.i. to infect naïve Vero cells for a second round. IFA was performed on these cells for viral E-positive cells.
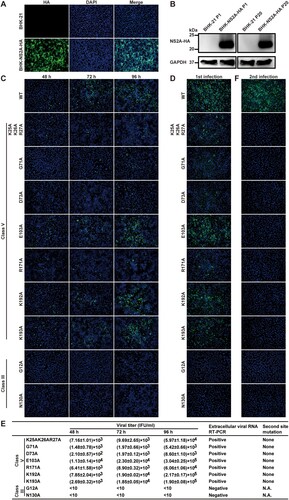
We tested trans complementation by transfecting class V genomic RNAs to BHK-NS2A-HA cells. The transfected cells developed an increasing number of E-positive cells from 48 to 96 h p.t. (C). Culture supernatants were collected at 96 h p.t. to infect naïve Vero cells. The infected cells developed E-positive cells for all class V mutants (D), demonstrating the rescue of infectious viruses through trans complementation. Quantification of the E-positive cells allowed us to determine the titers of rescued infectious viruses (E). Among the class V mutants, E103A, K192A, and K193A exhibited the highest trans complementation efficiencies, producing 1.9 × 105–3.04 × 105 IFU/ml viruses at 96 h p.i. (E). Sequencing of the rescued virion genomes confirmed the retention of the engineered class V mutations without any other changes in NS2A (E).
Next, supernatants from the first round of infection collected at 72 h p.i. (from D) were used to infect naïve Vero cells for a second round. In contrast to the first round of infection (D), no E-positive cells were detected in the second round of infection for any of the class V mutants (F). As a positive control, WT RNA produced high numbers of E-positive cells in both the first and second rounds of infections (D and F). Collectively, the results indicate that (i) the defect in virion assembly of class V mutants could be rescued through trans supply of WT NS2A protein and (ii) the rescued virions could only infect cells for a single round.
DENV-2 NS2A could not trans complement ZIKV class V mutants
We tested whether DENV NS2A could rescue class V mutants of ZIKV. Each class V mutant of ZIKV was transfected into a previously established BHK-21 cell line that constitutively expresses WT DENV-2 NS2A protein [Citation30]. For all class V mutants, no increase in E-positive cells or infections viruses was detected from the transfected cells from 24 to 96 h p.t. (data not shown). The results suggest that flavivirus NS2A modulates virion assembly in a virus-type specific manner.
WT NS2A could not rescue viral RNA synthesis of class III mutants
We tested whether the defect in viral RNA synthesis of class III mutants could be trans complemented. To test this possibility, we transfected class III genomic RNAs (G12A and N130A) into ZIKV BHK-NS2A-HA cells. Neither mutants generated any E-positive cells in the transfected BHK-NS2A-HA cells (C). Infection of naïve Vero cells with supernatants of the transfected cells did not produce any IFA-positive cells (D–F). The results suggest that the RNA synthesis defect of class III mutants could not be rescued through trans complementation. Due to the dual defects of class IV mutants in both viral RNA synthesis and virion assembly, we did not pursue trans complementation experiments for this class of mutants; such complementation experiments may not reveal clear mechanistic insights as compared with the class III and V mutants.
