
ABSTRACT
The Klebsiella pneumoniae complex comprises seven K. pneumoniae-related species, including K. variicola. K. variicola is a versatile bacterium capable of colonizing different hosts such as plants, humans, insects and animals. Currently, K. variicola is gaining recognition as a cause of several human infections; nevertheless, its virulence profile is not fully characterized. The clinical significance of K. variicola infection is hidden by imprecise detection methods that underestimate its real prevalence; however, several methods have been developed to correctly identify this species. Recent studies of carbapenemase-producing and colistin-resistant strains demonstrate a potential reservoir of multidrug-resistant genes. This finding presents an imminent scenario for spreading antimicrobial resistant genes among close relatives and, more concerningly, in clinical and environmental settings. Since K. variicola was identified as a novel bacterial species, different research groups have contributed findings elucidating this pathogen; however, important details about its epidemiology, pathogenesis and ecology are still missing. This review highlights the most significant aspects of K. variicola, discussing its different phenotypes, mechanisms of resistance, and virulence traits, as well as the types of infections associated with this pathogen.
Introduction
The Klebsiella pneumoniae complex is a member of the Klebsiella genus of the family Enterobacteriaceae. Recently, the taxonomy of the K. pneumoniae complex has expanded. It comprises K. pneumoniae, K. quasipneumoniae subsp. quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, K. variicola subsp. variicola (for practical purposes, we refer to this subspecies as K. variicola), K. variicola subsp. tropicalensis and K. africanensis bacterial species. K. quasivariicola is also included, but remains to be validly published [Citation1,Citation2]. All the members of this complex possess overlapping biochemical and phenotypic features; thus, classifying them by traditional microbiological methods is not practicable [Citation3–5]. Usually, all taxa of the K. pneumoniae complex are misassigned as K. pneumoniae, the major species within the complex. Different alternatives have been proposed to solve this inappropriate identification. The first alternatives developed were based on PCR [Citation6,Citation7]; however, the phylogenetic analysis of housekeeping genes has been routinely used and is highly accurate [Citation5,Citation8,Citation9]. Currently, MALDI-TOF MS (with a properly updated database) and gene and genome sequencing are feasible means of differentiating the bacterial species that constitute the K. pneumoniae complex [Citation2,Citation10,Citation11].
K. variicola is a Gram-negative, facultative anaerobic, and nonmotile bacillus. It forms circular, convex and mucoid colonies and grows at approximately 11–41°C [Citation12]. Infections caused by K. variicola have been reported in humans worldwide [Citation6,Citation8,Citation11,Citation13–15] and less frequently in wild and farm animal infections [Citation16,Citation17]. Additionally, K. variicola is frequently isolated from a wide range of plant ecosystems, playing a role in nitrogen fixation and plant growth promotion [Citation12,Citation18–23]. The inaccurate identification of the members of the K. pneumoniae complex has limited the study of K. variicola, leaving gaps in knowledge and clinical implications within health care systems. Therefore, the aim of this review is to discuss general aspects of K. variicola and highlight the most significant aspects of K. variicola as an emerging human pathogen with increasing antimicrobial resistance gene and virulence profiles.
Description of Klebsiella variicola as a novel bacterial species
Brisse et al. (2001) identified genetic diversity within K. pneumoniae clinical isolates. They assessed these isolates through a phylogenetic analysis of the parC and gyrA genes [Citation4]. The analysis revealed the existence of three distinct K. pneumoniae phylogroups named KpI, KpII and KpIII, with KpI being the most prevalent. In 2004, based on genetic and biochemical assays, K. variicola was proposed as a novel bacterial species recovered from plants and clinical isolates that was initially identified as K. pneumoniae. Likewise, it was observed that the phylogroup KpIII corresponded to K. variicola [Citation5]. The isolates identified as K. pneumoniae (using standard biochemical techniques) were subjected to various tests, such as adonitol fermentation, nitrogen fixation through acetylene reduction, phylogenetic analysis of housekeeping genes and DNA–DNA hybridization. The data obtained demonstrated the existence of a new bacterial species named “variicola”, which comes from the Latin word va.ri.i’co.la., meaning “inhabitant of different places”[Citation5]. Later, through phylogenetic analysis, Brisse et al. (2014) described that the phylogroup KpII corresponds to K. quasipneumoniae. Phylogenomic differences of these phylogroups that support them as distinct and separately evolving population species were subsequently confirmed [Citation3].
The phenotypic and biochemical characteristics of the K. pneumoniae complex are extremely similar under standard microbiological conditions [Citation12]. Like all members of the K. pneumoniae complex, K. variicola is positive for urease, ortho-nitrophenyl-β-galactoside (ONPG), the Voges-Proskauer test, and lysine decarboxylase but negative for indole and ornithine decarboxylase. Unlike the rest of the K. pneumoniae complex, both subspecies of K. variicola (K. variicola subsp. variicola and K. variicola subsp. tropicalensis) metabolize tricarballylic acid, L-galactonic acid-γ-lactone, L-sorbose, 5-keto-D-gluconic acid, 4-hydroxyl-L-proline and D-arabitol; likewise, the inability to ferment N-acetyl-neuraminic acid is also a particular trait of both subspecies. Moreover, some characteristics that differentiate K. variicola subsp. variicola from K. variicola subsp. tropicalensis are the ability to metabolize mono-methyl succinate, D-lactic acid methyl ester and 3-O-(ß-D-galactopyranosyl)-D-arabinose and the inability to metabolize D-psicose [Citation24,Citation25]. The assimilation of 5-keto-D-gluconate is supported by the presence of the idnO gene exclusively in both subspecies of K. variicola, and as expected, this gene is absent in K. pneumoniae and K. quasipneumoniae species [Citation26]. Great efforts have been made to gain a deeper understanding of the biochemical characteristics of the K. pneumoniae complex. This information may contribute to a more accurate differentiation of K. variicola.
K. variicola misidentification: a problem being resolved?
The identification of K. variicola is not routinely sought in clinical microbiology laboratories, and the misidentification of K. variicola as K. pneumoniae was documented several years ago [Citation27,Citation28]. Currently, the identification of new bacterial species and subspecies within the K. pneumoniae complex, such as K. quasipneumoniae, K. variicola subs tropicalensis, K. africanensis and K. quasivariicola, requires updating the developed protocols to increase the accuracy of identifying new species in the K. pneumoniae complex. Fortunately, molecular, genomic and proteomic methods are now available (). With the use of these different tools, it will be easier to better differentiate the species within the K. pneumoniae complex. An accurate identification of clinical and environmental isolates will help to understand its epidemiology. The available methods are described below.
Figure 1. Representation of the available methods that allow differentiating the K. pneumoniae complex. The black colour means that the indicated method distinguishes the species of the K. pneumoniae complex, and the grey colour implies the opposite. Microbiological methods refer to standard biochemical procedures. PCR refers to molecular methods based on the amplification of chromosomal β-lactamases or unique genes assembled in a multiplex PCR, or single PCR such as yggE. The phylogenetic analysis corresponds to the most commonly used gene, rpoB. KlebSeq is a sequencing tool for screening and epidemiological surveillance of the Klebsiella genus. Single nucleotide polymorphisms (SNPs) have been used for the differentiation of some species of the K. pneumoniae complex, but they are definitely feasible for all species of the complex. The average nucleotide identity (ANI) is a genomic tool used for species differentiation and is highly recommended for this purpose. With the recent update of the proteomic profile of the K. pneumoniae complex, it is possible to differentiate the species that constitute the K. pneumoniae complex by using MALDI-TOF. The Bruker reference library version 6.0.0.0 includes the reference spectra for proper identification of K. variicola from K. pneumoniae but not for the rest of the members of the K. pneumoniae complex. The asterisk symbol behind the species name means that the subspecies are included, for instance K. variicola subsp. variicola, K. variicola subsp. tropicalensis, K. quasipneumoniae subsp. quasipneumoniae and K. quasipneumoniae subsp. similipneumoniae.
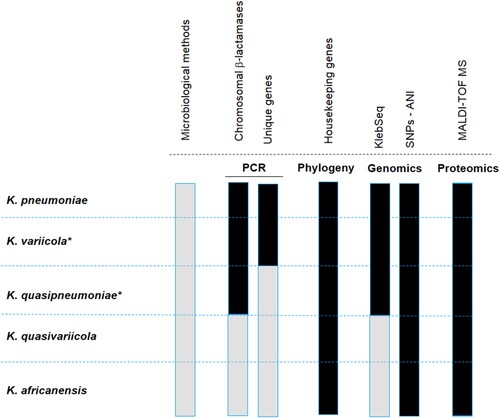
Microbiological methods
Automated systems such as Vitek 2XL, Phoenix, and MicroScan are widely used in microbiology and clinical laboratories for the identification of both Gram-negative and Gram-positive bacteria. These microbiological methods are based on colorimetric changes in miniaturized substrates. Because automated systems rely on the general biochemical characteristics of the K. pneumoniae complex, it is not possible to differentiate K. variicola or any species of the complex. Thus, automated systems will identify all isolates as K. pneumoniae ().
