
ABSTRACT
Many pathogens infect hosts through various immune evasion strategies. However, the molecular mechanisms by which pathogen proteins modulate and evade the host immune response remain unclear. Enterohemorrhagic Escherichia coli (EHEC) is a pathological strain that can induce mitogen-activated protein (MAP) kinase (Erk, Jnk and p38 MAPK) and NF-κB pathway activation and proinflammatory cytokine production, which then causes diarrheal diseases such as hemorrhagic colitis and hemolytic uremic syndrome. Transforming growth factor β-activated kinase-1 (TAK1) is a key regulator involved in distinct innate immune signalling pathways. Here we report that EHEC translocated intimin receptor (Tir) protein inhibits the expression of EHEC-induced proinflammatory cytokines by interacting with the host tyrosine phosphatase SHP-1, which is dependent on the phosphorylation of immunoreceptor tyrosine-based inhibition motifs (ITIMs). Mechanistically, the association of EHEC Tir with SHP-1 facilitated the recruitment of SHP-1 to TAK1 and inhibited TAK1 phosphorylation, which then negatively regulated K63-linked polyubiquitination of TAK1 and downstream signal transduction. Taken together, these results suggest that EHEC Tir negatively regulates proinflammatory responses by inhibiting the activation of TAK1, which is essential for immune evasion and could be a potential target for the treatment of bacterial infection.
Introduction
Humans have a sophisticated immune system to fight against pathogens. However, many pathogens employ unique strategies to evade, inhibit, or manipulate the innate immune response, leading to chronic infection [Citation1]. These strategies include infecting immune cells or delitescence, changing self-structures to subvert pathogen recognition, and inhibiting host signal transduction by structural or non-structural components.
Immune responses are regulated by immune receptors that contain either an immunoreceptor tyrosine-based activation motif (ITAM) or immunoreceptor tyrosine-based inhibitory motif (ITIM) [Citation2–4]. Tyrosine phosphorylation in the ITIMs creates binding sites for SH2-containing phosphatases, such as SHP-1 [Citation5–7]. SHP-1 is an intracellular protein tyrosine phosphatase, which may negatively regulate toll-like receptor mediated proinflammatory cytokine production by inhibiting the activation of transcription factor NF-κB and mitogen-activated protein kinase [Citation8,Citation9].
TAK1 was originally identified as a member of the transforming growth factor-β (TGF-β)-activated MAPK kinase kinase (MKKK) family [Citation10]. Multiple lines of evidence have shown that TAK1 is involved in distinct signalling pathways induced by various stimuli, such as environmental stress [Citation11], inflammatory cytokines [Citation12], lipopolysaccharide (LPS) [Citation13], and TGF-β [Citation14]. Phosphorylation of TAK1 at Thr-187, Ser-192 and Ser-412 is required for its activation [Citation15,Citation16]. Once activated, TAK1 can activate several downstream cellular signalling cascades, such as p38 MAPK, JNK, IKK, and Akt [Citation17], leading to the translocation of the transcription factors NF-κB and AP-1 [Citation18,Citation19] from the cytoplasm to nucleus and subsequent production of proinflammatory cytokines.
EHEC, which is associated with the consumption of contaminated food and water, constitutes a significant human health risk and may cause acute gastroenteritis, bloody diarrhea and hemorrhagic colitis. More than 10% of patients develop severe complications with 5% mortality [Citation20]. EHEC generates attaching and effacing (AE) lesions to colonize the intestine, damage the epithelium, and promote diarrheal illnesses [Citation21,Citation22]. The translocated intimin receptor (Tir) from EHEC is an important virulence protein that is injected into the plasma membrane of host cells via a type III secretion system (T3SS) [Citation23–26]. Proteases produced by EHEC and the prevalent human commensal Bacteroides thetaiotaomicron can modulate T3SS function by cleaving proteins in the EHEC T3SS translocon [Citation27]. Our previous results showed that the enteropathogenic E. coli (EPEC) Tir recruits SHP-1 to inhibit tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6) ubiquitination [Citation28–30]. Increasing evidence shows that these T3SS-bearing pathogens share similarities to target key cellular functions such as the cell cytoskeleton, trafficking, cell death/survival, and signalling pathways such as NF-κB and MAPK [Citation31]. However, whether EHEC Tir regulates the immune response to EHEC infection and the mechanisms involved in this process remains unknown. Therefore, we aimed to determine whether EHEC Tir might have important roles in regulating and evading the host immune response.
Materials and methods
Mice, bacterial growth, and cell culture
Ptpn6me-v/me-v (000811; Jackson Laboratories) mice on C57BL/6 background were maintained in clean and comfortable conditions at the Laboratory Animal Center of Fudan University. 3-4-week-old male mice were used in the experiments and littermates were used as controls. All animal studies were approved by the Institutional Animal Care and Use Committee of Fudan University.
EDL933 and EDL933 ΔTir were grown at 37°C in Luria–Bertani broth (10 g/L tryptone, 5 g/L yeast extract and 10 g/L NaCl). The bacterial mutants of Tir encoded in pK184 strains were grown in Luria–Bertani broth supplemented with 50 ng/ul kanamycin. Before infection, EDL933 and its mutants were propagated in Dulbecco’s modified Eagle’s medium (DMEM; HyClone) supplemented with 10 mM HEPES (pH 7.4) for 15 h at 37°C in 5% CO2.
HEK293 T cells, RAW264.7 cells and mouse primary peritoneal macrophages were cultured in DMEM supplemented with 10% (v/v) FBS (Gibco) and 100 U/ml penicillin and streptomycin. The HeLa cells were maintained in RPMI 1640 medium supplemented with 10% (v/v) FBS (Gibco) and 100 U/ml penicillin and streptomycin.
Reagents and antibodies
Plasmid construction
Flag-EDL933 Tir (wild-type (WT), Y490F, Y519F, Y490F, and Y519F) were constructed using standard molecular biology techniques, and the cDNAs were inserted into the Flag-pcDNA3.0 vector. Glutathione S-transferase (GST)-EDL933 Tir (WT, Y490F, Y519F, Y490F, and Y519F) were inserted into pGEX4T-1 from the Flag-tagged plasmids described above. SHP-1, SHP-1 (C453S), SHP-2, TAK1, and HA-EDL933 Tir were inserted into HA-pcDNA3.0. The primers are listed in Supplementary Table 1.
