
ABSTRACT
Yellow fever virus (YFV) infection is a major public concern that threatens a large population in South America and Africa. No specific anti-YFV drugs are available till now. Here, we report that rifapentine is a potent YFV inhibitor in various cell lines by high-throughput drugs screening, acting at both cell entry and replication steps. Kinetic test and binding assay suggest that rifapentine interferes the viral attachment to the target cells. The application of YFV replicon and surface plasmon resonance assay indicates that rifapentine suppresses viral replication by binding to the RNA-dependent RNA polymerase (RdRp) domain of viral nonstructural protein NS5. Further molecular docking suggests that it might interact with the active centre of RdRp. Rifapentine significantly improves the survival rate, alleviates clinical signs, and reduces virus load and injury in targeted organs both in YFV-infected type I interferon receptor knockout A129−/− and wild-type C57 mice. The antiviral effect in vivo is robust during both prophylactic intervention and therapeutic treatment, and the activity is superior to sofosbuvir, a previously reported YFV inhibitor in mice. Our data show that rifapentine may serve as an effective anti-YFV agent, providing promising prospects in the development of YFV pharmacotherapy.
Introduction
Mosquito-borne yellow fever virus (YFV) is the first reported haemorrhagic fever virus that belongs to the Flaviviridae family and the aetiological pathogen of yellow fever (YF), a serious infectious disease that is mostly prevalent in South America and Sub-Saharan Africa [Citation1]. Although an effective attenuated vaccine is available, the low vaccination coverage and poor vector control measurements limit its benefits and result in resurgence and periodic epidemics in risk areas [Citation2]. There is currently no specific antiviral drug for YF but only specific care to deal with related symptoms to improve the disease outcomes.
YF patients usually manifest common symptoms of fever, headache, muscle soreness, nausea and vomiting. However, approximately 20–60% of patients will progress to more severe cases with quickly developed multiorgan failure, haemorrhage, shock, or even death [Citation3,Citation4]. From 2016 until now, outbreaks of YF have increased substantially in popular areas such as Brazil and Angola, with high morbidity and mortality rates ranging from 40% to 60% [Citation5,Citation6]. In addition, importing YFV patients from endemic countries to areas free of the disease also poses a great threat of triggering new viral emergence in populations without any immune resistance [Citation7].
YFV is a single-stranded positive-sense RNA virus of approximately 11 kb, encoding 10 viral proteins including 3 structural proteins of Core, prM and E, and 7 nonstructural proteins of NS1, NS2A-B, NS3, NS4A-B and NS5 [Citation8,Citation9]. YFV infection consists of multiple steps. The virion firstly binds and enters host cells through unidentified host receptors followed by clathrin-mediated endocytosis. Viral RNA is then released in cytoplasm for the subsequent protein translation and genome replication. During these steps, viral proteinase and RNA-dependent RNA polymerase (RdRp) are essential to lead the whole process [Citation10,Citation11].
Several therapeutic interventions are reported to be effective in YFV-infected animal models [Citation12–14]. However, their inhibitory effect was mostly carried out in hamsters with adapted YFV strain which might not mimic the actual infection by the wild-type strain. Moreover, their specific mechanisms and targets were not studied thoroughly. Repurposing clinically approved drugs is a quick and economic way to discover optimal anti-YFV pharmacotherapy with readily availability [Citation15].
Rifapentine is a long-lasting, economic rifamycin antibiotic that is generally used to treat tuberculosis [Citation16]. Its congeneric compounds include rifampicin, rifamycin, rifabutin and rifaximin, which usually serve as inhibitors of DNA-dependent RNA polymerase to block bacterial infection [Citation17,Citation18]. Compared with rifampicin, the most widely used rifamycin antibiotic, rifapentine has a longer half-life and stronger inhibitory effect against tuberculosis [Citation19,Citation20].
In this study, we discovered that rifapentine potently inhibited YFV infection both in cell cultures and small animal models with low cytotoxicity. The inhibitory effect of rifapentine covers two stages of viral cell entry and RNA replication. Moreover, the intragastrical administration of rifapentine manifested optimal anti-YFV activity in type I interferon receptor knockout (A129−/−) and wild-type (C57) mice. Thus, rifapentine is a promising candidate for the treatment of YF.