Molecular methods: PCR-based on chromosomal β-lactamase genes
Previous analyses have revealed that the chromosomal presence of class A β-lactamase is a distinguishing characteristic among bacteria in the K. pneumoniae complex [Citation20,Citation29]. A multiplex PCR system based on the amplification of chromosomal β-lactamase was proposed to identify K. pneumoniae (blaSHV), K. quasipneumoniae (blaOKP) and K. variicola (blaLEN) [Citation30] (). This multiplex PCR system amplifies the blagenes along with their flanking region (deoR) and a gene coding for an ATPase, which is a conserved region within the chromosome. Although the authors included E. coli strains with and without a plasmid-borne blaSHV as controls, clinical isolates of K. variicola with plasmid-borne blaSHV have been reported [Citation7,Citation11,Citation31]. Therefore, cross-reactivity with another member of the K. pneumoniae complex that encodes mainly the broadly disseminated plasmid-borne SHV β-lactamase family should be considered. In addition, the fact that K. variicola harboured the OKP β-lactamase instead of the LEN β-lactamase makes it difficult to identify the members of the K. pneumoniae complex when applying gene-based systems based on β-lactamase proteins [Citation11].
PCR-based on unique genes
Yasuhara-Bell et al. (2015) designed a diagnostic molecular tool based on loop-mediated isothermal amplification (LAMP) for detecting K. variicola and K. oxytoca isolates from infected ironwood trees (). This assay was accomplished by whole-genome comparisons to obtain specific proteins for both bacterial species. Two proteins were selected: i) hemolysin-coregulated protein 1 (Hcp1) unique to K. variicola and ii) UGH, a glucuronyl hydrolase unique to K. oxytoca [Citation32]. Practically simultaneously, Garza-Ramos et al. (2015) [Citation7] and Berry et al. (2015) [Citation6] proposed the first efficient molecular methods for proper differentiation of K. variicola from K. pneumoniae. Berry et al. (2015) described a case of a bloodstream infection (BSI) caused by a “not detected” bacterium when using the Verigene system. The isolate was identified as K. pneumoniae through the Vitek 2XL system and was subsequently identified as K. variicola by phylogenetic analysis of the yggE gene [Citation6] (). Later, a collection of K. variicola isolates was identified by a yggE PCR-RFLP assay and MALDI-TOF MS. Since yggE PCR-RFLP had 94.6% coincidence with MALDI-TOF results, this finding suggests that the yggE gene may be suitable for K. variicola identification [Citation14]. Garza-Ramos et al. (2015) developed a multiplex PCR probe system based on comparative genomics of K. variicola and K. pneumoniae genomes to identify shared unique proteins of each bacterial species (). This multiplex PCR (M-PCR-1) consists of the amplification of three different amplicons: a phosphohydrolase (888 bp) for K. pneumoniae, a phosphoglycerate mutase (449 bp) for K. variicola, and the mtnC gene (340 bp) used as a molecular marker for both species. This probe system was used for screening isolates of K. variicola among 1,060 K. pneumoniae clinical isolates obtained from several Mexican hospitals. The authors found that K. variicola had a prevalence of 2.1%. Half of the isolates identified corresponded to multidrug-resistant isolates, which were determined to be extended-spectrum β-lactamase (ESBL)-producers. All K. variicola isolates were validated by phylogenetic analysis of rpoB genes, regarding which we consider that the multiplex-PCR probe system could identify K. variicola with high accuracy [Citation30]. This multiplex PCR (M-PCR-1) has been recently been used in screenings allowing the identification of K. variicola isolates in plants as well as in hospital- and community-acquired infections [Citation13,Citation23,Citation33,Citation34]. Even a K. quasipneumoniae clinical isolate (with a hypermucoviscous phenotype) could be indirectly determined because when an isolate is determined biochemically as K. pneumoniae (MicroScan Walkaway system in this case) but negative to probes used in the M-PCR-1, later it was confirmed that the clinical isolate corresponded to K. quasipneumoniae [Citation35].
The molecular methods are based on phylogenetic analysis of 16S rRNA and rpoB genes, with this last gene being the most widely used to properly identify isolates of K. variicola [Citation8,Citation36] (). Likewise, the use of the rpoB gene has been reported to be more accurate at Klebsiella species identification [Citation37]. Some strains of K. variicola have been identified from several sources through rpoB phylogenetic analysis, including animals such as dogs, birds, monkeys, and cows (mastitis) and plants such as coriander (food supply) [Citation16,Citation17,Citation38]. Moreover, a clinical strain of K. variicola causing fatal sepsis was initially misidentified by the MicroScan platform and later identified correctly through rpoB phylogenetic analysis [Citation15]. The phylogenetic analysis of concatenate housekeeping genes has been proposed [Citation3,Citation7], although it has not been tested with the new species of the K. pneumoniae complex.
Genomic-based and automated methods
The development of genomics has grown with next-generation sequencing platforms allowing the whole-genome sequencing (WGS) of bacterial pathogens. WGS has gained applications in health care diagnostics for screening and surveillance of bacterial pathogens, outbreak detection, and source tracing of major pathogens [Citation39]. Although the WGS approach is not currently cost-effective worldwide, other sequencing alternatives have been proposed not only for detecting but also for characterizing pathogens. The KlebSeq sequencing tool is a promising tool for screening and epidemiological surveillance of the Klebsiella genus, which includes the identification of some members of the K. pneumoniae complex (K. pneumoniae, K. variicola and K. quasipneumoniae) without a culturing step [Citation10] (). Hence, the species identification by means of KlebSeq is based on sequencing of target genes. In the case of K. variicola and K. quasipneumoniae, KlebSeq uses single nucleotide polymorphism (SNP) amplicon analysis for a specific gene. Although KlebSeq was designed for characterizing K. pneumoniae isolates, updating this platform to include the recently described Klebsiella species and information such as capsule type, antibiotic resistance and virulence genes for all the members that constitute the K. pneumoniae complex would contribute to understanding the epidemiological background of these species.
The use of SNP-base analysis for Klebsiella species identification was used previously by Holt et al. (2915) [Citation1]. They identified 175,120 SNPs within the core genome of 328 Klebsiella genomes with these SNPs a phylogenetic analysis revealed a split-network three clearly separated groups corresponding to K. pneumoniae, K. variicola and K. quasipneumoniae. Another option is the use of average nucleotide identity (ANI), a powerful tool for resolving genomic relatedness between strains (). ANI is calculated from pairwise comparisons of all sequences shared between two genomes [Citation40]. While ANI is not a K. variicola-specific genomic tool, several studies have reported its utility in resolving species differentiation. Clearly, misidentification at the microbiological level drives an incorrect genome classification, and several examples of misclassification have been described [Citation9,Citation26]. The WGS approaches are useful in pathogen and outbreak detection [Citation39]. There are several examples involving bacterial pathogens; however, for the Klebsiella pneumoniae complex, a clear example is the epidemiological study of 1,777 sequenced genomes; this approach allowed the identification of both K. variicola and K. quasipneumoniae ESBL- and carbapenemase-producing isolates [Citation11].
MALDI-TOF MS is a fast and cost-effective technique [Citation41,Citation42], while the Bruker microorganism reference library version 4.0.0.0 does not include a K. variicola profile [Citation11]. Moreover, the Bruker library version 6.0.0.0 includes reference spectra for K. variicola and K. pneumoniae. Recently specific biomarkers for all bacterial species of the K. pneumoniae complex were reported using MALDI-TOF MS [Citation2] (). The authors found different combinations of peaks in the spectra that highly correlate with the K. pneumoniae complex, mainly corresponding to ribosomal proteins. In the case of K. variicola, its identification is based on the L31 ribosomal protein. Thus, there is opportunity for updating MALDI-TOF databases to correctly differentiate the bacterial species of the K. pneumoniae complex and exploit the potential of MALDI-TOF platforms for discriminating this species [Citation43].
Several methods are available for correctly differentiating K. variicola within the K. pneumoniae complex; however, the best approaches are MALDI-TOF MS (with a properly updated database), PCR systems based on single or multiple genes (yggE or multiplex PCR probes) and ANI using the sequenced genome. Unless these methods are routinely adopted in clinical microbiology laboratories, misidentification will continue to be a problem.
Environmental distribution of K. variicola
In addition to the clinical environment where K. variicola has been identified, this bacterial species is also found in a wide diversity of natural niches. The mutualistic relationship between leaf-cutter ant colonies and fungus was the first example. The successful colony growth of leaf-cutter ants is supported by the symbiotic nitrogen fixation of K. variicola, suggesting that colonies obtain a substantial proportion of their nitrogen requirements from this bacterial association [Citation22]. Likewise, the misidentified K. variicola KP5-1 strain was isolated from the microbiota of wild southern green stink bugs (Nezara viridula). The potential of this insect as a vector of cotton disease-causing agents is well known. The opportunistic K. variicola has been documented to cause appreciable boll damage [Citation44]. In China, K. variicola KV321 was isolated from rhizosphere soil of Pisolithus tinctorius-Eucalyptus mycorrhiza. The isolate exhibited high nitrogen-fixation ability, and the antibiotic susceptibility tests demonstrated resistance to polymyxin, penicillin G, tetracycline, and erythromycin [Citation45]. Nitrogen cycling and diversity of the organisms that carry out nitrogen transformations in subterranean habitats is poorly studied. Some of the samples of bacterial mats collected from lava cave walls were identified as nitrogen-fixating K. pneumoniae by the presence of nifH genes. Analysis of reported nifH sequences revealed a possible misidentification of these strains, and the results revealed a K. variicola-like subset of the nitrogen-fixating bacterial community in the subterranean habitats [Citation46].