Transfection and immunoprecipitation
HEK293 T cells were transfected using the PEI method. Pervanadate (0.1 mM sodium orthovanadate and 10 mM H2O2) was added for 20 min at 37°C (later 48 h) before cells were lysed in lysis buffer (Beyotime) supplemented with 1% Protease Inhibitor Cocktail (B14002; Selleck.cn), 1 mM NaF and 1 mM Na3VO4. For ubiquitination detection in co-immunoprecipitation experiments, cell lysates were treated with 1% SDS and boiled at 95°C for 5 min prior to immunoprecipitation. Cell lysates were rotated overnight at 4 °C with anti-Flag M2 Affinity Gel. The sepharose samples were centrifuged, washed three times with washing buffer (PBS, 1% Triton X-100), and boiled with SDS loading buffer for 10 min and later analyzed by immunoblot.
Confocal microscopy
HeLa cells were transfected by Lipofectamine 2000 (11668–019, Invitrogen). The cells were infected for 4 h with EDL933 (ΔTir + HA-Tir) or EDL933ΔTir, treated by standard immunofluorescent staining techniques and examined with a Leica confocal microscope.
GST pull-down
The GST-tagged EDL933 Tir plasmids and its mutants Y490F, Y519F, and Y490F/Y519F were transformed into TKB1 and induced to express either phosphorylated WT EDL933 Tir or its point mutants by IPTG. Then, recombinant proteins were purified as GST fusion proteins and incubated with SHP-1 purified from BL-21 strain or mouse primary peritoneal macrophages. The mixture samples were centrifuged, washed three times with washing buffer (PBS, 1% Triton X-100), and boiled with SDS loading buffer for 10 min and later analyzed by immunoblot.
RNA interference
SHP-1 siRNA (5′-GAGAUGGCACCAUCAUCCACCUUAA-3’) was used for inhibiting the expression of endogenous SHP-1 and control siRNA (5’-UUCUCCGAACGUGUCACGUTT-3’) was used as a control siRNA. They were transfected into RAW264.7 cells by the Amaxa program D-032 with the Cell Line Nucleofector Kit.
Quantitative RT–PCR
Total RNA was extracted to measure the mRNA abundance of the indicated genes by standard molecular biology techniques. Briefly, Total RNA was extracted with 1 ml of TRIzol reagent according to the manufacturer's instructions (Invitrogen). Next, 1 μg total RNA was reverse-transcribed with the ReverTra Ace qPCR RT Kit (FSQ-101) according to the manufacturer's instructions (Toyobo). A LightCycler (LC480; Roche) and a SYBR RT–PCR kit (QPK-212; Toyobo) were used for quantitative real-time RT–PCR analysis. The data shown are the relative abundances of the indicated mRNAs derived from RAW264.7 or mouse primary peritoneal macrophages normalized to GAPDH. Gene-specific primer sequences are described in Supplementary Table 1.
RNA sequencing
A total amount of 1.5 μg RNA was used as input for sample preparations. Sequencing libraries were generated using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA). The clustering of the index-coded samples was performed on a cBot Cluster Generation System using the HiSeq 4000 PE Cluster Kit (Illumia) and the samples were sequenced on an Illumina Hiseq 4000 platform. In this process, paired-end reads of 150 bp were generated.
Oral infection, CFU counts, histology, and immunohistochemical analysis
3-4-week-old male C57Bl/6J mice were pretreated for 24 h with streptomycin and fasted for 12 h before the oral inoculation. Simultaneously, 1 × 109 colony forming units (CFU) EDL933 and its ΔTir mutants were prepared and orally gavaged for each mouse. Fecal specimens, colons and spleens were collected, weighed, homogenized in PBS and plated on MacConkey agar plates for CFU analysis. Some colon and spleen tissues were retained for qPCR to detect cytokine production, and the other colons were fixed in 4% (v/v) paraformaldehyde for evaluation of the tissue pathology by Hematoxylin and eosin (H&E) staining and F4/80, CD3, CD19-specific immunohistochemistry.
Results
Tir inhibits immune responses to EHEC in vivo
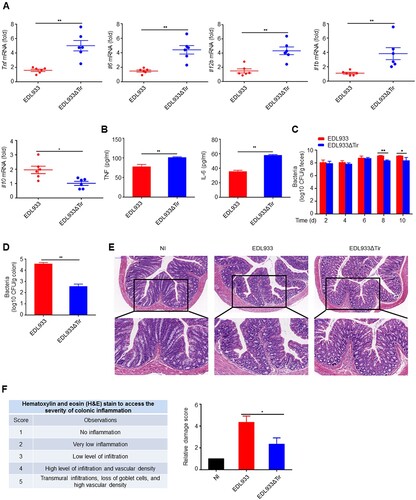
The attachment of EHEC to its host cell is triggered by the intracellular interaction of its translocated effector proteins Tir and EspFU. The Asn-Pro-Tyr motif of Tir is critical for this interaction [Citation32]. In order to determine whether the EHEC Tir protein inhibits the innate immune response in vivo, we constructed a mouse model where the mice were orally infected with EDL933 or EDL933ΔTir EHEC strains. We found that EDL933 induced a lower production of proinflammatory cytokines Tnf, Il6, Il12b and Il1b and a higher production of anti-inflammatory cytokines Il10 than EDL933ΔTir in mouse colons (a) and spleens (Supplemental Figure 1a) by quantitative real-time PCR (qPCR). In addition, EDL933-induced serum TNF and IL-6 were lower than the EDL933ΔTir strain did (b). We next detected the amount of bacteria in mouse feces and colons in the two groups. The number of bacterial CFUs from the feces (c) and colons (d) infected with the EDL933 strain was significantly higher than those infected with the EDL933ΔTir strain. Moreover, mice infected with EDL933 had a slower weight gain than mice infected with the EDL933ΔTir strain (Supplemental Figure 1b). Histological examination revealed that mice infected with EDL933 had more extensive neutrophil inflammatory infiltrates and structural disruption of the colonic epithelium (e-f). F4/80, CD3, and CD19-specific immunohistochemistry results show that mice infected with EDL933 had more macrophages, T cells and B cells, respectively (Supplemental Figure 1c). These results suggest that Tir may inhibit the intestinal immune response to EDL933 infection.
Figure 1. Tir inhibits immune responses to EHEC in vivo. a Quantitative RT–PCR analysis of Tnf, Il6, Il12b, or Il1b mRNA in colon from 3-4-week-old mice (n = 6 per group) infected orally with 1 × 109 CFU of EDL933 or EDL933ΔTir and sacrified at fourteenth day. b ELISA analysis of TNF or IL-6 from mouse serum in a. c-d Bacterial load in the feces (c) or colon (d) of mice infected as in a at the indicated times. e Histopathology of colons of mice left uninfected (NI) or infected as in a. Outlined areas at top are enlarged below. Original magnification, ×10 (top) or ×20 (bottom). f Hematoxylin and eosin (H&E) stain to access the severity of colonic inflammation. Data are representative of at least two independent experiments (mean ± SEM in a-d, f). *p < 0.05 and **p < 0.01 (Student’s t-test).