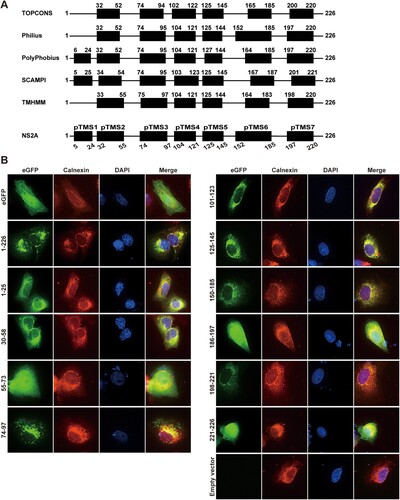
Probing the topology of ZIKV NS2A on the ER membrane
The distinct functions of amino acids in viral RNA synthesis and virion assembly prompted us to probe the topology of ZIKV NS2A on ER membrane. Bioinformatics analysis suggests that ZIKV NS2A may form seven transmembrane segments (pTMS1–7; A). To examine whether each pTMS associates with the ER membrane, we constructed a panel of plasmids encoding full-length or various NS2A fragments with a C-terminally fused eGFP reporter (B). The length of each segment was slightly modified from the bioinformatic prediction to allow some flexibility between pTMS and eGFP. Upon transfection into Vero cells, eGFP alone (without any NS2A fragment) exhibited an even distribution of fluorescence throughout the cell, whereas the full-length NS2A-eGFP (construct 1–226) displayed fluorescence co-localized with calnexin (an ER membrane protein) in the perinuclear region (B), indicating that ZIKV NS2A is an integral membrane protein. Expression of seven fragments spanning each pTMS, represented by constructs 1–25 (pTMS1), 30–58 (pTMS2), 74–97 (pTMS3), 101–123 (pTMS4), 125–145 (pTMS5), 150–185 (pTMS6), and 198–221 (pTMS7), exhibited fluorescence patterns that co-localized with calnexin in the perinuclear region (B). The results indicate that individual of the seven pTMSes is associated with the ER membrane. In contrast, expression of fragments between different pTMS, represented by constructs 55–73, 186–197, and 221–226, showed fluorescence throughout the cells (B), indicating that these fragments are not membrane-associated.
Figure 6. Membrane association of ZIKV NS2A transmembrane segments. (A) Bioinformatics analysis of ZIKV NS2A for transmembrane segments. ZIKV NS2A sequence was analyzed by bioinformatics programme TOPCONS, Philius, PolyPhobius, SCAMPI, and TMHMM. The compiled transmembrane segments are shown at the bottom. Seven predicted transmembrane segments (pTMS) are presented as thick bars. The amino acid positions of ZIKV NS2A are indicated. (B) Membrane association of individual pTMS. Expression plasmid pXJ-eGFP (without NS2A sequence) and pXJ-smNS2A-eGFP (smNS2A represents different NS2A segments) were transfected into Vero cell by using X-tremeGENE 9 DNA transfection reagent (Roche). For each plasmid construct, the NS2A segment is indicated by amino acid positions. At 24 h p.t., IFA was performed to monitor eGFP expression. ER and nuclei were stained with anti-Calnexin antibody and DAPI, respectively. The merged images are shown on the right panels.
An in vitro deglycosylation analysis was performed to determine the orientation of each NS2A pTMS on the ER membrane. A panel of plasmids were prepared to express truncated NS2A proteins that were C-terminally fused with an eGFP followed by an N-glycosylation acceptor peptide (Glyc; A). An SPG-C16 peptide (Gaussia luciferase signal peptide SPG and the C-terminal 16 amino acids of NS1) was fused to the N terminus of each NS2A-eGFP-Glyc construct to ensure a proper translocation of the expressed protein on the ER membrane. Western blot showed that, upon transfection into BHK-21 cells, all constructs expressed fusion proteins with expected molecular masses (B). Because N-glycosylation enzymes reside in the ER lumen, the glycosylation status could be used to indicate the location of the C-terminus of each NS2A-eGFP-Glyc protein (C). As a negative control, treatment of EGFP-Glyc (without any viral sequence) with PNGase F (an enzyme that removes high mannose and complex carbohydrates) did not change the mobility of the protein (D), indicating a cytosol localization of EGFP-Glyc. As a positive control, PNGaseF treatment increased the mobility of SPG-C16-EGFP-Glyc (construct 0), confirming that the C-terminus of this protein resides in the ER lumen. Among the NS2A constructs, PNGase F treatment increased the mobility of fusion proteins 1–26 and 1–56, but not 1–103, 1–123, 1–147, 1–192, and 1–226 (D). The results suggest that the N-terminal 56 amino acids of NS2A reside in the ER lumen, and the C-terminal region from residues 103–226 is located in the cytosol.