The potential capacities of K. variicola to be involved in or produce diseases are not limited only to wildlife animals. Bovine mastitis is the condition where the animal displays physical symptoms in the udder, such as swelling, heat, hardness, redness, pain and milk exhibiting a watery appearance, flakes, clots, or pus. The diseased cow presents a decrease in milk yield that depends on the specific pathogen causing the infection, where K. pneumoniae is one of the primarily microbial agents responsible for environment-derived mastitis. In Newfoundland, Canada, 4% of bovine mastitis cases from 11 farms were identified as being caused by K. variicola. The pathogenicity of K. variicola as an infectious agent in bovines is not well known [Citation17]. Diseases produced by K. variicola are widely extended to different organisms, such as ironwood (Casuarina equisetifolia subsp. equisetifolia), a nitrogen-fixing tree of considerable social, economic and environmental importance in zones of Asia, the Pacific, Africa, and Central America. The decline in ironwood was first reported in Guam in 2002 and associated with wetwood symptoms and is now affecting thousands of trees and the ecosystem. Bacterial samples obtained from diseased ironwood trees were identified as containing the common pathogen Ralstonia solanacearum associated with K. variicola and Klebsiella oxytoca. Causative agents of wetwood in the ironwood tree decline worldwide have not been completely determined, but scientists attribute the disease to a complex association between R. solanacearum, K variicola and K. oxytoca [Citation47].
Likewise, K. variicola can be found in several associations with its environment, such as contaminated soil, rivers, and wastewater [Citation19,Citation36,Citation48,Citation49]. Due to its potential for use in industrial processes, K. variicola has been used for the development of biotechnological and environmental protections, such as a source of renewable energy, fermentation and energy biocatalysts, waste degradation, wastewater treatment, bioremediation of pollutants and biodegradation [Citation19,Citation36,Citation48,Citation50,Citation51].
There is clear evidence that plants are a natural reservoir of K. variicola, and in some cases, this niche is shared with K. pneumoniae [Citation23]; however, this aspect is poorly studied. Recently, the term “phytonosis” was proposed in order to describe human infections caused by plant-borne bacteria, parallel to the term zoonosis for animal-borne bacterial pathogens [Citation20]. Later, phytonosis was described more as a case of kingdom-crossing bacteria [Citation52]. This term was used for the K. variicola X39 clinical isolate, which had the capacity to colonize maize roots, stems and leaves in a few days. This dual capacity (human infections and plant colonization) of K. variicola was determined previously by in vitro assays of plant growth promoting mechanisms, which were shared among plant and human isolates [Citation20]. Today, K. variicola can be considered a bacterial species that can infect humans as a pathogen and colonize plants as an endophyte and in few cases as pathogen.
Emergence of K. variicola as a human pathogen
Historically, K. pneumoniae has been the underlying cause of a wide range of healthcare-associated infections (HCaIs) and community-acquired infections (CAIs), with an increase in morbidity and mortality [Citation11,Citation53,Citation54]. K. variicola, as well as K. pneumoniae, is an opportunistic pathogen responsible for infections such as BSIs, respiratory tract infections and urinary tract infections (UTIs) [Citation1,Citation8,Citation11,Citation14,Citation31]. As previously mentioned, most of the cases in which K. variicola was the cause of the infection were first identified by biochemical testing as being caused by K. pneumoniae [Citation6,Citation11,Citation13,Citation15,Citation29,Citation31,Citation33].
BSIs represent a major challenge in health care systems, and timely pathogen detection is crucial for effective antibiotic therapy [Citation55]. Among the members of the K. pneumoniae complex, K. pneumoniae is the leading cause of BSI; however, patients diagnosed with a BSI caused by K. variicola have a high mortality rate [Citation8]. The first case of K. variicola BSI occurred as part of a pediatric outbreak. The BSI was caused by multidrug-resistant and ESBL-producing K. variicola clones; however, the clinical outcome was unknown [Citation7,Citation31]. Later, a report of 139 BSIs in a 30-day mortality retrospective study was described. K. variicola had the highest mortality rate (29.4%, 10/34 cases), in contrast with a 13.5% (13/96 cases) mortality rate associated with K. pneumoniae infections. Intriguingly, these isolates do not contain additional known virulence factors that could explain the mortality. In this study, K. variicola was isolated more frequently (24.4%, 34/139) than in other studies [Citation8]. In addition, several other reports have clearly demonstrated that K. variicola is associated with BSI [Citation6,Citation17,Citation56,Citation57]. Recently, an outbreak of neonatal sepsis caused by K. variicola was described by Farzana et al. (2018). They undertook an integral study of isolates causing sepsis in neonates and found a significant mortality attributable to K. variicola. This clone possessed virulence genes typically found in any other K. pneumoniae strain and, most importantly, the acquired antimicrobial resistance genes NDM-1 and CTX-M-15 [Citation56].
K. variicola has also been associated with infections in immunocompromised individuals. Some comorbidities, such as systemic lupus erythematous, cancer, diabetes mellitus, hepatobiliary diseases, solid organ transplantation and alcoholism, have been reported in several studies [Citation6,Citation15,Citation58,Citation59]. Berry et al. (2015) described a case of BSI caused by K. variicola in a female patient with systematic lupus erythematosus. In Japan, fatal sepsis was attributable to K. variicola (originally misidentified as K. pneumoniae). The patient died despite the immediate administration of antibiotics to which K. variicola was susceptible [Citation15]. Another report in which K. variicola was isolated from blood samples was described by Ledeboer et al. (2015). They reported that 25/26 blood cultures first reported as K. pneumoniae were subsequently identified as K. variicola. These isolates were collected in different clinical centres to validate a diagnostic tool specific for Gram-negative bacteria causing BSI [Citation6,Citation57].
K. variicola and K. pneumoniae can infect the same patient [Citation13]. In Mexico, an interesting case occurred in which a patient was infected by susceptible and carbapenem-producing K. pneumoniae at different time intervals. The susceptible isolates were recovered from a BSI and subsequently identified both molecularly and phylogenetically as K. variicola. Both isolates had different phenotypes and genotypes observed by pulsed-field gel electrophoresis (PFGE) analysis and displayed different plasmid profiles.
UTIs represent an important public health problem that is accompanied by a significant economic burden. UTIs are primarily caused by Gram-negative bacteria; K. pneumoniae is the second-most prevalent pathogen causing UTIs [Citation60]. Given the misidentification problem, K. variicola is an underrecognized pathogen capable of causing UTIs. Recently, a retrospective study carried out in a collection of K. variicola clinical isolates illustrated that 70% (39/56) of the K. variicola isolates were recovered from UTIs. From these data, the authors demonstrated the potential of some strains to cause UTIs using a murine model. In addition, the uropathogenicity of K. variicola was associated with novel usher fimbrial proteins and differential expression of type 1 fimbrial proteins [Citation14].
In health care systems, K. variicola produces a minor number of infections compared to those of K. pneumoniae; however, infections caused by K. variicola could be as severe as K pneumoniae infections [Citation8,Citation11]. Usually, K. variicola isolates display lower antibiotic resistance rates than K. pneumoniae [Citation7,Citation13]; however, this fact is not necessarily associated with a better treatment response [Citation6,Citation15].
Emergence of multidrug-resistant strains
K. variicola is intrinsically resistant to ampicillin due to the presence of the chromosomal β-lactamase LEN; however, it is susceptible to most antibiotic classes [Citation7,Citation11,Citation14,Citation20,Citation29]. This pattern has changed over time due to an increase in the number of reports of multidrug-resistant K. variicola isolates () [Citation7,Citation10,Citation14,Citation31,Citation61–66]. Likewise, reports of ESBL- and carbapenemase-producing K. variicola isolates have been increasing even though some strains were not isolated from clinical settings, which highlights a key role of the environment as reservoirs of antimicrobial resistance genes () [Citation38,Citation67].
Figure 2. Map of reported multidrug-resistant (MDR) and hypervirulent K. variicola (hvKv) isolates from different isolation sources. The red circle indicates clinical isolates, the green circle refers to K. variicola plant isolates and the blue circle represents environmental isolates. The numerals between parentheses indicate the number of isolates reported for both MDR and hvKv.