Materials and methods
Cell culture and viruses
Human hepatoma Huh7 cells (SCSP-526, Chinese Academy of Sciences, Shanghai, China), African green monkey Vero cells (ATCC CCL-81), baby hamster Syrian kidney BHK-21 cells (ATCC CCL-10) and human neuroblastoma SH-Sy5y cells (ATCC CRL-2266) were cultured in Dulbecco’s modified Eagle’s medium (Life Technologies, USA). Aedes albopictus C6/36 (ATCC CRL-1660) cells were cultured in 1640 medium (Life Technologies, USA). The medium was supplemented with 10% fetal bovine serum, 1× nonessential amino acids, 100 IU/mL streptomycin and penicillin, and 2 mM l-glutamine (Gibco, Carlsbad, CA, USA). C6/36 cells were incubated at 28°C, and other cells were incubated at 37°C in 5% CO2 [Citation21,Citation22].
YFV vaccinal (17D) and wide-type (WT) strains were passaged at a multiplicity of infection (MOI) of 0.01 in Vero cells for 48–72 h at 37°C. Virus titres were determined in Vero cell cultures by a plaque-forming assay as described previously [Citation23,Citation24]. Briefly, virus was diluted serially and incubated with the cells for 1 h before the supernatants were changed with semisolid medium containing 1.4% carboxymethyl cellulose and 1% FBS. After 4–5 days incubation, the cells were fixed and stained for plaque detection. The vaccine strain 17D was a gift from professor Zhigang Yi of Fudan University [Citation25], and the WT strain was isolated from the serum of a confirmed YF patient (FJYF03/2016, GenBank: KY587416.1). Information for other viruses was listed in supplementary data.
YFV replication detection
A replicon plasmid of YFV 17D strain carrying the NanoLuc reporter gene was constructed as described [Citation26]. The plasmid was used as the template for YFV replicon RNA transcription using a HiScribe T7 ARCA mRNA Kit (New England Biolabs, USA). The resultant RNA was then transfected into Huh7 cells using Lipofectamine™ 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA). Six hours after transfection, the supernatant was replaced with culture medium and the plates were incubated for 24 h at 37°C. Then, 10% NanoLuc reagent was added to the cells for 5 min incubation in the dark. Luminescence in live cells was detected using a ChemiScope 6000 Series Fluorescence (Promega) and Chemiluminescence imaging System (Clinx Science instruments Co., Ltd, Shanghai, China) before the cells were fixed with cold methanol to perform the subsequent IF assay.
Surface plasmon resonance (SPR) biosensor assay
A surface plasmon resonance (SPR) biosensor assay was performed in a BIAcoreTM 8K biosensor (GE Healthcare) as described previously [Citation27]. Compounds were diluted in PBS with 5% DMSO at the indicated concentrations. The synthesized and purified viral proteins were immobilized to a CM5 Series S sensor chip (GE, United States). The carboxylic groups of the chip were activated by EDC 0.2 M and NHS 0.05 M. The exceeding active groups were inactivated with ethanolamine 1 M. Analytes were prepared as 2-fold dilutions of indicated concentrations after the pilot experiments, and injected at a flow rate of 30 μl/min at 25°C [Citation28]. The concentrations for YFV NS5 ranged from 0.156 to 80 μM, and YFV NS5-RdRp from 0.195 to 100 μM in dilution of PBS containing 5% DMSO. The association time was 90 s. The signal responses of the compound solutions were recorded, and the signal of the solution without the compound was utilized as a blank control. The binding kinetics were analysed with BIAcore 8K evaluation software using a 1:1 Langmuir binding model introducing at least five-point serial concentrations to obtain the constant. The KD was calculated as described previously [Citation29].
Evaluation of in vivo infection
Liver, spleen, brain, kidney and serum samples from A129−/− or C57 mice were obtained at day 4 post YFV infection in each group. The virus titres in serum and tissue homogenates from the organs were detected by plaque formation tests. Tissue samples were also fixed in 10% buffered formalin for immunohistochemical (IHC) and immunofluorescent (IF) detection as well as histopathological assays (Servicebio, China).
Statistical analysis
Bar and line graphs showing the means ± SD or means ± SEM (in vivo study) of at least three independent experiments were plotted. Statistical analyses were performed using Prism 5 (GraphPad Software). The survival rates in the in vivo study were analysed utilizing Wilcoxon log-rank survival analysis. Other data were analysed with either Student’s t-test or one-way ANOVA. P value of <.05 was considered statistically significant.
Results
Screening for potential anti-YFV compounds
A total of 970 FDA-approved small-molecule drugs were screened for anti-YFV activity using the cell-based high-throughput screening we established ((a)). Twenty-eight compounds showed potent antiviral activity against YFV strain of FJYF03/2016 in Huh7 cells with inhibition rate being more than 90% at 10 μM (Table S1).