Figure 7. Probing the membrane topology of ZIKV NS2A by in vitro deglycosylation assay and in vitro proteinase K protection assay. (A) Schematic representation of C-terminally truncated ZIKV NS2A proteins. The amino acid positions of each truncated NS2A are indicated. A ‘SPG-C16’ sequence was fused to the N terminus of NS2A to ensure correct translocation of NS2A on the ER membrane. An ‘eGFP-Glyc’ sequence was fused to the C terminus of NS2A to probe its location at cytosol or ER lumen. See text for details. (B) BHK-21 cells were transfected with the expression plasmids described in (A). At 24 h p.t., the expression levels of various NS2A truncates were examined by Western blot using an eGFP antibody. The amino acid positions are indicated for each NS2A truncates. The molecular masses of marker proteins are shown to the left of Western blot image. (C) Diagram of glycosylation of eGFP-Glyc when located at ER lumen (left). No glycosylation occurs when eGFP-Glyc is located at cytosol (right). The glycosylation residue Asn is indicated. (D) In vitro deglycosylation assay. Plasmid-transfected cells were treated with PNGase F or PBS according to the protocol of in vitro deglycosylation assay (see Materials and Methods). The samples were examined for glycosylation status by Western blot using an eGFP antibody. (E) Diagram of in vitro proteinase K protection assay. When eGFP is located at ER lumen, it is protected from proteinase K cleavage (left); when located in the cytosol, eGFP is degraded by proteinase K (right). (F) In vitro proteinase K protection assay. Cell lysates from plasmid-transfected BHK-21 cells were treated with proteinase K or PBS according to the protocol of in vitro proteinase K protection assay (see Materials and Methods). The samples were detected for eGFP by Western blot using an eGFP antibody. Triton X-100 treatment is indicated.
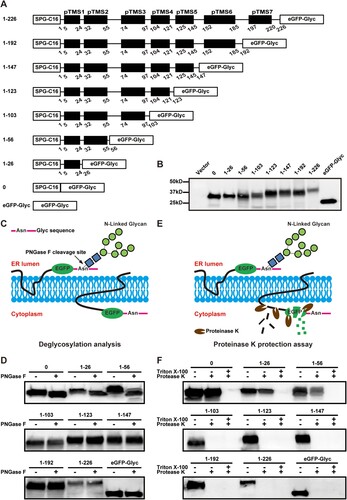
Next, we performed an in vitro proteinase K protection assay [Citation46] to validate the above in vitro deglycosylation results. BHK-21 cells were transfected with the same constructs depicted in A. At 24 h p.t., membrane fractions from the transfected cells were treated with proteinase K in the presence or absence of 0.5% Triton X-100 and analyzed by Western blot using an eGFP antibody. In the absence of Triton X-100, the eGFP residing in the cytosol would be susceptible to proteinase K digestion, whereas the eGFP residing in the ER lumen would be protected from proteinase K digestion (E). As shown in F, the eGFPs from constructs 0, 1–26, and 1–56 were protected from protease K digestion, again indicating that the first 56 residues are located in the ER lumen. In contrast, the eGFPs from constructs 1–103, 1–123, 1–147, 1–192, 1–226, and control eGFP-Glyc were completely degraded by the protease K treatment (F), supporting that the C-terminal region from residues 103–226 is located in the cytosol. As expected, in the presence of Triton X-100, the eGFPs from all constructs were degraded by proteinase K (F). The results of proteinase K assay are in full agreement with those of deglycosylation assay.
A topology model for ZIKV NS2A
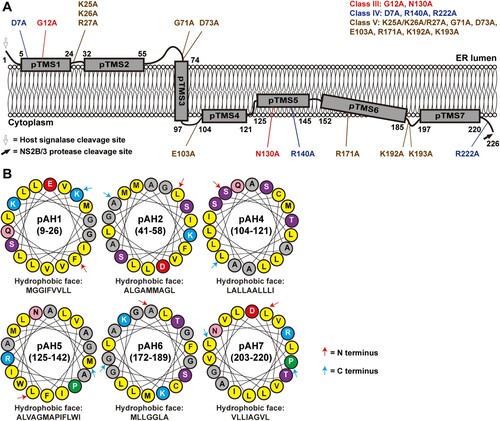
The above biochemical results support a topology model for ZIKV NS2A on the ER membrane (A): (i) The N-terminal region (including pTMS1 and pTMS2) resides in the ER lumen; (ii) the region spanning residues 74–97 (pTMS3) traverses the ER membrane in a lumen-to-cytosol direction; and (iii) the C-terminal region from residues 103–226 (including pTMS4 to pTMS7) is located in the cytosol side. Since pTMS1, pTMS2, pTMS4, PTMS5, PTM6, and pTMS7 associate with ER membraned when expressed individually (B), they may interact with the ER membrane in a peripheral manner. In support of this notion, informatics analyses using Jpred 4 and HeliQuest programmes suggest that amino acids 9–26 (pTMS1), 41–58 (pTMS2), 104–121 (pTMS4), 125–142 (pTMS5), 172–189 (pTMS6), and 203–220 (pTM7) may form amphipathic helices with hydrophobic and hydrophilic surfaces (B), where the hydrophobic surface may interact with the ER membrane.