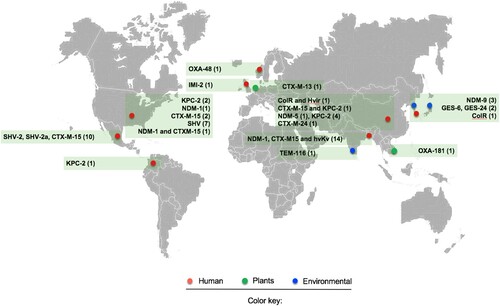
The first case reported in a clinical setting goes back to a pediatric hospital outbreak that occurred in 1996 in Mexico. The isolates were positive for ESBL of the SHV-type, for which the β-lactamase gene was located on a conjugative plasmid [Citation7,Citation31]. Afterward, an IMI-2 carbapenemase-producing K. variicola strain was obtained from a soft tissue infection of a patient in the intensive therapy unit in the UK. The strain was resistant to ertapenem, meropenem and imipenem (MIC ≥32 mg/L) but susceptible to cefotaxime and ceftazidime (MIC ≥0.25 mg/L). The IMI-2 carbapenemase was located on a transferable plasmid with a size of 77,843 bp and from the IncFIIγ family; however, this plasmid did not encode any other known resistance genes. In silico analysis revealed high similarities with other published IncFIIγ plasmids, of which many, such as the NDM-, IMI-, IMP- or KPC-type families, also harboured a carbapenemase gene () [Citation62].
More recently, Long et al. (2017) demonstrated that K. pneumoniae, K quasipneumoniae and K. variicola share chromosomal and mobile genes, which encode virulence factors and antimicrobial resistance genes. In this study, 15/1,777 and 13/1,777 of clinical isolates were identified as K. quasipneumoniae and K. variicola, respectively. K. variicola isolates were identified as ESBL-producing (9/13) or carbapenemase-producing (3/13) isolates. The ESBLs identified were SHV-type in 7/13 (SHV-5, −12, -30 alleles) isolates and CTX-M-15 in 2/13 isolates. The carbapenemases identified were KPC-2 in 2/13 isolates and NDM-1 in 1/13 isolates. One K. variicola isolate contained both the CTX-M-15 and NDM-1 genes [Citation11]. A recent report of genome sequence-based analysis of carbapenemase-producing isolates among the K. pneumoniae complex was described [Citation64]. The authors reported the presence of KPC-2 (4/5) and NDM-5 (1/5) in clinical isolates of K. variicola; this report is the first description of K. variicola producing the NDM-5 allele. The analysis of the distribution of antimicrobial resistant genes by means of genomic approaches has revealed the presence of an OXA-48-producing isolate recovered from a hospitalized patient in Norway () [Citation68].
Colistin resistance mediated by chromosomal mechanisms in K. variicola was recently described [Citation63]. Joo E et al. (2018) analyzed fecal samples from healthy Korean adults to evaluate fecal carriage of antibiotic-resistant pathogenic Enterobacteriaceae. A total of 1,417 fecal samples were analyzed, 4.5% (64/1417) from which Gram-negative isolates were obtained, and chief among them (14%, 9/64) were Klebsiella spp. The distribution of species within these isolates was 44.4% (4/9) identified as K. pneumoniae, 33.3% (3/9) identified as K. oxytoca and 22.2% (2/9) identified as K. variicola. One K. variicola isolate displayed colistin resistance (MIC 32 mg/L) without the presence of the mcr-1 gene, and because of this result, the authors considered a chromosomal mechanism; however, no further analysis was performed. Later, Lu et al. (2018) described the first report of hypervirulence associated with colistin resistance (see detailed information below) [Citation66]. Colistin resistance was mediated by chromosomal modification of a two-component regulatory system (PhoP-PhoQ). The substitution of D150G in PhoP, which is known to mediate colistin resistance, was found () [Citation69].
With respect to environmental settings, Zurfluh et al. (2015) reported the first detection of K. variicola harbouring OXA-181 carbapenemase in fresh vegetables (coriander) imported from Asia to Switzerland [Citation38]. The OXA-181 carbapenemase gene was encoded on an IncX3-type plasmid of 51,480 bp and flanked by two IS26-like elements without the capacity of transference. In addition, the emergence of K. variicola producing NDM-9 was described in a South Korean urban river; the NDM-9 gene was located on a 108-kb IncFIII(Y) plasmid, and a mercury resistance operon was located upstream of NDM-9 flanked by IS26-TnAS3 and IS15, IS26 and IS15D1 [Citation67]. Moreover, the presence of GES-6 and GES-24 in wastewater samples has also been reported [Citation70]. Interestingly, the genetic context of these carbapenemase genes was found in two distinct integrons, In1439, which contains the array blaGES-24-aacA4, and In1442, which contains blaGES-6-aacA4-blaOXA-17 (). These data highlight the existence of possible routes for the dissemination of carbapenemase-producing K. variicola in the environment through food and water [Citation38]. Dissemination of carbapenemases is accompanied by other antibiotic resistance genes within highly transferable mobile genetic elements that enhance the possibility of propagation among microorganisms in the environment.
Plasmids play an important role in the dissemination of antimicrobial resistance and virulence genes [Citation71]. K. variicola shares a similar distribution of plasmids with the other members of the K. pneumoniae complex, suggesting that horizontal gene transfers between these members occur and favour the spread of both antimicrobial resistance and virulence genes. Particularly, the FIBk, FIIk, and FII replicon types were found mostly in both K. variicola and K. quasipneumoniae isolates [Citation11].
The genomic studies described above have contributed to understanding the distribution of antimicrobial resistant genes in K. variicola. The recent acquisition of antimicrobial resistance genes highlights the possibility of horizontal gene transfer as the main mechanism in the adaptation of K. variicola. These studies demonstrate the importance of precise identification since this bacterial species is becoming a concern because of its potential to acquire and disseminate antimicrobial resistance genes in clinical and environmental settings. The map presented in is likely an underestimate of resistant K. variicola infections, as the majority are still misclassified in clinical laboratories.
Virulence and pathogenicity traits
Most of the virulence factors displayed in K. variicola correspond to factors described previously in K. pneumoniae. K. variicola exhibits four major virulence factors: capsule, lipopolysaccharide (LPS), siderophores and fimbriae. It has been suggested that K. variicola can be as virulent as K. pneumoniae strains [Citation8,Citation11]. The first study that analyzed the virulence factors and determined the pathogenicity of K. variicola was performed in a single strain (K. variicola 342) obtained from maize [Citation18]. This isolate was initially misidentified as K. pneumoniae [Citation7,Citation26]. The authors proved that K. variicola 342 was able to produce UTIs and lung infections in an animal model. Likewise, the authors examined the presence or absence of virulence factors identified in previous studies by signature-tagged mutagenesis or IVET approaches using K. pneumoniae strains. They found the genes wecA, msrA, and orfKPK-3791 and a type IV transfer system to be present in the K. variicola 342 genome but absent from the multidrug-resistant strain K. pneumoniae MGH78578. Nonetheless, these genes were not fully characterized [Citation18].
Screening of virulence-associated genes was carried out in sets of K. variicola genomes in independent works [Citation14,Citation20,Citation72]. As expected, the virulence genes that were ubiquitous among K. variicola were those involved in type 3 and 1 fimbriae biosynthesis (mrk, fim), metabolism of urea (ureA), biosynthesis of the core lipopolysaccharide (wabG) and iron uptake systems, primarily the enterobactin (entB) and KfuABC iron systems. The presence of the aerobactin siderophore receptor (iutA) was distributed among all K. variicola genomes, but no genes encoding the proteins responsible for the biosynthesis of the siderophore itself were found (iucABCD cluster) [Citation20]. Other virulence genes found in a minority of genomes were glycerate pathway genes, allantoin utilization genes and the iron uptake systems yersiniabactin (ybtAESTX cluster) and salmochelin (iroN) [Citation14,Citation20]. The main virulence genes found in K. variicola are chromosomally located; however, hypervirulent strains of K. variicola harbour virulence genes in large plasmids, such as the siderophores aerobactin, salmochelin, and yersiniabactin and the regulators of the mucoid phenotype rmpA and rmpA2 (see detailed information below) [Citation66].
Interestingly, recent findings about K. variicola uropathogenicity have demonstrated significant genetic variability in the type 1 fimbriae system that changes its expression, and nine novel fimbrial usher proteins (Kva and Kvi families) distributed among distinct K. variicola clades are involved in their role in uropathogenicity [Citation14]. Martinez-Romero et al. (2017) [Citation20] also described a new fimbrial protein (FimV) found in the set of unique proteins of the K. variicola pangenome. FimV was distributed among K. variicola genomes except for the BIDMC90, KTE92 and B1 genomes. Its role in pathogenicity is unknown.
K. variicola has a heterogeneous distribution of canonical K. pneumoniae virulence-associated genes, but in most cases, they are rarely found in K. variicola except for those classical virulence factors (capsule, adhesins and siderophores). K. variicola possesses its own virulome, which is not completely determined, as is the case for the UTI-associated virulence genes that were elucidated [Citation14]. However, it is unclear whether specific virulence mechanisms are linked to severe infections, mainly BSIs, which are the most common infections produced by K. variicola. It is likely that there are other novel virulence genes responsible for this type of infection [Citation8,Citation56]. Like that of K. pneumoniae and K. quasipneumoniae, the genome of K. variicola is considered an open genome, which implies that the organism has the capacity to continue incorporating genes that allow it to adapt to different environments by conferring resistance to antibiotics to which it previously was susceptible, as well as expanding its pathogenicity by incorporating virulence factors [Citation1].