EHEC Tir inhibits cytokine production in vitro
To identify whether EHEC Tir is involved in bacteria-induced innate immune responses and proinflammatory cytokine production in vitro, we performed qPCR experiments to investigate the dynamic expression of proinflammatory cytokines in mouse primary peritoneal macrophages and RAW264.7 cells in response to the EDL933 or EDL933ΔTir strains. We found that the EDL933ΔTir strain induced more Tnf, Il6, and Il12b than the EDL933 strain in mouse primary peritoneal macrophages (a) and RAW264.7 cells (Supplemental Figure 2a). The viability of RAW264.7 cells infected with EDL933 or EDL933ΔTir were the same (Supplemental Figure 2b). In addition, enzyme-linked immunosorbent assays (ELISA) showed that EDL933-induced secretion of TNF, IL-6, and IL-12b was also lower than that of EDL933ΔTir in the supernatant of mouse primary peritoneal macrophages (b). These results suggest that the EHEC Tir protein inhibits cytokine production in vitro. To confirm whether EDL933 Tir inhibits cytokine production specifically, we infected mouse primary peritoneal macrophages with EDL933, EDL933ΔTir or EDL933(ΔTir + HA-Tir) (an EDL933ΔTir strain transformed with a HA-tagged Tir encoded by the vector pK184) strains to detect the production of Tnf, Il6 and Il12b by qPCR (c). We found that EDL933(ΔTir + HA-Tir) induced lower levels of proinflammatory cytokines than EDL933ΔTir, and both groups had similar levels of cell viability and phagocytic function (Supplemental Figure 2c-d). These results suggest that EHEC Tir specifically inhibits EHEC-triggered induction of proinflammatory cytokines.
Figure 2. EHEC Tir inhibits cytokine production in vitro. a Quantitative RT–PCR analysis of Tnf, Il6, and Il12b mRNAs in mouse primary peritoneal macrophages infected with EDL933 or EDL933ΔTir for the indicated times. b ELISA analysis of TNF, IL-6, and IL-12b in the supernatants of mouse primary peritoneal macrophages infected with EDL933 or EDL933ΔTir for the indicated times. c Quantitative RT–PCR analysis of Tnf, Il6, and Il12b mRNAs in mouse primary peritoneal macrophages infected with medium, EDL933, EDL933ΔTir or EDL933(ΔTir + HA-Tir). d Immunoassay of the indicated proteins in mouse primary peritoneal macrophages infected with EDL933 or EDL933ΔTir. e Image J analysis of the greyscale value of western blot bands in panel d, n = 3. f Immunoassay of lysates of or mouse primary peritoneal macrophages infected with EDL933ΔTir or EDL933(ΔTir + HA-Tir). Data are representative of at least three independent experiments (mean ± SEM in a–c). *p < 0.05 and **p < 0.01 (Student′s t-test).
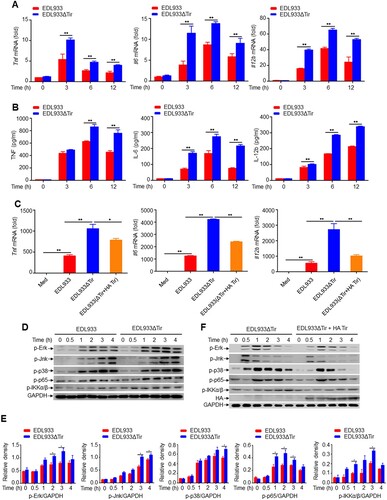
We next detected whether EDL933 Tir modulated the activation of the MAP kinase and NF-κB signalling pathways, which regulate downstream proinflammatory cytokine production. Consistent with the cytokine results, EDL933ΔTir induced higher phosphorylation of Erk, Jnk, p38 MAPK, NF-κB p65, and IKKα/β than EDL933 in mouse primary peritoneal macrophages (d-e), which was inhibited by the EDL933(ΔTir + HA-Tir) strain (f) when the Tir protein was inserted into the host cell and phosphorylated (Supplemental Figure 3a-b). RNA sequencing results showed that some of the EDL933ΔTir-induced genes such as Ror1, Prkdc, and Mras were associated with inflammation (Supplemental Figure 3c). These results suggested that Tir might be an important protein in suppression of EHEC-induced cytokine production.
EHEC Tir recruits phosphatase SHP-1
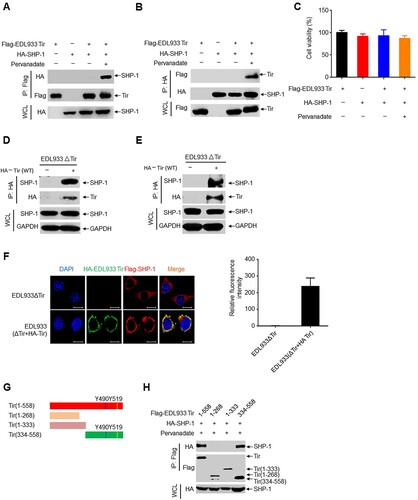
Using bioinformatics methods, we unexpectedly found that EHEC Tir contains two ITIMs. ITIMs can interact with SHP-1 upon tyrosine phosphorylation [Citation33], which inhibits intracellular calcium mobilization and subsequent cell activation [Citation34]. Co-immunoprecipitation experiments showed that EDL933 Tir interacted with SHP-1 in the presence of the tyrosine phosphatase inhibitor pervanadate (a-b), while both groups had similar levels of cell viability (c). Similarly, EDL933 Tir also immunoprecipitated with SHP-2 (Supplemental Figure 4a-b). However, EDL933 Tir did not interact with the SHP-1 (C453S) mutant, indicating that the interaction is dependent on SHP-1 phosphatase activity (Supplemental Figure 4c). Furthermore, we infected RAW264.7 cells or mouse primary peritoneal macrophages with EDL933ΔTir or EDL933(ΔTir + HA-Tir) to determine whether EDL933 Tir interacted with endogenous SHP-1. The results showed that Tir interacted with endogenous SHP-1 in RAW264.7 cells (d) and mouse primary peritoneal macrophages (e). Next, we determined the intracellular co-localization of EHEC Tir and SHP-1 by immunofluorescent confocal microscopy. HeLa cells were transfected with Flag-SHP-1 and then infected with EDL933ΔTir or EDL933(ΔTir + HA-Tir). The results showed that HA-Tir (green) colocalized with Flag-SHP-1 (red) (f). To detect whether EHEC Tir and SHP-1 bind each other directly, we used competent TKB1 E. coli cells (encoding a tyrosine kinase to induce expression of tyrosine-phosphorylated proteins) to express the recombinant GST fusion EHEC Tir and incubated it with purified recombinant histidine-tagged SHP-1 or endogenous SHP-1 from RAW264.7 cells. The results showed that EDL933 Tir proteins interacted directly with His-SHP-1, the endogenous SHP-1 from RAW264.7 cells, and mouse primary peritoneal macrophages (Supplemental Figure 4d-f). To identify the binding region of EHEC Tir and SHP-1, we constructed a series of deletion mutants of EDL933 Tir and SHP-1 ( g, supplemental 4 g). The results showed that SHP-1 bound only to amino acid residues 334–558 of EDL933 Tir [containing the two ITIMs (Tyr490 and Tyr519)] but not to amino acid residues 1–333, indicating that ITIMs play an important role in the EDL933 Tir–SHP-1 interaction ( h). Hence, the SHP-1-binding domain of EDL933 Tir may lie in residues 334–558. Similarly, EDL933 Tir bound to amino acid residues 107–595 or 221–595, but not 1–242 of SHP-1 (Supplemental Figure 4 h), suggesting that the C-terminus of SHP-1 is essential for the binding of Tir. Taken together, these results suggest that EHEC Tir interacts with tyrosine phosphatase SHP-1.