Figure 8. A topological model of ZIKV NS2A. (A) A model of ZIKV NS2A membrane topology. See text for details. Class III, IV, and V mutations are depicted on the topology model. (B) Helical wheel representations of NS2A fragment 9–26, 41–58, 104–121, 125–142, 172–189, and 203–220. The amphiphilic helix regions were analyzed using Heliquest server (heliquest.ipmc.cnrs.fr/). The best 18-residue windows are presented. The amino acids forming the hydrophobic surface of each helix wheel are depicted. Yellow, gray, blue, red, purple, pink, and green circles indicate nonpolar, glycine/alanine, positively charged, negatively charged, polar, amide, and proline amino acids, respectively. The first and last residues in each helical wheel are indicated by a red and blue arrow, respectively.
Discussion
Flavivirus NS2A functions in viral RNA replication, virion assembly, evasion of innate immune response, and disease pathogenesis in a virus-specific manner [Citation35]. This is evidenced by its high sequence diversity (A), distinct membrane topology [Citation35,Citation46], and association with different disease pathways [Citation41,Citation42]. Thus, it is critical to perform a systematic genetic and biochemical characterization of ZIKV NS2A to delineate its structure and function. Our biochemical results showed that, unlike other flaviviruses, ZIKV NS2A has only a single transmembrane segment that traverses the ER membrane, the other six pTMSes are peripherally associated with the ER membrane (A). Our functional analysis revealed five classes of mutants with distinct replication phenotypes, among which class III, IV, and V mutants could not produce infectious viruses through different mechanisms (B). The critical mutations from class III to V are not clustered in specific regions of NS2A (A), suggesting that multiple domains of NS2A are essential for viral replication.
The membrane topology of ZIKV NS2A is in stark contrast with the topologies of other flavivirus NS2A proteins: DENV-2 NS2A has five membrane-traversing segments [Citation46] and YFV NS2A has three membrane-traversing segments [Citation35]. Interestingly, these topologically diverse flavivirus NS2A proteins are able to converge their functions in viral RNA replication, virion assembly, and evasion of immune response. High resolution, three-dimensional structures of flavivirus NS2A are needed to address this question. It should be noted that the membrane topology of NS2A presented here is in the absence of other viral proteins. In the context of viral replication, the NS2A protein is associated with various viral and cellular proteins that would modulate the structure of NS2A and perform biological functions.
Our mutagenesis study has identified distinct NS2A mutations that selectively abolish ZIKV RNA synthesis (class III) or virion assembly (class V), among which only the virion assembly-defective mutants could be rescued through trans complementation (). The results suggest that there are two species of NS2A molecules in ZIKV-infected cells. One NS2A species is located at the viral replication complex that could only be formed in cis; another NS2A species is located at virion assembly complex that could be formed in trans or cis. These results are in agreement with the previous studies from DENV and YFV NS2A [Citation30,Citation35,Citation36]. Compared with the viral replication complex, the virion assembly complex is much less well defined for flaviviruses. Previously studies showed that second-site mutations in prM, E, NS2B, and NS3 could rescue the defect of virion assembly caused by DENV-2 and YFV NS2A mutations, suggesting that these viral proteins participate in virion assembly. Besides the genetic evidence, the role of NS3 in virion assembly is also supported by (i) mutations in the NS3 helicase domain alone affect YFV assembly [Citation49], (ii) the helicase domain of DENV or YFV NS3 directly binds to NS2A [Citation35,Citation36], (iii) the cleavage between C and prM is tightly modulated by NS2B/NS3 protease and cellular signalase during YFV and Murray Valey encephalitis virus assembly [Citation50–52]. These data, together with the result that recombinant NS2A of Kunjin binds to viral 3’UTR RNA [Citation29], suggest a flavivirus assembly model. During flavivirus assembly, the NS2A protein recruits NS2B/NS3 through binding to the NS3 helicase domain. Once the NS2A-NS2B/NS3 complex reaches the virion assembly site, the NS3B/NS3 protease cleaves the C protein from the C/prM/E polyprotein. The C protein assembles onto genomic RNA (presented by NS2A) to form nucleocapsid, which is subsequently enveloped by prM/E complex to form virions. Since flavivirus assembly has to be coupled to viral RNA replication [Citation53], the newly synthesized genomic RNA has to be transported from the replication site to the virion assembly site [Citation54], possibly through the NS2A protein.