Classical, hypermucoviscous and hypervirulent strains
Currently, there are two well-recognized K. pneumoniae phenotypes: classic (cKpn) and hypervirulent (hvKpn) () [Citation73]. cKpn strains occur mainly in hospitals and long-term care facilities. Most classic strains are resistant to a wide range of antibiotic classes, thus clinical management is challenging. Otherwise, hvKpn differs from classical strains due to its capacity to cause infections in healthy and immunocompromised populations in the community [Citation74]. summarizes the set of genetic and phenotypic traits that define the cKpn and hvKpn variants. The increased siderophore production (mainly aerobactin (iucA) and salmochelin (iroB)), capsule overproduction (hypermucoviscous phenotype) driven by rmpA and/or rmpA2 and the presence of new proposed biomarkers such as peg-344 gene are associated with severe disease or death in murine model assessments [Citation74]. Both K. pneumoniae variants are supported by genomic analysis; thus, it is clear that cKpn and hvKpn form two well-separated lineages [Citation75,Citation76]. Moreover, the last few years have seen increasing reports of hypermucoviscous K. pneumoniae (hmvKpn) negative for the rmpA/rmpA2 genes ()[Citation73,Citation77,Citation78], but these bacteria are poorly characterized, putting into the question the key role of the hypermucoviscous phenotype without a hypervirulence background.
Table 1. Genetic and phenotypic traits of K. variicola and K. pneumoniae distinct phenotypes.
Similar to the phenotypes observed in K. pneumoniae, three distinct phenotypes may be displayed by K. variicola: classic (cKv), hypermucoviscous (hmvKv) and hypervirulent (hvKv) (). This classification is based on reports in the literature rather than genomic traits; however, with the increasing number of K. variicola genomes, it would be better to have a deep understanding of the correlation between phenotypic and genomic traits. We defined i) cKv as both multidrug-resistant and susceptible isolates that lacked excessive capsule production (nonhypermucoviscous); ii) hmvKv as one isolate that was positive for the string test (hypermucoviscous) however, unlike hypervirulent K. pneumoniae, this isolate was negative for the rmpA/rmpA2 genes; and iii) hvKv as isolates in which higher or similar mortality rates were observed compared to those of hypervirulent strains of K. pneumoniae (). Intriguingly, in both cases, the isolates were reported as hypervirulent, although by different mechanisms (see detailed information below) [Citation43,Citation49,Citation52]. The emergence of these phenotypes in K. variicola should be considered when the isolates are being characterized.
Classic K. variicola
K. variicola was first identified in plant and clinical settings. As mentioned previously, K. variicola isolates are broadly antimicrobial susceptible; however, reports of ESBL-producing and carbapenemase-producing K. variicola have increased. In general, both antimicrobial susceptible and multidrug-resistant isolates could be considered classical strains of K. variicola. cKva isolates contain virulence factors shared with K. pneumoniae; however, cKva has been demonstrated to be pathogenic in BSIs, which could indicate undiscovered virulence factors [Citation7,Citation8,Citation14,Citation54,Citation57,Citation64]. Clinically important multidrug-resistant clones are mentioned in .
Hypermucoviscous K. variicola
The hypermucoviscous (hmv) phenotype was first described in hypervirulent K. pneumoniae (hvKpn). This phenotypic trait is defined by the formation of a viscous filament of ≥5 mm (positive string test) [Citation81]; however, a modified string test has been proposed to avoid discrepancies between clinical laboratories. The tested strain should be inoculated onto agar plates with 5% sheep blood and evaluated for the formation of a filament >10 mm long [Citation82]. The first hmv K. variicola isolate was described by Garza-Ramos et al. (2015) [Citation34]. In this report, the authors described the draft genome sequence of the clinical isolate K. variicola 8917 obtained from hospitalized elderly patients. Virulence-associated genes, capsule type and the sequence type (ST) were predicted from the genome sequence. Classical virulence factors were found, and among them were siderophores, iron uptake systems, pili and adhesins (). However, the rmpA and rpmA2 genes associated with the hypermucoviscous phenotype in hvKpn clones were absent. The capsular type was determined by using wzc K-typing corresponding to the wzc-932 allele. However, recent findings regarding the genomic diversity of the K-locus suggest that molecular typing based on a single gene, either wzc or wzi, is not accurate enough; instead, a complete K-locus analysis is suggested to increase the accuracy of K-typing [Citation83]. Considering these results, we analyzed the complete K-locus using the KAPTIVE tool and identified that K. variicola 8917 corresponded to the KL114 capsular type. The ST2 was determined using a multilocus sequence typing (MLST) K. variicola scheme [Citation80]. K. variicola 8917 bears an ∼200-kb plasmid (pKV8917), in which no antibiotic resistance genes were identified. Nevertheless, pKV8917 harbours the tellurium resistance genes terZABCDE and terW (ter operon), which are associated with hvKpn [Citation84]. pKV8917 plasmid curing causes the loss of the hmv phenotype; therefore, pKV8917 harbours the locus responsible for the hmv phenotype in K. variicola 8917 (unpublished data). To date, no other reports regarding K. variicola with the hmv phenotype have been described.
Other species besides K. variicola and K. pneumoniae have been described as exhibiting the hmv phenotype: K. quasipneumoniae subs. similipneumoniae and K. quasipneumoniae subsp. quasipneumoniae [Citation35,Citation85]. Furthermore, in our bacterial collection, we identified one E. coli isolate, one K. oxytoca isolate and numerous K. pneumoniae isolates with the hmv phenotype (unpublished data). In all these isolates, the rmpA and rmpA2 genes were missing. This phenomenon questions the existence of another mechanism to express hypermucoviscosity [Citation35,Citation85]. Therefore, the mechanism by which these strains acquired this phenotype has yet to be determined. The hmv phenotype itself is considered a virulence trait [Citation86]; however, in a nonhypervirulent background, its implication in human infection has been poorly characterized.
Hypervirulent K. variicola
The hypervirulent (hv) phenotype was initially described in the K. pneumoniae ST23 clone. This emerging clone has been a threat to human health [Citation87–89]. Until a few years ago, it was thought that hypervirulence was restricted to K. pneumoniae; however, years later, K. quasipneumoniae subsp. similipneumoniae and K. quasipneumoniae subsp. quasipneumoniae were found to acquire the hv phenotype by means of recombination events [Citation90,Citation91]. Hypervirulence associated with a multidrug-resistant phenotype has also been described, although it appears less frequently [Citation54,Citation79,Citation92]. Nonetheless, the trend is toward the convergence of both phenotypes becoming a major challenge for clinical management [Citation79,Citation93–95]. We identified two reports of hypervirulent K. variicola in the literature (). The first report (R1) corresponds to an hvKv and colistin-resistant strain (WCHKV030666), recovered from blood cultures of a hospitalized Chinese patient [Citation79]. Genomic analysis revealed the presence of a large plasmid, pVir_030666 (236 kb), in which multiple virulence genes commonly found in strains of hvKpn were identified. Among them, the plasmid-borne genes were the regulators of hmv phenotype rmpA and rmpA2 and three siderophore systems, namely, aerobactin, salmochelin and yersiniabactin. Commonly, the yersiniabactin siderophore is chromosomally located [Citation96]; however, in this case, the siderophore system was found on the virulence plasmid. The strain WCHKV030666 lacked the hmv phenotype owing to a frameshift mutation in the rmpA2 gene; however, the role of the rpmA gene in this genetic background is not clear. Interestingly, when the authors compared the sequence of the pVir_030666 plasmid against pLVPK [Citation97], the well-studied virulence plasmid of hvKpn suggested that more virulence genes are carried by pVir_030666 due to differences in genetic structure and size. Like other virulence plasmids, pVir_030666 is not self-transmissible; this trait is associated with an incomplete conjugation system plus the presence of an IS3 in the traG gene rather than the absence of the complete conjugation system, which is very common in other virulence plasmids [Citation66].
The second report of hypervirulent K. variicola (R2) corresponded to a multidrug-resistant (MDR) clone causing a fatal outbreak in neonates with high mortality associated [Citation56] (). Unlike the strain described in the work by Lu et al. (2018) [Citation66], the outbreak clone possessed only classical virulence factors such as an iron acquisition system, fimbriae and urease utilization genes. The K. variicola clone lacked the rmpA and rmpA2 genes, and the in vivo pathogenicity of K. variicola was tested in the G. mellonella model and compared to that of the hypervirulent strain of K. pneumoniae ST23 (A58300). The results suggested that K. variicola was more pathogenic than K. pneumoniae ST23. In this case, the hypervirulence may have been driven by new virulence determinants distinct from those reported in hvKpn strains. Moreover, the presence of the carbapenemase NDM-1 and the β-lactamase CTX-M-15 were identified, which is consistent with the convergence of multidrug resistance and hypervirulence [Citation56]. In addition, the authors found inconsistencies when determining the capsular type, which suggests a novel KL locus with respect to the described K. pneumoniae serotypes.