Figure 1. Antiviral activity of rifamycins on YFV and other flaviviruses. (a) Schematic diagram of high-throughput drugs screening for anti-YFV compounds and further mechanistic study. (b) Cell viability detection of rifamycins on Huh7 cells. Anti-YFV activity of rifamycins in Huh7 cells infected with YFV-17D (MOI = 0.1) by RT-qPCR (c) and IF (d). (e) Effect of rifapentine on other flaviviruses. Huh7 cells were infected with HCV, DENV, ZIKV, JEV, or WNV (MOI = 0.1) with indicated concentrations of rifapentine or DMSO, and the infection was analysed by IF. Results were exhibited as infection rate compared to DMSO treated-group. Data were shown as mean ± SD of three independent experiments. *p < .05, **p < .01 compared to DMSO group.
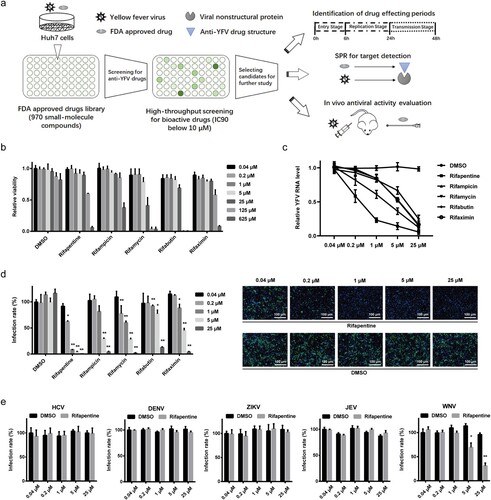
Among these compounds, two rifamycin antibiotics (rifampicin and rifapentine) manifested optimal inhibitory effects. Further investigations of the five congeneric compounds (rifapentine, rifampicin, rifamycin, rifabutin and rifaximin) showed that all rifamycin antibiotics had low cytotoxicity ((b)), and suppressed YFV infection significantly with IC50 values ranging from approximately 0.5 to 4.3 μM ((c,d), ). Among them, rifapentine displayed the best therapeutic effect, as its selective index (SI) (280.2) exceeded other compounds (). The antiviral activity of rifapentine was also evaluated in other cell lines including BHK-21, SH-Sy5y and C6/36 cells, and it showed extensive inhibitory effects in multiple cell lines originating from different targeted tissues (Table S2).
Table 1. Selective index of rifamycin antibiotics.
We also tested the effect of rifapentine on other flaviviruses to check whether its antiviral activity was unique to YFV. The results suggested that rifapentine had no obvious inhibitory effect on hepatitis C virus (HCV), dengue virus (DENV), Zika virus (ZIKV) or Japanese encephalitis virus (JEV), except for West Nile virus (WNV) with slight suppressive activity above 5 μM ((e)).
Rifapentine blocked YFV entry through the viral binding step
To reveal the inhibition stage of rifapentine on YFV, Huh7 cells were infected with the virus and treated with rifapentine during different periods. As shown in (a), compared with sofosbuvir, an acknowledged viral replication inhibitor, rifapentine exhibited prominent suppressive activity during all therapeutic windows except pretreatment, reducing the possibility that it hindered YFV infection by affecting host cell environment.
Figure 2. Rifapentine impaired viral binding. (a) Huh7 cells were infected with YFV-17D (MOI = 0.1) and treated with 25 μM rifapentine during different periods of viral infection. At 48 h post-infection, cells were analysed by IF. (b) Huh7 cells infected with YFV-17D were treated with rifapentine (25 μM), bafilomycin A1 (25 nM) or NH4Cl (25 mM) for 2 h during different periods as shown in the panel. At 24 h post-infection, YFV infection was assessed by IF. (c, d) Huh7 cells were incubated with YFV-17D at 4°C for 90 min with indicated concentrations of rifapentine. Part of the cells were cultured at 37°C for 24 h before being detected by IF (c). The other cells were lysed to determine the bound virus by RT-qPCR (d). (e) The virions were incubated with rifapentine (25 μM) for 6 h followed by sucrose density centrifugation to remove the compound. Viral infectivity was tested by reinfecting naïve Huh7 cells and analysed by IF. (f) Huh7 cells were incubated with labelled TF and rifapentine for 6 h before the cellular fluorescent intensity was detected. (g) Huh7 cells inoculated with YFV-17DDiD were treated with rifapentine (25 μM), Bafilomycin A1(25 nM) or NH4Cl (25 mM). Fluorescence units were collected every 15 min for total 32 cycles. Results were plotted as relative fluorescence units after subtraction of the background (uninfected culture). Data were shown as mean ± SD of three independent experiments. **p < .01 compared to DMSO group.