The trans complemented class V mutants could potentially be used as vaccine candidates, as recently reported for DENV-2 [Citation36]. Since the trans complemented virions are able to infect cells for only a single cycle, such vaccine platform is safer than the multi-round infectious live-attenuated vaccines [Citation55,Citation56]. This is particularly important for ZIKV vaccine when immunizing immune-compromised individuals and pregnant women [Citation57]. However, for development of class V mutants as a vaccine platform, the trans complementation efficiency needs to be improved to higher titers before they could be tested for in vivo efficacy.
In summary, we report the genetic and biochemical characterization of ZIKV NS2A protein. The results indicate that different flavivirus NS2A proteins have distinct membrane topologies, but converge to function in viral RNA synthesis and virion assembly. Together with previous results, we propose a model for flavivirus assembly in which NS2A plays a central role in mediating viral structural and nonstructural proteins as well as genomic RNA during virion formation.
Acknowledgments
We thank Mariano Garcia-Blanco, Shelton Bradrick, and other colleagues at University of Texas Medical Branch for helpful discussions during the course of this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Hamel R, Liegeois F, Wichit S, et al. Zika virus: epidemiology, clinical features and host-virus interactions. Microbes Infect. 2016;18(7–8):441–449. doi:10.1016/j.micinf.2016.03.009. PubMed PMID: WOS:000378848400001; English.
- Chouin-Carneiro T, Vega-Rua A, Vazeille M, et al. Differential susceptibilities of Aedes aegypti and Aedes albopictus from the Americas to Zika virus. Plos Neglect Trop D. 2016;10(3):e0004543. doi:10.1371/journal.pntd.0004543. PubMed PMID: WOS:000373272500062; English.
- Musso D, Roche C, Robin E, et al. Potential sexual transmission of Zika virus. Emerging Infect Dis. 2015;21(2):359–361. doi:10.3201/eid2102.141363. PubMed PMID: WOS:000348491400030; English.
- Besnard M, Lastere S, Teissier A, et al. Evidence of perinatal transmission of Zika virus, French Polynesia, December 2013 and February 2014. Eurosurveillance. 2014;19(13):13–16. doi:10.2807/1560-7917.Es2014.19.13.20751. PubMed PMID:WOS:000334029600004; English.
- Musso D, Nhan T, Robin E, et al. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Eurosurveillance. 2014;19(14):6–8. doi:10.2807/1560-7917.Es2014.19.14.20761. PubMed PMID: WOS:000334350200002; English.
- Dick GWA, Kitchen SF, Haddow AJ. Zika virus (I). Isolations and serological specificity. T Roy Soc Trop Med H. 1952;46(5):509–520. doi:10.1016/0035-9203(52)90042-4. PubMed PMID: WOS:A1952XT98500007; English.
- Duffy MR, Chen TH, Hancock WT, et al. Zika virus outbreak on yap Island, federated states of micronesia. New Engl J Med. 2009;360(24):2536–2543. doi:10.1056/Nejmoa0805715. PubMed PMID: WOS:000266813800008; English.
- Musso D, Gubler DJ. Zika virus. Clin Microbiol Rev. 2016;29(3):487–524. doi:10.1128/Cmr.00072-15. PubMed PMID: WOS:000375057900003; English.