In summary, this section has discussed K. variicola distinct phenotypes and highlights the importance of genomic evidence in order to establish associations between phenotypic traits and genomic traits. K. variicola mostly displays the classic phenotype, and although K. variicola is susceptible to most antibiotic classes, an increasing number of MDR strains have been reported. The virulence factors and the implications for the pathogenesis of K. variicola are not well studied. Thus, to understand the pathogenesis and virulence, studies that include not only associations between virulence factors but also STs and capsular types are needed. Some authors suggest that infections caused by cKv are as severe as those caused by cKpn. Since hvKpn variants were described much controversy regarding the hmv phenotype has been generated. It is known that hypermucoviscosity is not enough to define hypervirulent strains; however, the main role of this phenotype in strains that lack hypervirulent properties is not clear. Interestingly, hypervirulence in K. variicola may not be determined by the same traits as those present in hvKpn. As mentioned above, hvKv displays a genetic and genomic context distinct from that of hvKpn, and even more clinical parameters differ in hvKv. None of the K. variicola strains reported as hypervirulent exhibit the hypermucoviscous phenotype, but the only hypermucoviscous strain does not appear to be hypervirulent.
MLST scheme for typing K. variicola isolates
The MLST is a powerful tool for epidemiological investigation of bacterial pathogens, allowing research on the genetic relatedness of bacterial isolates [Citation98]. The MLST scheme for K. pneumoniae is used for typing isolates within the K. pneumoniae complex, such as K. variicola and K. quasipneumoniae. This section discusses the available MLST schemes and also addresses some pitfalls of using the K. pneumoniae MLST scheme and the advantages of having a specific K. variicola MLST scheme.
The K. pneumoniae MLST scheme works for K. variicola, and different studies mentioned in this review have applied the K. pneumoniae MLST scheme for typing K. variicola isolates [Citation1,Citation8,Citation11,Citation14,Citation56]. Although a vast proportion of K. variicola genomes (54% 78/145) do not have an ST assignation or have a novel allele combination, previous authors have not discussed any possible reason why these isolates do not have a defined ST [Citation11,Citation14]. It is alarming that some important MDR clones are not assigned to an ST because this may limit molecular epidemiology of MDR clones among K. variicola. Moreover, Long et al. [Citation11] suggest that the K. pneumoniae MLST typing scheme would misidentify K. variicola as K. pneumoniae, as the database contains information about K. pneumoniae isolates, which may contribute to the problems in identification.
K. variicola, like the other members of the K. pneumoniae complex, are distinct species [Citation1,Citation2]. For an epidemiological purpose, a specific MLST typing scheme may be needed to trace the genetic relationship and evolutionary history within K. variicola isolates. Recently, a research group developed a MLST typing scheme for K. variicola (http://mlstkv.insp.mx) [ Citation80]. This scheme is based on 7 housekeeping genes; one of them (pyrG gene) is a unique gene in K. variicola, which may promote the proper identification of K. variicola. This MLST uses the whole-genome sequence or the sequences of the seven housekeeping genes to determine the ST. In addition, when the whole genome sequence is used, the ANI has been implemented to identify which bacterial species of the K. pneumoniae complex are being submitted for ST assignment. In this case, it only assigns an ST for K. variicola genomes. This study included 254 genomes and isolates with a total of 166 distinct sequence types (STs). A global distribution for some STs was observed, and in some cases, isolates obtained from different sources belonged to the same ST. Interestingly, kingdom-crossing bacteria from plants to humans were identified, establishing this as a possible route of transmission of K. variicola. In addition, Clonal Complex 1 (CC1) was identified as the clone with the greatest distribution.
Additionally, the authors compared both typing schemes (K. pneumoniae and K. variicola MLST schemes) using K. variicola genomes. Interestingly, they found that by applying the K. pneumoniae MLST scheme in K. variicola isolates, it was difficult to establish genetic relationships. This was observed when isolates of K. variicola have a genetic relationship using K. variicola MSLT, and using the K. pneumoniae MLST, are completely dispersed. This is because the isolates of K. variicola are being related with isolates of K. pneumoniae than to those of their own species [Citation80]. In summary, the comparison revealed that K. variicola isolates were dispersed among K. pneumoniae isolates, making it difficult to determine true relationships [Citation80].
The unsuitability of standard techniques for distinguishing K. variicola from the rest of the K. pneumoniae complex has masked its epidemiological context. However, recent genomic and phylogenomic studies as well as molecular epidemiology analysis [Citation80] () have been conducted on K. variicola isolates. The authors consider the K. variicola MLST scheme is a strong molecular epidemiological tool that allows following the presence of MDR clones and the evolution of this bacterial species in different environmental niches [Citation99].
Genomic population structure
Recent insights into the population structure of K. variicola have revealed two genomic lineages (L1 and L2) that are not restricted by geographic area, host or antimicrobial resistance patterns. L2 is composed of the largest number of genomes (143/145) distributed among 26 clades and two distant genomes conforming to L1 [Citation14]. Previous phylogenomic analyses have demonstrated that K. variicola is derived from a common ancestor and possesses a genome with a low frequency of recombination events [Citation1,Citation11]; however, recombination between K. pneumoniae complex members occurs and may be driven by the extensive genetic diversity within these species [Citation11,Citation14]. With the increasing number of new uploaded K. variicola genomes, differences in the population structure are likely to be observed as new lineages and subspecies.
Concluding remarks and future perspectives
K. variicola is an important human pathogen, and over the years, it has been misidentified as K. pneumoniae. This misidentification has caused unawareness about significant aspects of this bacterial species. Great efforts have been made to differentiate K. variicola from the K. pneumoniae complex and to highlight the importance of this species as a cause of severe infections, such as BSIs and UTIs, two major HCaIs commonly attributed to K. pneumoniae. K. variicola is becoming a public health concern not only for the infections that it can cause but also due to its potential to acquire antimicrobial and virulence genes, complicating the clinical management of the infections produced by K. variicola. There are several methods to correctly identify K. variicola; however, they have not been routinely adopted, and as it currently stands, incorrect classification continues to be a problem. K. variicola possesses a particular set of virulence genes that are not completely determined; the investigation of new or additional virulence factors will contribute to depicting its virulome. Most of the typing schemes (capsule and MLST) are based on data obtained for K. pneumoniae; however, it would be feasible to design specific tools for characterizing K. variicola due to the distinctive characteristics of this species compared to those of the K. pneumoniae complex. In addition, the wide distribution of K. variicola in the environment and its potential use in industrial processes is clear evidence that the organism has its own molecular typing system. The implementation of methods that allow for proper differentiation of K. variicola from the K. pneumoniae complex should be implemented both at the clinical level and in experimental laboratories. Considering the different risks involved in facing an infection caused by K. variicola, correct identification of the microorganism that causes a particular infection will benefit patient outcomes. Regarding the information in the present review, we consider K. variicola to be an emerging pathogen in humans.
Acknowledgements
Rodríguez-Medina N, is a doctoral student of Programa de Doctorado en Ciencias Biomedicas from Universidad Nacional Autonoma de México and she was awarded a scholarship by the CONACyT. This work was supported by grant 256988 from SEP-CONACyT (Secretaría de Educación Pública-Consejo Nacional de Ciencia y Tecnología).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Holt KE, Wertheim H, Zadoks RN, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci USA. 2015;112:E3574–E3581.
- Rodrigues C, Passet V, Rakotondrasoa A, et al. Identification of Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola and related phylogroups by MALDI-TOF Mass Spectrometry. Front Microbiol. 2018;9:3000.
- Brisse S, Passet V, Grimont PA. Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int J Syst Evol Microbiol. 2014;64:3146–3152.
- Brisse S, Verhoef J. Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. Int J Syst Evol Microbiol. 2001;51:915–924.
- Rosenblueth M, Martinez L, Silva J, et al. Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst Appl Microbiol. 2004;27:27–35.
- Berry GJ, Loeffelholz MJ, Williams-Bouyer N. An investigation into Laboratory misidentification of a bloodstream Klebsiella variicola infection. J Clin Microbiol. 2015;53:2793–2794.
- Garza-Ramos U, Silva-Sanchez J, Martinez-Romero E, et al. Development of a multiplex-PCR probe system for the proper identification of Klebsiella variicola. BMC Microbiol. 2015;15:64.
- Maatallah M, Vading M, Kabir MH, et al. Klebsiella variicola is a frequent cause of bloodstream infection in the Stockholm area, and associated with higher mortality compared to K. pneumoniae. PLoS One. 2014;9:e113539.
- Martinez-Romero E, Rodriguez-Medina N, Beltrán-Rojel M, et al. Klebsiella variicola and Klebsiella quasipneumoniae with capacity to adapt to clinical and plant settings. Salud Publica Mex. 2017;60:29–40.
- Bowers JR, Lemmer D, Sahl JW, et al. Klebseq, a diagnostic tool for surveillance, detection, and Monitoring of Klebsiella pneumoniae. J Clin Microbiol. 2016;54:2582–2596.