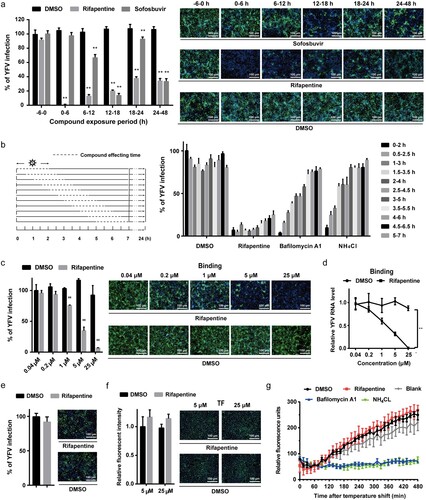
We then performed a kinetic test to elucidate the inhibitory pattern of this compound. In contrast to membrane fusion inhibitors of bafilomycin A1 and NH4Cl, rifapentine displayed obstructive effect on YFV infection throughout the entire phase of viral entry and post-entry stages ((b)), indicating that the inhibitory effect might cover more than one target in YFV life cycle.
YFV entry can be divided into binding, endocytosis and membrane fusion processes [Citation30]. We first investigated the effect of rifapentine on viral binding. As shown in (c,d), rifapentine impeded YFV attachment to the cell surface in a dose-dependent manner. In contrast, rifapentine did not affect WNV binding (Figure S1). However, virions incubated with rifapentine and subjected to sucrose density centrifugation exerted no obvious differences in infectivity ((e)), indicating that the inhibitory effect might occur during the virus-host interactions instead of the alteration of virion itself by the compound.
Subsequently, we detected the effect of rifapentine on virus endocytosis and membrane fusion. We used a fluorescence-labelled transferrin (TF) to evaluate whether rifapentine affected the classic clathrin-mediated endocytosis [Citation5]. No major changes were observed in TF uptake after rifapentine treatment, even at 25 μM ((f)). Finally, we utilized the DiD-labelled YFV to dynamically detect virion-host cell fusion. This hydrophobic fluorophore is de-quenched when diluted during viral fusion. In contrast with bafilomycin A1 and NH4Cl, of which the fluorescence signal reduced significantly compared to the control group, rifapentine displayed no inhibitory effect during this period ((g)).
Rifapentine suppressed YFV replication but did not affect viral assembly and release
The above kinetic assay suggested that rifapentine might also act during post-entry stage of YFV life cycle, and YFV initiated robust RNA synthesis approximately 12 h after virus inoculation (Figure S2). Since intracellular YFV RNA level was suppressed in a dose-dependent manner under rifapentine treatment ((a)), we therefore assessed its role in post-entry steps. A YFV replicon plasmid carrying the nanoluciferase (NanoLuc) reporter gene was constructed, and its transfection efficiency was verified ((b)). The transfected replicon cells were then utilized to evaluate the effect of rifapentine on YFV replication by detecting live cell luminescence and viral nonstructural protein expression. As shown in (c,d), rifapentine robustly abolished YFV replication from 1 to 25 μM, better than the approved sofosbuvir.
Figure 3. Rifapentine suppressed YFV replication. (a) Huh7 cells infected by YFV-17D (MOI = 0.1) were incubated with rifapentine for 24 h, and YFV RNA levels were determined by qPCR. (b) Schematic diagram of YFV replicon plasmid was shown. The transfection efficiency of various concentrations of YFV replicon RNA in Huh7 cells was detected 24 h later by measuring cellular Nano-Luc intensity. (c, d) Huh7 cells were transfected with YFV replicon RNA, and treated with rifapentine or sofosbuvir for 24 h. Cells were then detected by Nano-Luc reagent and IF assay. Results were plotted as relative ratio of grey scale or fluorescence intensity compared to DMSO-treated group. (e) Huh7 cells were transfected with YFV RNA, and treated with rifapentine. At 48 h after transfection, cells were subjected to three cycles of freeze and thaw to get the assembled virions. Supernatants were collected to assess the released virions. Results were exhibited as % of YFV infection compared to DMSO-treated group. Data were shown as mean ± SD of three independent experiments. *p < .05, **p < .01 compared to DMSO group.
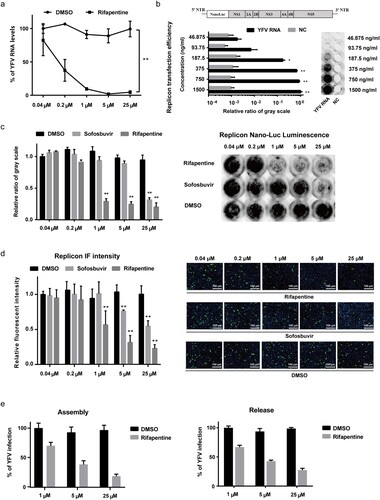
To determine whether rifapentine affects YFV assembly and release, we transfected Huh7 cells with YFV RNA and treated the cells with rifapentine 6 h post transfection. The intracellular assembled virions were then collected by repeated freeze and thaw of the cells, and extracellular released virions were collected in the supernatants of infected cells. The results indicated that rifapentine exhibited barely additional suppressive effect other than the original inhibition detected in replicon cells even at 25 μM ((e)). Thus, it did not influence YFV assembly and release.