- Musso D, Nilles EJ, Cao-Lormeau VM. Rapid spread of emerging Zika virus in the Pacific area. Clin Microbiol Infec. 2014;20(10):O595–O596. doi:10.1111/1469-0691.12707. PubMed PMID: WOS:000345825900002; English.
- Campos GS, Bandeira AC, Sardi SI. Zika virus outbreak, Bahia, Brazil. Emerg Infect Dis. 2015;21(10):1885–1886. doi:10.3201/eid2110.150847. PubMed PMID: WOS:000362158000038; English.
- Hennessey M, Fischer M, Staples JE. Zika virus spreads to new areas - region of the Americas, May 2015-January 2016. Am J Transplant. 2016;16(3):1031–1034. doi:10.1111/ajt.13743. PubMed PMID: WOS:000371240500036; English.
- Xia HJ, Xie XP, Shan C, et al. Potential mechanisms for enhanced Zika epidemic and disease. Acs Infect Dis. 2018;4(5):656–659. doi:10.1021/acsinfecdis.8b00004. PubMed PMID: WOS:000432478400002; English.
- Brasil P, Pereira JP, Moreira ME, et al. Zika virus infection in pregnant women in Rio de Janeiro. New Engl J Med. 2016;375(24):2321–2334. doi:10.1056/Nejmoa1602412. PubMed PMID: WOS:000390036500004; English.
- Schuler-Faccini L, Ribeiro EM, Feitosa IML, et al. Possible association between Zika virus infection and microcephaly — Brazil, 2015. Mmwr-Morbid Mortal W. 2016;65(3):59–62. doi:10.15585/Mmwr.Mm6503e2. PubMed PMID: WOS:000369119300005; English.
- Oehler E, Watrin L, Larre P, et al. Zika virus infection complicated by Guillain-Barre syndrome - case report, French Polynesia, December 2013. Eurosurveillance. 2014;19(9):4–6. PubMed PMID:WOS:000332933900002; English. doi: 10.2807/1560-7917.ES2014.19.9.20720
- Dos Santos T, Rodriguez A, Almiron M, et al. Zika virus and the Guillain-Barre syndrome - case series from seven countries. New Engl J Med. 2016;375(16):1598–1601. doi:10.1056/Nejmc1609015. PubMed PMID: WOS:000393102500029; English.
- Heymann DL, Hodgson A, Sall AA, et al. Zika virus and microcephaly: why is this situation a PHEIC? Lancet. 2016;387(10020):719–721. doi:10.1016/S0140-6736(16)00320-2. PubMed PMID: WOS:000370418000004; English.
- Shan C, Xie X, Shi PY. Zika virus vaccine: progress and challenges. Cell Host Microbe. 2018;24(1):12–17. doi:10.1016/j.chom.2018.05.021. PubMed PMID: 30008291; PubMed Central PMCID: PMC6112613.
- Xie X, Zou J, Shan C, et al. Small molecules and antibodies for Zika therapy. J Infect Dis. 2017;216(suppl_10):S945–S950. doi:10.1093/infdis/jix406. PubMed PMID: 29267911; PubMed Central PMCID: PMC5853826.
- Barrows NJ, Campos RK, Powell ST, et al. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe. 2016;20(2):259–70. doi:10.1016/j.chom.2016.07.004. PubMed PMID: 27476412; PubMed Central PMCID: PMC4993926.
- Sapparapu G, Fernandez E, Kose N, et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature. 2016;540(7633):443–447. doi:10.1038/nature20564. PubMed PMID: 27819683; PubMed Central PMCID: PMC5583716.
- Zou J, Shi PY. Strategies for Zika drug discovery. Curr Opin Virol. 2019;35:19–26. doi:10.1016/j.coviro.2019.01.005. PubMed PMID: 30852345.
- Mlakar J, Korva M, Tul N, et al. Zika virus associated with microcephaly. New Engl J Med. 2016;374(10):951–958. doi:10.1056/Nejmoa1600651. PubMed PMID: WOS:000371660000008; English.
- Lindenbach BD, Thiel H-J, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, Griffin DE, et al., editors. Fields virology. 5th ed. Vol. 1. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. p. 1101–1152.
- Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, et al. Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci U S A. 2003;100(24):14333–14338. doi:10.1073/pnas.2335168100. PubMed PMID: WOS:000186803800104; English.
- Xia HJ, Luo HL, Shan C, et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat Commun. 2018;9. doi:10.1038/s41467-017-02816-2. PubMed PMID: WOS:000423431900001; English.
- Falgout B, Markoff L. Evidence that flavivirus Ns1-Ns2a cleavage is mediated by a membrane-bound host protease in the endoplasmic-reticulum. J Virol. 1995;69(11):7232–7243. PubMed PMID: WOS:A1995RZ10000077; English.
- Chambers TJ, Mccourt DW, Rice CM. Production of yellow-fever virus proteins in infected-cells - identification of discrete polyprotein species and analysis of cleavage kinetics using region-specific polyclonal antisera. Virology. 1990;177(1):159–174. doi:10.1016/0042-6822(90)90470-C. PubMed PMID: WOS:A1990DK26100017; English.
- Mackenzie JM, Khromykh AA, Jones MK, et al. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology. 1998;245(2):203–215. doi:10.1006/viro.1998.9156. PubMed PMID: WOS:000074186800004; English.
- Xie XP, Zou J, Puttikhunt C, et al. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J Virol. 2015;89(2):1298–1313. doi:10.1128/Jvi.02882-14. PubMed PMID: WOS:000347178900035; English.
- Wu RH, Tsai MH, Chao DY, et al. Scanning mutagenesis studies reveal a potential intramolecular interaction within the C-terminal half of dengue virus NS2A involved in viral RNA replication and virus assembly and secretion. J Virol. 2015;89(8):4281–4295. doi:10.1128/Jvi.03011-14. PubMed PMID: WOS:000352218400022; English.
- Kummerer BM, Rice CM. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol. 2002;76(10):4773–84. PubMed PMID: 11967294; PubMed Central PMCID: PMC136122. doi: 10.1128/JVI.76.10.4773-4784.2002
- Leung JY, Pijlman GP, Kondratieva N, et al. Role of nonstructural protein NS2A in flavivirus assembly. J Virol. 2008;82(10):4731–4741. doi:10.1128/Jvi.00002-08. PubMed PMID: WOS:000255619600005; English.
- Liu WJ, Chen HB, Khromykh AA. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J Virol. 2003;77(14):7804–7813. doi:10.1128/Jvi.77.14.7804-7813.2003. PubMed PMID: WOS:000183899200012; English.
- Vossmann S, Wieseler J, Kerber R, et al. A basic cluster in the N terminus of yellow fever virus NS2A contributes to infectious particle production. J Virol. 2015;89(9):4951–4965. doi:10.1128/Jvi.03351-14. PubMed PMID: WOS:000352219600023; English.
- Shan C, Xie XP, Zou J, et al. Using a virion assembly-defective dengue virus as a vaccine approach. J Virol. 2018;92(21). doi:10.1128/JVI.01002-18. PubMed PMID: WOS:000447139100013; English.
- Tu YC, Yu CY, Liang JJ, et al. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J Virol. 2012 Oct;86(19):10347–10358. doi:10.1128/Jvi.00525-12. PubMed PMID: WOS:000308740700010; English.
- Liu WJ, Wang XJ, Clark DC, et al. A single amino acid substitution in the west Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J Virol. 2006;80(5):2396–2404. doi:10.1128/Jvi.80.5.2396-2404.2006. PubMed PMID: WOS:000235388400032; English.
- Wen JL, Hua BC, Xiang JW, et al. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J Virol. 2004;78(22):12225–12235. doi:10.1128/Jvi.78.22.12225-12235.2004. PubMed PMID: WOS:000224781400014; English.
- Wu RH, Tsai MH, Tsai KN, et al. Mutagenesis of dengue virus protein NS2A revealed a novel domain responsible for virus-induced cytopathic effect and interactions between NS2A and NS2B transmembrane segments. J Virol. 2017;91(12). doi:10.1128/JVI.01836-16. PubMed PMID: WOS:000402169500027; English.