- Long SW, Linson SE, Ojeda SM, et al. (2017). Whole-Genome Sequencing of Human Clinical Klebsiella pneumoniae Isolates Reveals Misidentification and Misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. mSphere. 2.
- Lin L, Wei C, Chen M, et al. Complete genome sequence of endophytic nitrogen-fixing Klebsiella variicola strain DX120E. Stand Genomic Sci. 2015;10:22.
- Garza-Ramos U, Moreno-Dominguez S, Hernandez-Castro R, et al. Identification and Characterization of imipenem-resistant Klebsiella pneumoniae and susceptible Klebsiella variicola isolates obtained from the same patient. Microb Drug Resist. 2016;22:179–184.
- Potter RF, Lainhart W, Twentyman J, et al. Population structure, antibiotic resistance, and uropathogenicity of Klebsiella variicola. MBio. 2018;9. doi:10.1128/mBio.02481-18.
- Seki M, Gotoh K, Nakamura S, et al. Fatal sepsis caused by an unusual Klebsiella species that was misidentified by an automated identification system. J Med Microbiol. 2013;62:801–803.
- Brisse S, Duijkeren E. Identification and antimicrobial susceptibility of 100 Klebsiella animal clinical isolates. Vet. Microbiol. 2005;105:307–312.
- Podder MP, Rogers L, Daley PK, et al. Klebsiella species associated with bovine mastitis in Newfoundland. PLoS. One. 2014;9:e106518.
- Fouts DE, Tyler HL, DeBoy RT, et al. Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Genet. 2008;4:e1000141.
- Liu W, Hou J, Wang Q, et al. Isolation and characterization of plant growth-promoting rhizobacteria and their effects on phytoremediation of petroleum-contaminated saline-alkali soil. Chemosphere. 2014;117:303–308.
- Martinez-Romero E, Rodriguez-Medina N, Beltrán-Rojel M, et al. Genome misclassification of Klebsiella variicola and Klebsiella quasipneumoniae isolated from plants, animals and humans. Salud Publica Mex. 2017;60:56–62.
- Martinez-Romero E, Silva-Sanchez J, Barrios H, et al. Draft genome sequences of Klebsiella variicola plant isolates. Genome Announc. 2015;3. doi:10.1128/genomeA.01015-15.
- Pinto-Tomas AA, Anderson MA, Suen G, et al. Symbiotic nitrogen fixation in the fungus gardens of leaf-cutter ants. Science. 2009;326:1120–1123.
- Reyna-Flores F, Barrios-Camacho H, Dantan-Gonzalez E, et al. Draft genome sequences of Endophytic isolates of Klebsiella variicola and Klebsiella pneumoniae obtained from the same Sugarcane plant. Genome Announc. 2018;6. doi:10.1128/genomeA.00147-18.
- Blin C, Passet V, Touchon M, et al. Metabolic diversity of the emerging pathogenic lineages of Klebsiella pneumoniae. Environ Microbiol. 2017;19:1881–1898.
- Rodrigues C, Passet V, Rakotondrasoa A, et al. Description of Klebsiella africanensis sp. nov., Klebsiella variicola subsp. tropicalensis subsp. nov. and Klebsiella variicola subsp. variicola subsp. nov. Res Microbiol. 2019;170:165–170.
- Chen M, Li Y, Li S, et al. Genomic identification of nitrogen-fixing Klebsiella variicola, K. pneumoniae and K. quasipneumoniae. J Basic Microbiol. 2016;56:78–84.
- Hazen TH, Zhao L, Sahl JW, et al. Characterization of Klebsiella sp. strain 10982, a colonizer of humans that contains novel antibiotic resistance alleles and exhibits genetic similarities to plant and clinical Klebsiella isolates. Antimicrob Agents Chemother. 2014;58:1879–1888.
- Kumar V, Sun P, Vamathevan J, et al. Comparative genomics of Klebsiella pneumoniae strains with different antibiotic resistance profiles. Antimicrob Agents Chemother. 2011;55:4267–4276.
- Haeggman S, Lofdahl S, Paauw A, et al. Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother. 2004;48:2400–2408.
- Fonseca EL, Ramos ND, Andrade BG, et al. A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn Microbiol Infect Dis. 2017;87:315–317.
- Garza-Ramos U, Martinez-Romero E, Silva-Sanchez J. SHV-type extended-spectrum beta-lactamase (ESBL) are encoded in related plasmids from enterobacteria clinical isolates from Mexico. Salud Publica Mex. 2007;49:415–421.
- Yasuhara-Bell J, Ayin C, Hatada A, et al. Specific detection of Klebsiella variicola and K. oxytoca by loop-mediated isothermal amplification. J Plant Pathol Microbiol. 2015;6. doi:10.4172/2157-7471.1000271.
- Garza-Ramos U, Barrios-Camacho H, Moreno-Dominguez S, et al. Phenotypic and molecular characterization of Klebsiella spp. isolates causing community-acquired infections. New Microbes New Infect. 2018;23:17–27.
- Garza-Ramos U, Silva-Sanchez J, Barrios H, et al. Draft genome sequence of the first hypermucoviscous Klebsiella variicola clinical isolate. Genome Announc. 2015;3. doi:10.1128/genomeA.01352-14.
- Garza-Ramos U, Silva-Sanchez J, Catalan-Najera J, et al. Draft genome sequence of a hypermucoviscous extended-spectrum-beta-lactamase-producing Klebsiella quasipneumoniae subsp. similipneumoniae clinical isolate. Genome Announc. 2016;4. doi:10.1128/genomeA.00475-16.
- Liu W, Wang Q, Hou J, et al. Whole genome analysis of halotolerant and alkalotolerant plant growth-promoting rhizobacterium Klebsiella sp. D5A. Sci Rep. 2016;6:26710.
- Martinez J, Martinez L, Rosenblueth M, et al. How are gene sequence analyses modifying bacterial taxonomy? The case of Klebsiella. Int Microbiol. 2004;7:261–268.
- Zurfluh K, Poirel L, Nordmann P, et al. First detection of Klebsiella variicola producing OXA-181 carbapenemase in fresh vegetable imported from Asia to Switzerland. Antimicrob Resist Infect Control. 2015;4:38.
- Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4:148ra116.
- Goris J, Konstantinidis KT, Klappenbach JA, et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91.
- Emonet S, Shah HN, Cherkaoui A, et al. Application and use of various mass spectrometry methods in clinical microbiology. Clin Microbiol Infect. 2010;16:1604–1613.
- van BA, Welker M, Pincus D, et al. Matrix-Assisted Laser Desorption Ionization time-of-Flight Mass Spectrometry in clinical microbiology: What Are the Current Issues? Ann Lab Med. 2017;37:475–483.
- Fontana L, Bonura E, Lyski Z, et al. The Brief case: Klebsiella variicola-identifying the misidentified. J Clin Microbiol. 2019;57. doi:10.1128/JCM.00826-18.
- Medrano EG, Forray MM, Bell AA. Complete genome sequence of a Klebsiella pneumoniae strain isolated from a known cotton insect boll vector. Genome Announc. 2014;2. doi: 10.1128/genomeA.00850-14
- Jiang SF, Liu Y, Xiao MY, et al. Draft genome sequence of Klebsiella variicola strain KV321 isolated from rhizosphere soil of Pisolithus tinctorius-Eucalyptus mycorrhiza. Genome Announc. 2016;4. doi:10.1128/genomeA.00676-16.
- Hathaway JJ, Sinsabaugh RL, Dapkevicius ML, et al. Diversity of Ammonia Oxidation (amoA) and nitrogen fixation (nifH) genes in lava Caves of Terceira, Azores, Portugal. Geomicrobiol J. 2014;31:221–235.
- Ayin CM, Schlub RL, Yasuhara-Bell J, et al. Identification and characterization of bacteria associated with the decline of ironwood (Casuarina equisetifolia) in Guam. Australas Plant Pathol. 2015;44(22). doi:10.1007/s13313-014-0341-4.
- Afzal AM, Rasool MH, Waseem M, et al. Assessment of heavy metal tolerance and biosorptive potential of Klebsiella variicola isolated from industrial effluents. AMB Express. 2017;7:184.
- Mukherjee N, Bartelli D, Patra C, et al. Microbial diversity of source and Point-of-Use water in Rural Haiti - A Pyrosequencing-based Metagenomic Survey. PLoS One. 2016;11:e0167353.
- Das A, Osborne JW. Enhanced bioremoval of lead by earthworm-Lumbricus terrestris co-cultivated with bacteria-Klebsiella variicola. J Photochem Photobiol B. 2017;175:65–72.
- Feruke-Bello YM, Babalola G, Odeyemi O. A comparative study of the wild and mutated heavy metal resistant Klebsiella variicola generated for cadmium bioremedation. Bioremedation J. 2018;22:1–9.
- Guo Y, Zhai Y, Zhang Z, et al. Complete genomic analysis of a kingdom-crossing Klebsiella variicola isolate. Front Microbiol. 2018;9:2428.
- Martin RM, Bachman MA. Colonization, infection, and the Accessory genome of Klebsiella pneumoniae. Front Cell Infect Microbiol. 2018;8:4.