Figure 3. EHEC Tir recruits phosphatase SHP-1. a-b Immunoprecipitation (IP) and immunoblot (IB) analysis of Flag-EDL933 Tir and HA-SHP-1 in HEK293 T cells untreated or treated with pervanadate, probed with antibody to Flag or HA (left margin). c CCK-8 analysis of cell viability in HEK293 T cells treated as in a and b. d-e Immunoassay of RAW 264.7 cells (d) or mouse primary peritoneal macrophages (e) infected with EDL933ΔTir (−) or EDL933(ΔTir + HA-Tir) (+). f Immunofluorescence microscopy of HeLa cells transfected with Flag-SHP-1 and infected with EDL933ΔTir (top row) or EDL933(ΔTir + HA-Tir) (bottom row), stained with antibody to HA (green), Flag (red) and the DNA-binding dye DAPI (blue). Scale bars, 10 μm. Relative fluorescence intensity of Flag-SHP-1 was calculated with Image J from three different areas, each of which contains three cells (right graph). n = 3. g EDL933 Tir deletion mutants. h Co-immunoprecipitation assay of Flag-EDL933 Tir deletion mutants and HA-SHP-1 in HEK293 T cells. Data are representative of at least three independent experiments.
SHP-1 inhibits TAK1 phosphorylation and ubiquitination
TAK1 is widely accepted as a key regulator in proinflammatory cytokine signalling, which can be activated by TNF, interleukin-1 (IL-1), and toll-like receptor (TLR) ligands [Citation35,Citation36]. LPS from Gram-negative bacteria stimulates the TLR4-MyD88-TRAF6-TAK1 axis and leads to the activation of the MAPK and NF-κB pathways, which cooperatively regulate the transcription of genes involved in the inflammatory immune response [Citation37–39]. Phosphorylation and ubiquitination of TAK1 regulate this pathway. Using lysates of HEK293 T cells expressing Flag-tagged TAK1 and HA-tagged SHP-1, we determined that SHP-1 immunoprecipitated with TAK1 in a co-immunoprecipitation assay ( a-b). An in vitro GST pull-down assay showed that TAK1 interacted directly with purified recombinant His-SHP-1, endogenous SHP-1 from RAW264.7 cells and mouse primary peritoneal macrophages (Supplemental Figure 5a-c). However, TAK1 did not interact with the SHP-1 (C453S) mutant, indicating that the interaction is dependent on SHP-1 phosphatase activity (Supplemental Figure 5d). Similarly, SHP-2 co-immunoprecipitated with TAK1 (Supplemental Figure 5e). Furthermore, overexpression of EDL933 Tir in HEK293 T cells significantly enhanced the association of TAK1 and SHP-1 (c) but did not enhance the association of TRAF6 and SHP-1 (Supplemental Figure 5f). The endogenous interaction between SHP-1 and TAK1 was enhanced in mouse primary peritoneal macrophage cells infected with EDL933(ΔTir + HA-Tir) (d).
Figure 4. SHP-1 inhibits TAK1 phosphorylation and ubiquitination. a-c Co-immunoprecipitation assay of Flag-TAK1, HA-SHP-1 or Myc-EDL933 Tir expressed in HEK293 T cells. Immunoassay of mouse primary peritoneal macrophages uninfected (NI) or infected with EDL933ΔTir or EDL933(ΔTir + HA-Tir). d Immunoassay of mouse primary peritoneal macrophages uninfected (NI) or infected with EDL933ΔTir or EDL933(ΔTir + HA-Tir). e-g Immunoassay of HEK293 T cells expressing various vectors (above lanes). h-j Immunoassay of mouse primary peritoneal macrophages infected with indicated strains (above lanes). k-m Immunoassay of HEK293 T cells expressing TAK1 point mutants (T187A, S192A, S412A) and ubiquitin (total, K48 or K63 ubiquitin). Data are representative of at least three independent experiments.
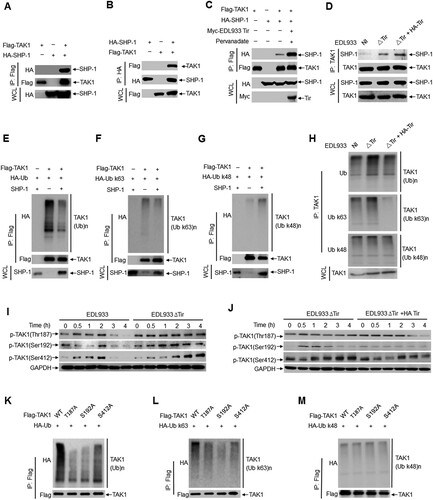
TAK1 depends on its phosphorylation and polyubiquitination to modulate downstream signalling. As EDL933 Tir enhanced the association of TAK1 and SHP-1, we examined the effect of Tir and SHP-1 on the phosphorylation and polyubiquitination of TAK1. The results showed that overexpression of SHP-1 resulted in less total ubiquitination (e) and K63-linked polyubiquitination of TAK1 (f) but had no effect on the K48-linked polyubiquitination ( g) in HEK293 T cells. Infection of mouse primary peritoneal macrophages with EDL933(ΔTir + HA-Tir) elicited less total ubiquitination and K63-linked polyubiquitination of TAK1 than infection with EDL933ΔTir but did not affect K48-linked polyubiquitination ( h).