- Melian EB, Edmonds JH, Nagasaki TK, et al. West Nile virus NS2A protein facilitates virus-induced apoptosis independently of interferon response. J Gen Virol. 2013;94:308–313. doi:10.1099/vir.0.047076-0. PubMed PMID: WOS:000315546100008; English.
- Yoon KJ, Song G, Qian XY, et al. Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell. 2017;21(3):349–358.e6. doi:10.1016/j.stem.2017.07.014. PubMed PMID: WOS:000409527700012; English.
- Shan C, Xie XP, Muruato AE, et al. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe. 2016;19(6):891–900. doi:10.1016/j.chom.2016.05.004. PubMed PMID: WOS:000377447000019; English.
- Xie XP, Zou J, Shan C, et al. Zika virus replicons for drug discovery. Ebiomedicine. 2016;12:156–160. doi:10.1016/j.ebiom.2016.09.013. PubMed PMID: WOS:000386878500031; English.
- Miller S, Sparacio S, Bartenschlager R. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J Biol Chem. 2006;281(13):8854–8863. doi:10.1074/jbc.M512697200. PubMed PMID: WOS:000236247100065; English.
- Xie XP, Gayen S, Kang CB, et al. Membrane topology and function of dengue virus NS2A protein. J Virol. 2013;87(8):4609–4622. doi:10.1128/Jvi.02424-12. PubMed PMID: WOS:000316671000043; English.
- Hooper R, Churamani D, Brailoiu E, et al. Membrane topology of NAADP-sensitive two-pore channels and their regulation by N-linked glycosylation. J Biol Chem. 2011;286(11):9141–9149. doi:10.1074/jbc.M110.189985. PubMed PMID: WOS:000288247700045; English.
- Rossi SL, Fayzulin R, Dewsbury N, et al. Mutations in West Nile virus nonstructural proteins that facilitate replicon persistence in vitro attenuate virus replication in vitro and in vivo. Virology. 2007;364(1):184–195. doi:10.1016/j.virol.2007.02.009. PubMed PMID: WOS:000247198200021; English.
- Patkar CG, Kuhn RJ. Yellow fever virus NS3 plays an essential role in virus assembly independent of its known enzymatic functions. J Virol. 2008;82(7):3342–3352. doi:10.1128/Jvi.02447-07. PubMed PMID: WOS:000254139800015; English.
- Lobigs M, Lee E. Inefficient signalase cleavage promotes efficient nucleocapsid incorporation into budding flavivirus membranes. J Virol. 2004;78(1):178–186. doi:10.1128/Jvi.78.1.178-186.2004. PubMed PMID: WOS:000187451700018; English.
- Stocks CE, Lobigs M. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J Virol. 1998;72(3):2141–2149. PubMed PMID: WOS:000071997600052; English.
- Amberg SM, Rice CM. Mutagenesis of the NS2B-NS3-mediated cleavage site in the flavivirus capsid protein demonstrates a requirement for coordinated processing. J Virol. 1999;73(10):8083–8094. PubMed PMID: WOS:000082554300019; English.
- Khromykh AA, Varnavski AN, Sedlak PL, et al. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J Virol. 2001;75(10):4633–4640. doi:10.1128/Jvi.75.10.4633-4640.2001. PubMed PMID: WOS:000168309300017; English.
- Welsch S, Miller S, Romero-Brey I, et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5(4):365–375. doi:10.1016/j.chom.2009.03.007. PubMed PMID: WOS:000265571600009; English.
- Xie XP, Yang YJ, Muruato AE, et al. Understanding Zika virus stability and developing a chimeric vaccine through functional analysis. Mbio. 2017;8(1). doi:10.1128/mBio.02134-16. PubMed PMID: WOS:000395835000086; English.
- Shan C, Muruato AE, Nunes BTD, et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat Med. 2017;23(6):763–767. doi:10.1038/nm.4322. PubMed PMID: WOS:000402768000019; English.
- Xie XP, Kum DPB, Xia HJ, et al. A single-dose live-attenuated Zika virus vaccine with controlled infection rounds that protects against vertical transmission. Cell Host Microbe. 2018;24(4):487–499.e5. doi:10.1016/j.chom.2018.09.008. PubMed PMID: WOS:000446888300008; English.