- Paczosa MK, Mecsas J. Klebsiella pneumoniae: Going on the Offense with a strong Defense. Microbiol Mol Biol Rev. 2016;80:629–661.
- Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–S116.
- Farzana R, Jones LS, Rahman MA, et al. Outbreak of hypervirulent Multi-Drug resistant Klebsiella variicola causing high mortality in neonates in Bangladesh. Clin Infect Dis. 2019;68:1225–1227.
- Ledeboer NA, Lopansri BK, Dhiman N, et al. Identification of Gram-negative bacteria and genetic resistance determinants from positive blood Culture Broths by Use of the Verigene Gram-negative blood Culture multiplex Microarray-based molecular assay. J Clin Microbiol. 2015;53:2460–2472.
- Liao CH, Lai CC, Hsu MS, et al. Correlation between time to positivity of blood cultures with clinical presentation and outcomes in patients with Klebsiella pneumoniae bacteraemia: prospective cohort study. Clin Microbiol Infect. 2009;15:1119–1125.
- Meatherall BL, Gregson D, Ross T, et al. Incidence, risk factors, and outcomes of Klebsiella pneumoniae bacteremia. Am J Med. 2009;122:866–873.
- Mazzariol A, Bazaj A, Cornaglia G. Multi-drug-resistant Gram-negative bacteria causing urinary tract infections: a review. J Chemother. 2017;29:2–9.
- Hazen TH, Mettus R, McElheny CL, et al. Diversity among blaKPC-containing plasmids in Escherichia coli and other bacterial species isolated from the same patients. Sci Rep. 2018;8:10291.
- Hopkins KL, Findlay J, Doumith M, et al. IMI-2 carbapenemase in a clinical Klebsiella variicola isolated in the UK. J Antimicrob Chemother. 2017;72:2129–2131.
- Joo EJ, Kim SJ, Baek M, et al. Fecal carriage of antimicrobial-resistant Enterobacteriaceae in healthy Korean adults. J Microbiol Biotechnol. 2018;28:1178–1184.
- Liu L, Feng Y, Tang G, et al. Carbapenem-resistant isolates of the Klebsiella pneumoniae complex in Western China: The common ST11 and the Surprising hospital-specific types. Clin Infect Dis. 2018;67:S263–S265.
- Long SW, Linson SE, Ojeda SM, et al. Whole-Genome sequencing of a human clinical isolate of the novel species Klebsiella quasivariicola sp. nov. Genome Announc. 2017;5. doi:10.1128/genomeA.01057-17.
- Lu Y, Feng Y, McNally A, et al. Occurrence of colistin-resistant hypervirulent Klebsiella variicola. J Antimicrob Chemother. 2018;73:3001–3004.
- Di DY, Jang J, Unno T, et al. Emergence of Klebsiella variicola positive for NDM-9, a variant of New Delhi metallo-beta-lactamase, in an urban river in South Korea. J Antimicrob Chemother. 2017;72:1063–1067.
- Brusselaers N, Monstrey S, Snoeij T, et al. Morbidity and mortality of bloodstream infections in patients with severe burn injury. Am J Crit Care. 2010;19:e81–e87.
- Poirel L, Jayol A, Nordmann P. Polymyxins: Antibacterial Activity, susceptibility testing, and resistance mechanisms encoded by plasmids or Chromosomes. Clin Microbiol Rev. 2017;30:557–596.
- Gomi R, Matsuda T, Yamamoto M, et al. Characteristics of carbapenemase-producing Enterobacteriaceae in wastewater revealed by genomic analysis. Antimicrob Agents Chemother. 2018;62. doi:10.1128/AAC.02501-17.
- Ramirez MS, Traglia GM, Lin DL, et al. Plasmid-Mediated antibiotic resistance and virulence in Gram-Negatives: the Klebsiella pneumoniae Paradigm. Microbiol Spectr. 2014;2:1–15.
- Andrade BG, de Veiga RN, Marin MF, et al. The genome of a clinical Klebsiella variicola strain reveals virulence-associated traits and a pl9-like plasmid. FEMS Microbiol Lett. 2014;360:13–16.
- Catalan-Najera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: Two different but complementary Klebsiella spp. phenotypes? Virulence. 2017;8:1111–1123.
- Russo TA, Olson R, Fang CT, et al. Identification of biomarkers for differentiation of hypervirulent Klebsiella pneumoniae from classical K. pneumoniae. J Clin Microbiol. 2018;56. doi:10.1128/JCM.00776-18.
- Bialek-Davenet S, Criscuolo A, Ailloud F, et al. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis. 2014;20:1812–1820.
- Struve C, Roe CC, Stegger M, et al. Mapping the evolution of hypervirulent Klebsiella pneumoniae. MBio. 2015;6:e00630.
- Cubero M, Grau I, Tubau F, et al. Hypervirulent Klebsiella pneumoniae clones causing bacteraemia in adults in a teaching hospital in Barcelona, Spain (2007–2013). Clin Microbiol Infect. 2016;22:154–160.
- Lee CH, Liu JW, Su LH, et al. Hypermucoviscosity associated with Klebsiella pneumoniae-mediated invasive syndrome: a prospective cross-sectional study in Taiwan. Int J Infect Dis. 2010;14:e688–e692.
- Dong N, Lin D, Zhang R, et al. Carriage of blaKPC-2 by a virulence plasmid in hypervirulent Klebsiella pneumoniae. J Antimicrob Chemother. 2018;73:3317–3321.
- Humberto B-C, Alejandro A-V, Marilu B-R, et al. Molecular epidemiology of Klebsiella variicola obtained from different sources. Sci Rep. 2019; under review.
- Hadano Y. String test. BMJ Case Rep. 2013;2013. doi:10.1136/bcr-2012-008328.
- Lee HC, Chuang YC, Yu WL, et al. Clinical implications of hypermucoviscosity phenotype in Klebsiella pneumoniae isolates: association with invasive syndrome in patients with community-acquired bacteraemia. J Intern Med. 2006;259:606–614.
- Wick RR, Heinz E, Holt KE, et al. Kaptive Web: User-Friendly capsule and lipopolysaccharide Serotype Prediction for Klebsiella genomes. J Clin Microbiol. 2018;56. doi:10.1128/JCM.00197-18.
- Passet V, Brisse S. Association of tellurite resistance with hypervirulent clonal groups of Klebsiella pneumoniae. J Clin Microbiol. 2015;53:1380–1382.
- Arena F, Henrici De AL, Pieralli F, et al. Draft genome sequence of the first hypermucoviscous Klebsiella quasipneumoniae subsp. quasipneumoniae isolate from a bloodstream infection. Genome Announc. 2015;3. doi:10.1128/genomeA.00952-15.
- Lin YC, Lu MC, Tang HL, et al. Assessment of hypermucoviscosity as a virulence factor for experimental Klebsiella pneumoniae infections: comparative virulence analysis with hypermucoviscosity-negative strain. BMC Microbiol. 2011;11:50.
- Pomakova DK, Hsiao CB, Beanan JM, et al. Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: an emerging and under-recognized pathogenic variant. Eur J Clin Microbiol Infect Dis. 2012;31:981–989.
- Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4:107–118.
- Turton JF, Payne Z, Micah K, et al. Capsular type K54, clonal group 29 and virulence plasmids: an analysis of K54 and non-K54 closely related isolates of Klebsiella pneumoniae. Epidemiol Infect. 2018;146:1813–1823.
- Breurec S, Melot B, Hoen B, et al. Liver abscess caused by infection with community-acquired Klebsiella quasipneumoniae subsp. quasipneumoniae. Emerg Infect Dis. 2016;22:529–531.
- Shankar C, Nabarro LEB, Muthuirulandi Sethuvel DP, et al. Draft genome of a hypervirulent Klebsiella quasipneumoniae subsp. similipneumoniae with novel sequence type ST2320 isolated from a chronic liver disease patient. J Glob Antimicrob Resist. 2017;9:30–31.
- Lam MMC, Wyres KL, Wick RR, et al. Convergence of virulence and MDR in a single plasmid vector in MDR Klebsiella pneumoniae ST15. J Antimicrob Chemother. 2019. doi:10.1093/jac/dkz028.
- Surgers L, Boyd A, Girard PM, et al. ESBL-Producing Strain of hypervirulent Klebsiella pneumoniae K2, France. Emerg Infect Dis. 2016;22:1687–1688.
- Wyres KL, Holt KE. Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. 2016;24:944–956.
- Wyres KL, Wick RR, Judd LM, et al. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019;15:e1008114.
- Lam MMC, Wick RR, Wyres KL, et al. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb Genom. 2018;4:1–14.
- Chen YT, Chang HY, Lai YC, et al. Sequencing and analysis of the large virulence plasmid pLVPK of Klebsiella pneumoniae CG43. Gene. 2004;337:189–198.
- Urwin R, Maiden MC. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;11:479–487.
- Duran-Bedolla J, Rodriguez-Medina N, Garza-Ramos U, et al. Klebsiella variicola: a pathogen and eco-friendly bacteria with applications in biological and industrial processes. Apppl Environ Microbiol. 2019; under review.