Rifapentine bound to YFV NS5 through potential interactions with the central RdRp domain
Rifamycins are antibiotic compounds that inhibit bacterial DNA-dependent RNA polymerase (DdRp). Due to the inhibitory effect of rifapentine on YFV replication, we speculated that this compound might produce antiviral functions by targeting viral RdRp.
To validate the target of rifapentine on YFV replication, the full-length viral NS5 protein which contains RdRp, was expressed using E. coli strain BL21/DE3. The soluble product was used to monitor its interaction with rifapentine using a BIAcore surface plasmon resonance (SPR) affinity assay. The results suggested that rifapentine specifically bound to YFV NS5 on the sensor surface in a dose-dependent manner, and had a better affinity than sofosbuvir ((a,b)). According to general criteria, the binding between rifapentine and YFV NS5 was strong (KD: 20.8 μM) without nonspecific binding (Table S3). However, the noncovalent binding between sofosbuvir and NS5 was weak (KD: 6140 μM). We also tested the affinity of rifapentine with WNV NS5. The interaction was weaker (KD: 54.6 μM) than what we observed in YFV NS5, but still stronger than sofosbuvir (Figure S3(a,b) and Table S4). As YFV NS5 contains a methyltransferase (MTase) domain in the N-terminus and an RdRp domain in the C-terminus, we then prepared the truncated NS5 containing only the RdRp domain. The biological activity of rifapentine with YFV RdRp was intriguingly strong with clear binding, in contrast to sofosbuvir ((c,d) and Table S5). These results confirmed the direct binding between rifapentine and the RdRp domain of NS5.
Figure 4. Rifapentine inhibited YFV replication by binding to viral RdRp domain. (a, b, c, d) SPR assay was utilized to examine the binding by BIAcore 8K system. YFV NS5 or RdRp protein was immobilized on the CM5 chip, and serial dilutions of rifapentine and sofosbuvir were detected. The KD values was analysed by Biacore 8K software. (e, f) The binding of rifapentine to YFV RdRp was simulated by computer docking. Three-dimensional ligand-interaction maps of YFV RdRp domains bound with rifapentine were generated (e). (f) Residues lining the pocket for rifapentine (incarnadine sticks) were shown as green sticks. Individual residues were labelled according to their numbering in YFV NS5 (PDB: 6qsn), hydrogen bonds were indicated with red dashed lines.
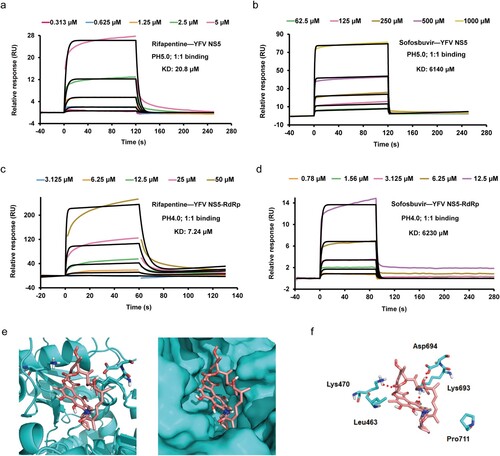
Therefore, further analysis was conducted to validate the potential functional mechanism. Molecular docking was performed to calculate the interplay between the compound and the active site of NS5 RdRp domain. The 3D structure model of the YFV RdRp in complex with rifapentine indicated optimal structural quality. As shown in (e), rifapentine interacted best in the active centre of RdRp which plays versatile roles in viral RNA replication (ΔG: −15.32). Potential hydrogen bonds and hydrophobic interactions formed between the macrolide skeleton of the compound and protein were listed in (f). Rifapentine could also interact with WNV RdRp (Figure S3(c)) (ΔG: −52.766). However, only two hydrophobic interactions were found between them (Figure S3(d)). These results further elucidated the inhibitory effect of rifapentine on YFV replication.
Rifapentine exerted potent antiviral activity in type I interferon receptor-deficient (A129−/−) mice
Subsequently, we assessed the antiviral activity of rifapentine in vivo using mice with basically mature adaptive immunity [Citation31]. Five-week-old A129−/− mice were infected with YFV at inoculation doses of 5.0 × 104, 5.0 × 103 or 5.0 × 102 PFU. Rifapentine treatment began either 24 h prior to or post infection by gavage. Sofosbuvir was also used in the assay. Pretreatment with rifapentine significantly enhanced the survival rate of infected mice with the three different infection dosages respectively, while posttreatment showed an obvious effect only in the 5.0 × 104 or 5.0 × 103 group ((a)). Sofosbuvir pretreatment manifested a slight protective activity in the highest infection-dose group ((a)). Body weight of survival mice by rifapentine treatment recovered to almost the normal level, indicating the preferable therapeutic effect of the compounds in vivo ((b)).