Several threonine and serine residues in TAK1, including Thr-187, Ser-192, and Ser-412, are phosphorylated upon cytokine stimulation to induce TAK1 activation [Citation15,Citation40,Citation41]. By immunoblot analysis, we found that the levels of phosphorylated TAK1 (p-TAK1 Thr187, Ser192, Ser412) were significantly increased in mouse primary peritoneal macrophages (i) and RAW264.7 cells (Supplemental Figure 5 g) infected with EDL933ΔTir compared to those infected with EDL933. Furthermore, the levels of phosphorylated TAK1 were significantly decreased in mouse primary peritoneal macrophages (j) infected with EDL933(ΔTir + HA-Tir) compared to those infected with EDL933ΔTir.
As EHEC Tir promotes SHP-1 to interact with TAK1 and inhibit TAK1 phosphorylation and K63-linked polyubiquitination, we wanted to determine whether SHP-1 inhibits TAK1 polyubiquitination by regulating its own phosphorylation. To test this hypothesis, we constructed three TAK1 phosphorylation point mutants (T187A, S192A, S412A). The results showed that TAK1(T187A) and TAK1(S192A) decreased total ubiquitination (k) and K63-linked polyubiquitination ( l) but not K48-linked polyubiquitination ( m) of TAK1. Thus, these results suggest that phosphorylation of TAK1 promotes K63-linked polyubiquitination. In other words, Tir promotes SHP-1 to inhibit K63-linked polyubiquitination of TAK1 by inhibiting TAK1 phosphorylation. However, the mechanism by which TAK1 phosphorylation regulates its own ubiquitination needs to be further elucidated.
EHEC Tir–SHP-1 interaction and cytokine inhibition depend on ITIM tyrosine phosphorylation
Host cellular ITIM-containing proteins must be phosphorylated on both ITIMs to interact with SHP-1 [Citation42]. We constructed EDL933 Tir mutants containing two sites of ITIM in which either one or both ITIMs were replaced with phenylalanine to determine whether Tir and SHP-1 require tyrosine phosphorylation in ITIMs (a). The results showed that the Y490F and Y519F mutants resulted in less co-immunoprecipitation with SHP-1 than WT Tir (b). Similarly, we observed that EDL933 Tir mutants had a lower interaction with SHP-2 (Supplemental Figure 6a-b). A GST pull-down assay showed that tyrosine phosphorylation of GST-Tir disappeared when the tyrosine residues in ITIMs were transformed to phenylalanine (c). Consistently, only EDL933 Tir proteins with fully phosphorylated tyrosine (d) in the ITIMs associated with purified recombinant histidine-tagged SHP-1 (e) or endogenous SHP-1 (f) from mouse primary peritoneal macrophages, while the tyrosine residue mutants had a lower association. These results suggest that EHEC Tir requires tyrosine phosphorylation in ITIMs to interact with SHP-1.
Figure 5. EHEC Tir–SHP-1 interaction and cytokine inhibition are dependent on ITIM tyrosine phosphorylation. a EDL933 Tir ITIM mutants. Red indicates tyrosine in the wild-type (WT) site that can be tyrosine-phosphorylated; blue indicates phenylalanine in the mutant residue that cannot be tyrosine-phosphorylated. b Immunoassay of lysates of HEK293 T cells expressing SHP-1 and wild-type EDL933 Tir or its ITIM mutants in A (top). c GST fusion mutants of EDL933 Tir. p-Y and Yp indicate phosphorylated tyrosine. Tyrosine phosphorylation of GST-Tir (second lane) or its ITIM mutants purified from TKB1 that expresses tyrosine kinase. d-f GST precipitation assay of GST-EDL933 Tir or its ITIM mutants (d) with purified His-SHP-1 (e) or endogenous SHP-1 from primary macrophage (f). g-h Quantitative RT–PCR analysis of Tnf, Il6 or Il12b mRNAs (g) and immunoassay the protein of HA-Tir (h) in mouse primary peritoneal macrophages left uninfected (Med) or infected for 6 h with EDL933(ΔTir + HA-Tir), EDL933(ΔTir + Y490F), EDL933(ΔTir + Y519F) or EDL933(ΔTir + Y490F, Y519F). i Immunoassay of lysates of mouse primary peritoneal macrophages infected with EDL933(ΔTir + HA-Tir) or EDL933(ΔTir + Y490F, Y519F) for the indicated times. Data are representative of at least three independent experiments (mean ± SEM in g). **p < 0.01 (Student’s t-test).
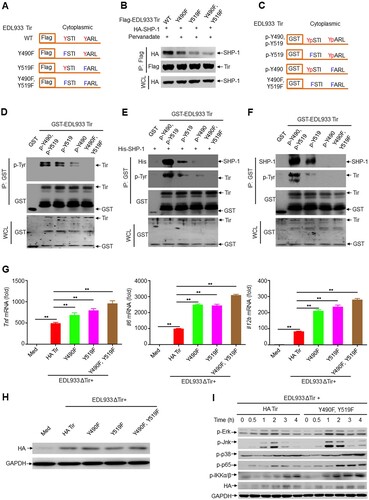
We constructed the bacterial strains EDL933(ΔTir + HA-Tir), EDL933(ΔTir + Y490F), EDL933(ΔTir + Y519F), and EDL933(ΔTir + Y490F, Y519F) ( h). The infection results showed that EDL933(ΔTir + Y490F) and EDL933(ΔTir + Y519F) induced much higher production of Tnf, Il6 and Il12b than the EDL933(ΔTir + HA-Tir) strain in mouse primary peritoneal macrophages, and EDL933(ΔTir + Y490F, Y519F) induced even higher cytokine production ( g). Moreover, we obtained similar results in RAW264.7 cells (Supplemental Figure 6c). In addition, EDL933(ΔTir + Y490F), EDL933(ΔTir + Y519F), and EDL933(ΔTir + Y490F, Y519F) induced higher secretion of TNF, IL-6, and IL-12b than EDL933(ΔTir + HA-Tir) in the primary macrophage supernatants (Supplemental Figure 6d). Consistently, EDL933(ΔTir + Y490F, Y519F) induced much higher phosphorylation of Erk, Jnk, p38 MAPK, NF-κB p65, and IKKα/β than EDL933(ΔTir + HA-Tir) in mouse primary peritoneal macrophages (i). Thus, the Tir–SHP-1 interaction and cytokine inhibition are dependent on ITIM tyrosine phosphorylation.
ITIMs of EHEC Tir specifically inhibit intestinal immunity
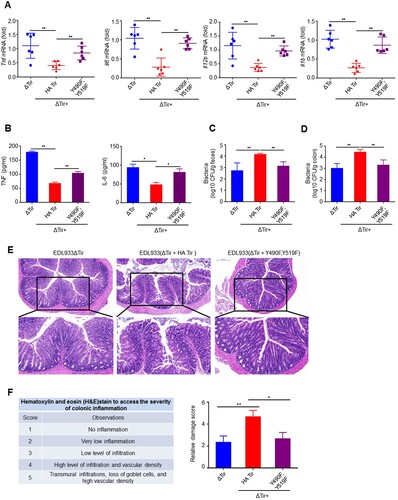
With the infection mouse model above, we found that EDL933(ΔTir + HA-Tir) induced lower Tnf, Il6, Il12b, and Il1b than EDL933ΔTir, which was nearly reversed by EDL933(ΔTir + Y490F, Y519F) in the colons (a) and spleens (Supplemental Figure 7a). In addition, EDL933(ΔTir + HA-Tir) inhibition of TNF and IL-6 secretion was rescued by EDL933(ΔTir + Y490F, Y519F) in the serum (b). In contrast, the EDL933(ΔTir + Y490F, Y519F) CFUs were significantly less than those of EDL933(ΔTir + HA-Tir) in the feces (c) and colon (d), which was comparable to EDL933ΔTir. Conversely, mice infected with EDL933ΔTir or EDL933(ΔTir + Y490F, Y519F) had greater increases in body weight than EDL933(ΔTir + HA-Tir) infected mice (Supplemental Figure 7b). Histological examination of mice infected with EDL933(ΔTir + Y490F, Y519F) revealed less extensive neutrophil inflammatory infiltrates and structural disruption of the colonic epithelium than EDL933(ΔTir + HA-Tir) infected mice (e-f). These results suggest that tyrosine phosphorylation of the ITIMs of Tir may have inhibited intestinal immunity during EDL933 infection.
Figure 6. ITIMs in EHEC Tir specifically inhibit intestinal immunity. a Quantitative RT–PCR analysis of Tnf, Il6, Il12b or Il1b mRNAs in colon from 3-4-week-old mice (n = 6 per group) infected orally with 1 × 109 CFU of EDL933ΔTir, EDL933(ΔTir + HA-Tir) or EDL933(ΔTir + Y490F, Y519F). b ELISA assay of TNF or IL-6 from mouse serum in a. c-d Bacterial load in the feces (c) or colon (d) of mice infected as in a at the indicated times. e Histopathology of colons of mice left uninfected (NI) or infected as in a. Outlined areas at top are enlarged below. Original magnification, ×10 (top) or ×20 (bottom). f Hematoxylin and eosin (H&E) stain to access the severity of colonic inflammation. Data are representative of at least two independent experiments (mean ± SEM in a–d, f). *p < 0.05 and **p < 0.01 (Student’s t-test).
Inhibition of cytokine production by EHEC Tir is mediated by SHP-1
SHP-1-deficient mice have confirmed an integral role for SHP-1 in negatively regulating cell activation to modulate the immune response [ Citation43,Citation44]. To determine the role of SHP-1 in Tir-mediated inhibition of cytokine production, we transfected RAW264.7 cells with negative control small interfering RNA (siRNA) (NC) or SHP-1-specific siRNA by electroporation (a). We infected cells with EDL933 or EDL933ΔTir and observed that the SHP-1 siRNA group infected with EDL933 induced much higher production of Tnf, Il6, Il12b, and Il1b than the NC group, whereas EDL933ΔTir induced relatively high cytokine production in both the NC and SHP-1 siRNA groups (b, supplemental 8a).
Figure 7. Inhibition of cytokine production by EHEC Tir is mediated by SHP-1. a Immunoassay of lysates of RAW264.7 cells transfected with negative control siRNA (NC) or SHP-1-specific siRNA. b Quantitative RT–PCR analysis of Tnf, Il6 or Il12b mRNAs in RAW264.7 cells transfected with siRNA and then left uninfected (Med) or infected with EDL933 or EDL933ΔTir. c Expression of SHP-1 in mouse primary peritoneal macrophages from wild-type (WT) or Ptpn6 me-v/me-v (mev/mev) mice. d Immunoassay of lysates of WT or mev/mev primary peritoneal macrophages infected with EDL933 or EDL933ΔTir for the indicated times. e Quantitative RT–PCR analysis of Tnf, Il6 or Il12b mRNAs in WT or mev/mev primary peritoneal macrophages infected for 6 h with EDL933 or EDL933ΔTir. Data are representative of at least three independent experiments (mean ± SEM in b, e). **p < 0.01 (Student’s t-test).
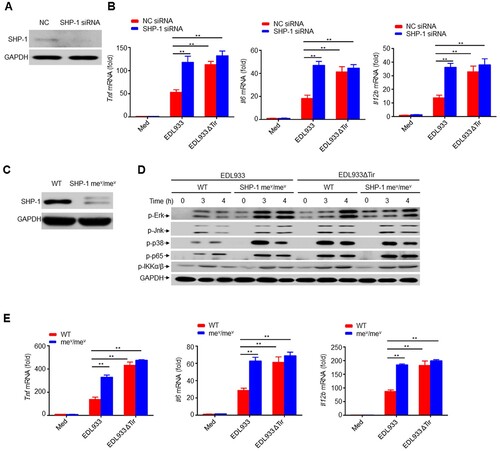
We isolated mouse primary peritoneal macrophages from viable motheaten strain (Ptpn6me-v/me-v) mice, which have low expression of SHP-1 (c), and infected mouse primary peritoneal macrophages with EDL933 or EDL933ΔTir, respectively. The SHP-1 mev/mev group infected with EDL933 had significantly higher phosphorylation levels of Erk, Jnk, p38 MAPK, NF-κB p65, and IKKα/β, as well as significantly higher production of Tnf, Il6, Il12b, and Il1b than the WT group, whereas EDL933ΔTir induced relatively high phosphorylation of these kinases and cytokine production in both WT and SHP-1 mev/mev cells (d-e, supplemental 8b). Thus, SHP-1 could be a critical regulator in Tir-mediated inhibition of cytokine production.
Discussion
The EHEC effector protein Tir, which is injected into the plasma membrane of host cells via bacterial T3SS and localized at sites of bacterial attachment [Citation25] adopts a hairpin loop conformation and serves as a receptor for the bacterial surface adhesin intimin [Citation23]. EHEC generates AE lesions to colonize the intestine, damage the epithelium and promote diarrheal illnesses [Citation21,Citation22]. These lesions are characterized by localized replacement of microvilli with organized filamentous (F-) actin ‘pedestals’ beneath intimately adherent bacteria [Citation45,Citation46]. The binding of intimin and the central extracellular domain of Tir promotes clustering of the N- and C-terminal cytoplasmic regions and initiates localized actin assembly beneath the plasma membrane of intestinal epithelial cells [Citation26]. However, the regulatory role of EHEC Tir in immune cells remains unknown. By analyzing an EHEC infection model, we uncovered a pathway by which EHEC Tir inhibited immune responses by recruiting SHP-1 in an ITIM tyrosine phosphorylation-dependent manner. SHP-1 inhibits TAK1 activity to down-regulate signal transduction and subsequent cytokine production.