Figure 5. Antiviral activity of rifapentine in YFV-infected A129−/− mice. (a and b) 5-week A129−/− mice (n = 8) were inoculated with 100 μl WT YFV of 5 × 104, 5 × 103 or 5 × 102 PFU respectively by intraperitoneal injection. Treatment of rifapentine or sofosbuvir (20 mg/kg/day) was given daily either one day prior to (Pre-Rifapentine or Pre-Sofosbuvir) or after infection (Post-Rifapentine) until day 21 by gavage. The survival of the mice was analysed by log-rank test (a). Body weight changes (b) and clinical scores (c) were documented and calculated daily. Viral load in the serum (d) or tissues of brain (e), spleen (f), liver (g) and kidney (h) were determined by plaque formation test. Viral protein expression in the brains of infected mice was tested by IHC and IF assay (i). Pathological damage in brains of infected mice was analysed by histological section using Haematoxylin and Eosin (HE) staining (j). Data were shown as mean ± SEM. *p < .05, **p < .01 compared to DMSO group.
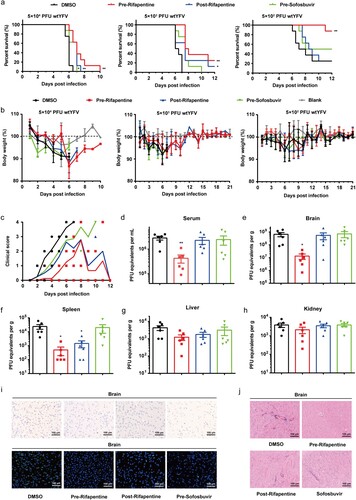
Infected mice displayed clinical manifestations including ruffled fur, trembling and hunched posture. Nevertheless, treatment with rifapentine slowed down or reduced the occurrence of clinical signs ((c)). YFV levels in the serum and organs of A129−/− mice were also tested. Compared with the control group, pretreatment with rifapentine reduced virus titres significantly in serum ((d)), brain and spleen ((e,f)). However, we did not recognize distinctive changes in the liver and kidney ((g,h)). Moreover, immunohistochemical (IHC) and immunofluorescent (IF) assays of the organs were also performed, and viral proteins were only detected in the brains. Though all three treated group showed obvious anti-YFV activity, pretreatment has the most potent inhibitory effect ((i)). Histopathological analysis revealed that some major alterations including the infiltration of inflammatory cells appeared in infected mice while mild injury was observed in treated mice ((j) and S4(a)).
Rifapentine enhanced the survival rate in YFV-infected normal mice
We then evaluated the antiviral activity of rifapentine in normal C57 mice with a higher inoculation dose of wild-type virus to achieve 100% mortality within one week. Consistent with the experiments carried out in A129−/− mice, rifapentine significantly improved the survival of mice, and the pretreatment group increased the survival rate to approximately 40% ((a)). The body weight of surviving mice all recovered to normal levels and the mice achieved long-term survival ((b)).
Figure 6. Antiviral activity of rifapentine in YFV-infected C57 mice. 3-week C57 mice (n = 10) were inoculated with 100 μl WT YFV of 6 × 108 PFU by intraperitoneal injection. Treatment of rifapentine or sofosbuvir (20 mg/kg/day) was given daily until day 21 by gavage. The survival of mice was analysed by log-rank test (a). Changes in body weight (b) and clinical scores (c) were recorded daily. Viral load was detected in brains by plaque formation test (d). Viral protein expression in brains of infected mice was tested by IHC and IF assay (e). Pathological damage in brains was analysed using HE staining (f). Data were shown as mean ± SEM. *p < .05, **p < .01 compared to DMSO group.