Innate immune responses are achieved by the activation of several pathogen-recognition receptors (PRPs), including TLRs, retinoic acid inducible gene I (RIG-I)-like receptors (RLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs). In mammals, PRPs utilize TAK1 to activate NF-κB through myeloid differentiation factor 88 (MyD88) in macrophages [Citation47]. Although EHEC Tir is essential for the assembly of F-actin pedestals [Citation48], relatively little is known about how Tir regulates innate inflammatory responses. The data obtained in our study demonstrate that Tir inhibits EHEC-induced expression of the proinflammatory cytokines TNF and IL-6 in vitro and in vivo. Our observation of lower bacterial burdens in mice infected with EDL933 ΔTir may be due to the higher expression of TNF and IL-6. Thus, in addition to its role in pedestal formation, EHEC Tir also contributes to the pathogenicity of EHEC by inhibiting host proinflammatory responses.
TAK1 is widely accepted as a key regulator in proinflammatory cytokine signalling, which can be activated by TNF, IL-1, and TLR ligands [Citation35,Citation36]. Several threonine and serine residues in TAK1, including Thr-187, Ser-192, and Ser-412, are phosphorylated upon cytokine stimulation [Citation15,Citation40,Citation41]. We found that EHEC Tir inhibited TAK1 phosphorylation at Thr-187, Ser-192, and Ser-412. EHEC Tir enhanced the interaction between TAK1 and SHP-1, which then inhibited K63-linked polyubiquitination. These data indicate that EHEC Tir may inhibit TAK1 activation through SHP-1. However, little is known about the relationship between the phosphorylation and ubiquitination of TAK1. We found that two TAK1 point mutants, TAK1(T187A) and TAK1(S192A), decreased total and K63-linked polyubiquitination of TAK1 and that EHEC Tir down-regulated TAK1 phosphorylation at Thr-187 and Ser-192. Thus, we propose that Tir might promote SHP-1 to inhibit K63-linked TAK1 polyubiquitination by inhibiting its phosphorylation. However, the mechanism by which TAK1 phosphorylation regulates its own ubiquitination must be further elucidated.
In summary, the results of this study show that EHEC Tir interacted with the host cellular tyrosine phosphatase SHP-1 in an ITIM phosphorylation-dependent manner to inhibit proinflammatory cytokines and intestinal immunity. The association of Tir with SHP-1 facilitated the recruitment of SHP-1 to TAK1 and inhibited phosphorylation of TAK1, which is negatively related to TAK1, MAPK, and NF-κB activation. Moreover, Tir may promote SHP-1 to inhibit K63-linked TAK1 polyubiquitination by mechanically inhibiting its phosphorylation (Fig. S9). Taken together, these results not only provide a new molecular basis for the immune evasion mechanisms of pathogens, but also supply important theoretical evidence for the development of clinical strategies for the treatment of infectious diseases.
Supplemental Material
Download MS Word (29.8 MB)Acknowledgements
We thank Dr. X. Cao (Chinese Academy of Medical Sciences) for providing Ptpn6me-v/me-v mice, Dr. J. Leong (University of Massachusetts Medical School) for Tir cDNA, EDL933 and EDL933 ΔTir strains.
R.Z. performed experiments, analyzed the data and wrote the manuscript; Z.C., D.H., Y.W., Y.Z., X.Y, L.L. and H.L. performed experiments; Q.Z., Y.C. and B.G. designed this study; D.Y. designed this study, analyzed the data, wrote the manuscript and supervised the project. All authors discussed the results and commented on the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Reddick LE, Neal MA. Bacteria fighting back: How pathogens target and subvert the host innate immune system. Mol Cell. 2014;54:321–328. doi: 10.1016/j.molcel.2014.03.010
- Beutler BA. TLRs and innate immunity. Blood. 2008;113:1399–1407. doi: 10.1182/blood-2008-07-019307
- Daëron M, Jaeger S, Du Pasquier L, et al. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. 2008;224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x
- Blank U, Launay P, Benhamou M, et al. Inhibitory ITAMs as novel regulators of immunity. Immunol Rev Nov. 2009;232:59–71. doi: 10.1111/j.1600-065X.2009.00832.x
- Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000;12:361–378. doi: 10.1006/smim.2000.0223
- Neel BG, Gu H, Pao L. The ‘shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4
- Hamerman JA, Tchao NK, Lowell CA, et al. Enhanced toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6:579–586. doi: 10.1038/ni1204
- An HZ, Hou J, Zhou J, et al. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat Immunol. 2008;9:542–550. doi: 10.1038/ni.1604
- Khaled AR, Butfiloski EJ, Sobel ES, et al. Functional consequences of the SHP-1 defect in motheaten viable mice: role of NF-κB. Cell Immunol. 1998;185:49–58. doi: 10.1006/cimm.1998.1272
- Ajibade AA, Wang HY, Wang RF. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013;34:307–316. doi: 10.1016/j.it.2013.03.007
- Shirakabe K, Yamaguchi K, Shibuya H, et al. TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem. 1997;272:8141–8144. doi: 10.1074/jbc.272.13.8141
- Ninomiya-Tsuji J, Kishimoto K, Hiyama A, et al. The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465
- Lee J, Mira-Arbibe L, Ulevitch RJ. TAK1 regulates multiple protein kinase cascades activated by bacterial lipopolysaccharide. J Leukoc Biol. 2000;68:909–915.
- Shibuya H, Yamaguchi K, Shirakabe K, et al. TAB1: an activator of the TAK1 MAPKKK in TGF-β signal transduction. Science. 1996;272:1179–1182. doi: 10.1126/science.272.5265.1179
- Singhirunnusorn P, Suzuki S, Kawasaki N, et al. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-β-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–7368. doi: 10.1074/jbc.M407537200
- Ouyang C, Nie L, Gu M, et al. Transforming growth factor (TGF)-β-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit α (PKACα) and X-linked protein kinase (PRKX). J Biol Chem. 2014;289:24226–24237. doi: 10.1074/jbc.M114.559963
- Lin CC, Kuo C-T, Cheng C-Y, et al. IL-1β promotes A549 cell migration via MAPKs/AP-1 and NF-κB-dependent matrix metalloproteinase-9 expression. Cell Signal. 2009;21:1652–1662. doi: 10.1016/j.cellsig.2009.07.002
- Kim SI, Kwak JH, Zachariah M, et al. TGF-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-β1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. AJP: Renal Physiology. 2007;292:F1471–F1478.
- Chen WP, Tzeng H-J, Ku H-C, et al. Thaliporphine ameliorates cardiac depression in endotoxemic rats through attenuating TLR4 signaling in the downstream of TAK1 phosphorylation and NF-κB signaling. Naunyn Schmiedebergs Arch Pharmacol. 2010;382:441–453. doi: 10.1007/s00210-010-0562-1
- Phillips AD. Enterohaemorrhagic Escherichia coli O157:H7 target Peyer’s patches in humans and cause attaching/effacing lesions in both human and bovine intestine. Gut. 2000;47:377–381. doi: 10.1136/gut.47.3.377
- Donnenberg MS, Whittam TS. Pathogenesis and evolution of virulence in enteropathogenic and enterohemorrhagic Escherichia coli. J Clin Invest. 2001;107:539–548. doi: 10.1172/JCI12404
- Gaytán MO, Martínez-Santos VI, Soto E, et al. Type three secretion system in attaching and effacing pathogens. Front Cell Infect Microbiol ECollection. 2016;6:1–25.
- Kenny B, DeVinney R, Stein M, et al. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell. 1997;91:511–520. doi: 10.1016/S0092-8674(00)80437-7
- Oh KH, Jung S-M, Shin E, et al. Comparison of enterohemorrhagic Escherichia coli (EHEC) O157 and EHEC non-O157 isolates from patients with diarrhea in Korea. Jpn J Infect Dis. 2017;70:320–322. doi: 10.7883/yoken.JJID.2016.178
- Vingadassalom DA, Kazlauskas A, Skehan B, et al. Insulin receptor tyrosine kinase substrate links the E. coli O157:H7 actin assembly effectors Tir and EspF(U) during pedestal formation. Proc Natl Acad Sci U.S.A. 2009;106:6754–6759. doi: 10.1073/pnas.0809131106
- Campellone KG, Rankin S, Pawson T, et al. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J Cell Biol. 2004;164:407–416. doi: 10.1083/jcb.200306032
- Elizabeth AC, Meredith MC, Aman K, et al. Microbiota and pathogen proteases modulate type III secretion activity in Enterohemorrhagic Escherichia coli. mBio. 2018;9:e02204–18.
- Yan D, Quan H, Wang L, et al. Enteropathogenic Escherichia coli Tir recruits cellular SHP-2 through ITIM motifs to suppress host immune response. Cell Signal. 2013;25:1887–1894. doi: 10.1016/j.cellsig.2013.05.020
- Yan D, Wang X, Luo L, et al. Inhibition of TLR signaling by a bacterial protein containing immunoreceptor tyrosine-based inhibitory motifs. Nat Immunol. 2012;13:1063–1071. doi: 10.1038/ni.2417
- Zhuang X, Chen Z, He C, et al. Modulation of host signaling in the inflammatory response by enteropathogenic Escherichia coli virulence proteins. Cell Mol Immunol. 2017;14:237–244. doi: 10.1038/cmi.2016.52
- Laurie P, Philippe JS, Armelle P. Host cell targeting by enteropathogenic bacteria T3SS effectors. Trends Microbiol. 2018;26:266–283. doi: 10.1016/j.tim.2018.01.010
- Jens CG, Schlüter K, Carius Y, et al. Structural basis for complex formation between human IRSp53 and the translocated intimin receptor Tir of Enterohemorrhagic E. coli. Structure. 2011;19:1294–1306. doi: 10.1016/j.str.2011.06.015
- Cambier JC. Inhibitory receptors abound? Proc Natl Acad. Sci U.S.A. 1997;94:5993–5995. doi: 10.1073/pnas.94.12.5993
- Getahun A, Cambier JC. Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol Rev. 2015;268:66–73. doi: 10.1111/imr.12336
- Sakurai H, Shigemori N, Hasegawa K, et al. TGF-β-activated kinase 1 stimulates NFκ-B activation by an NFκ-inducing kinase-independent mechanism. Biochem Biophys Res Commun. 1998;243:545–549. doi: 10.1006/bbrc.1998.8124
- Sakurai H, Miyoshi H, Toriumi W, et al. Functional interactions of transforming growth factor β-activated kinase 1 with IκB kinases to stimulate NF-kappaB activation. J Biol Chem. 1999;274:10641–10648. doi: 10.1074/jbc.274.15.10641
- Broglie P, Matsumoto K, Akira S, et al. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J Biol Chem. 2010;285:2333–2339. doi: 10.1074/jbc.M109.090522
- Kajino T, Ren H, Iemura S-I, et al. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem. 2006;281:39891–39896. doi: 10.1074/jbc.M608155200
- Takaesu G, Ninomiya-Tsuji J, Kishida S, et al. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol Cell Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001
- Yu Y, Ge N, Xie M, et al. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFκB and AP-1 activation as well as IL-6 gene expression. J Biol Chem. 2008;283:24497–24505. doi: 10.1074/jbc.M802825200
- Pathak S, Borodkin VS, Albarbarawi O, et al. O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO J. 2012;31:1394–1404. doi: 10.1038/emboj.2012.8
- Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol. 2006;36:1646–1653. doi: 10.1002/eji.200636195
- Sharma Y, Bashir S, Bhardwaj P, et al. Protein tyrosine phosphatase SHP-1: resurgence as new drug target for human autoimmune disorders. Immunol Res. 2016;64:804–819. doi: 10.1007/s12026-016-8805-y
- Kruger J, Butler JR, Cherapanov V, et al. Deficiency of Src homology 2-containing phosphatase 1 results in abnormalities in murine neutrophil function: studies in motheaten mice. J Immunol. 2000;165:5847–5859. doi: 10.4049/jimmunol.165.10.5847
- Campellone KG, Leong JM. Tails of two Tirs: actin pedestal formation by enteropathogenic E. coli and enterohemorrhagic E. coli O157: H7. Curr Opin Microbiol. 2003;6:82–90. doi: 10.1016/S1369-5274(03)00005-5
- Celli J, Deng W, Finlay BB. Enteropathogenic Escherichia coli (EPEC) attachment to epithelial cells: exploiting the host cell cytoskeleton from the outside. Cell Microbiol. 2000;2:1–9. doi: 10.1046/j.1462-5822.2000.00033.x
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006
- Campellone KG. Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFU and actin pedestal assembly. FEBS J. 2010;277:2390–2402. doi: 10.1111/j.1742-4658.2010.07653.x