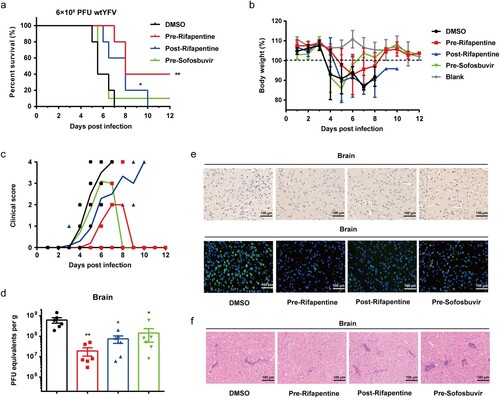
The clinical symptoms were also monitored, and the mice in rifapentine-treated group had fewer injury features ((c)). Viral titres were evaluated in serum and targeted organs. However, they were only detected in the brains and decreased in all three treatment groups compared with vehicle-treated control, among which pretreatment of rifapentine resulted in the most potent effect, reducing virus levels by approximately 30-fold ((d)). The IHC and IF assays also confirmed the above findings, with fewer viral protein-positive cells in the brain tissue in rifapentine-treated group ((e)). Histopathological changes of the targeted organs were assessed. Large amounts of pericapillary space dilation and perivascular cuffing caused by inflammatory cell infiltrations were detected in the brain tissue of all the groups except rifapentine pretreatment one ((f) and S4(b)).
Discussion
YFV is one of the major flaviviruses that can cause unexpected severe disease in human. The main methods to restrain YF involve vector control and vaccination [Citation32]. However, recent outbreaks in epidemic areas and the ongoing extensive global transmission demonstrate that measurements might not be sufficient. For the first time, we found the potential of a rifamycin antibiotic, rifapentine as a robust anti-YFV agent, blocking virion binding and suppressing viral RNA replication through interfering the function of the key RNA polymerase that leads YFV replication.
The rifamycin compounds have been widely used to treat tuberculosis for a long time and show favourable safety and compliance. There are several reports regarding the antiviral activity of rifamycin derivatives on DNA viruses, oncornaviruses and retroviruses [Citation33–35]. However, no studies has suggested the biological activity of rifamycins against flaviviruses. We found that the antiviral mechanism of rifapentine on YFV infection is unique and powerful. Nevertheless, resistant mutants which might emerge under long durations of drug pressure should be monitored carefully and vigilantly.
In cell culture, rifapentine significantly inhibited YFV vaccinal and wild-type strain infection in multiple cell lines, such as Huh7, SH-Sy5y and BHK-21 cells. It is worth noting that Huh7 and SH-Sy5y cells are derived from YFV-targeted organs, the failure of which would lead to fatal outcomes of infected patients. Therefore, rifapentine might benefit YFV-infected cases who undergo disease progression or develop multi-organ failure.
YFV entry is the initiation of virus infection. However, the ubiquitous viral entry receptors have not been elucidated clearly [Citation30,Citation36]. Therefore, we only identified the early effecting stage of rifapentine as the initial binding. Nevertheless, the blockage of viral entry provided rifapentine with a unique preventive property, making it an ideal intervention strategy for YFV infection [Citation30].
Viral replication controls the speed of the entire infection cycle. YFV NS5 is the key enzyme for replication [Citation37], thus becoming an ideal target for antiviral agents [Citation38]. Our study suggested that rifapentine directly bound to the RdRp domain of YFV NS5. Although sofosbuvir, an approved RdRp inhibitor, presented anti-YFV efficacy in vitro, its noncovalent affinity with YFV NS5 or the RdRp domain was weak. A possible explanation is that the interaction might involve other unknown elements to assist the binding or the affinity cannot be detected by BIAcore analysis. Molecular docking further speculated that the possible interaction might occur in the active site of YFV RdRp domain. Similar but weaker interactions between rifapentine and WNV NS5 were also detected in SPR and docking analysis, explaining its modest inhibitory effect on WNV infection. Further studies should be made to investigate the structure similarity and identify the key amino acid sites between YFV and WNV NS5.
In animal models, pretreatment and posttreatment with rifapentine efficiently inhibited infection at multiple virus inoculation doses, of which pretreatment group achieved the most powerful antiviral activity, improving the survival rate by approximately 40%, lowering viremia, organ infection and injury in infected mice. These phenomena were most likely due to the potent inhibitory effect of rifapentine on both viral entry and replication in vivo, preventing the establishment of infection on naïve cells and reducing existing virus titres in serum and targeted organs as well. Additionally, the regimen of 20 mg/kg/d by gavage was basically equivalent to human doses in treating tuberculosis, thus becoming potentially safe and well-tolerated anti-YFV therapy in future. Sofosbuvir achieved a relatively good inhibitory effect in YFV replicon cells. However, its efficacy in vivo was quite limited compared with rifapentine, probably because the antiviral targets of rifapentine are implicated at two different stages of YFV infection.
In conclusion, we identified rifapentine as a potent anti-YFV agent in vitro and in vivo. The compound not only blocked virion binding to the target cells but also hindered viral replication by binding the RdRp domain of NS5. Rifapentine may serve as an optimal therapeutic antiviral agent against YFV infection.
Ethics approval
This study and experimental protocol have been approved by our ethics committee of Naval Medical University.
Supplemental Material
Download MS Word (1.5 MB)Supplemental Material
Download MS Word (49.5 KB)Supplemental Material
Download MS Word (22.3 KB)Acknowledgements
We thank professor Zhigang Yi of Fudan University for helping with YFV infection of vaccinal strain. We thank professor Jianfeng Xie for helping with YFV isolation of wild-type strain.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Related Research Data
References
- Yellow fever in Africa and the Americas, 2016. Wkly Epidemiol Rec. 2017;92(32):442–452.
- Jentes ES, Poumerol G, Gershman MD, et al. The revised global yellow fever risk map and recommendations for vaccination, 2010: consensus of the informal WHO working group on geographic risk for yellow fever. Lancet Infect Dis. 2011;11(8):622–632.
- Carrington CV, Auguste AJ. Evolutionary and ecological factors underlying the tempo and distribution of yellow fever virus activity. Infect Genet Evol. 2013;13:198–210.
- Monath TP, Vasconcelos PF. Yellow fever. J Clin Virol. 2015;64:160–173.
- Douam F, Ploss A. Yellow fever virus: knowledge gaps impeding the fight against an old foe. Trends Microbiol. 2018;26(11):913–928.
- de Oliveira Figueiredo P, Stoffella-Dutra AG, Barbosa Costa G, et al. Re-emergence of yellow fever in Brazil during 2016–2019: challenges, lessons learned, and perspectives. Viruses. 2020;12(11):1233.
- Lataillade LG, Vazeille M, Obadia T, et al. Risk of yellow fever virus transmission in the Asia-Pacific region. Nat Commun. 2020;11(1):5801.
- Monath TP. Yellow fever: an update. Lancet Infect Dis. 2001;1(1):11–20.
- Higuera A, Ramirez JD. Molecular epidemiology of dengue, yellow fever, Zika and chikungunya arboviruses: an update. Acta Trop. 2019;190:99–111.
- Best SM. The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J Virol. 2017;91(3):e01970–16.
- Selisko B, Wang C, Harris E, et al. Regulation of flavivirus RNA synthesis and replication. Curr Opin Virol. 2014;9:74–83.
- Julander JG, Morrey JD, Blatt LM, et al. Comparison of the inhibitory effects of interferon alfacon-1 and ribavirin on yellow fever virus infection in a hamster model. Antiviral Res. 2007;73(2):140–146.
- Julander JG, Shafer K, Smee DF, et al. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother. 2009;53(1):202–209.
- Julander JG, Bantia S, Taubenheim BR, et al. BCX4430, a novel nucleoside analog, effectively treats yellow fever in a hamster model. Antimicrob Agents Chemother. 2014;58(11):6607–6614.
- Faria NR, Kraemer MUG, Hill SC, et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science. 2018;361(6405):894–899.
- Alfarisi O, Alghamdi WA, Al-Shaer MH, et al. Rifampin vs. rifapentine: what is the preferred rifamycin for tuberculosis? Expert Rev Clin Pharmacol. 2017;10(10):1027–1036.
- Campbell EA, Korzheva N, Mustaev A, et al. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104(6):901–912.
- Rothstein DM. Rifamycins, alone and in combination. Cold Spring Harb Perspect Med. 2016;6(7):a027011.
- Dorman SE, Goldberg S, Stout JE, et al. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. J Infect Dis. 2012;206(7):1030–1040.
- Munsiff SS, Kambili C, Ahuja SD. Rifapentine for the treatment of pulmonary tuberculosis. Clin Infect Dis. 2006;43(11):1468–1475.
- Guo F, Wu S, Julander J, et al. A novel benzodiazepine compound inhibits yellow fever virus infection by specifically targeting NS4B protein. J Virol. 2016;90(23):10774–10788.
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76(24):13001–13014.
- Guo F, Zhao X, Gill T, et al. An interferon-beta promoter reporter assay for high throughput identification of compounds against multiple RNA viruses. Antiviral Res. 2014;107:56–65.
- de Freitas CS, Higa LM, Sacramento CQ, et al. Yellow fever virus is susceptible to sofosbuvir both in vitro and in vivo. PLoS Negl Trop Dis. 2019;13(1):e0007072.
- Yi Z, Sperzel L, Nurnberger C, et al. Identification and characterization of the host protein DNAJC14 as a broadly active flavivirus replication modulator. PLoS Pathog. 2011;7(1):e1001255.
- Patkar CG, Larsen M, Owston M, et al. Identification of inhibitors of yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob Agents Chemother. 2009;53(10):4103–4114.
- Dal Piaz F, Cotugno R, Lepore L, et al. Chemical proteomics reveals HSP70 1A as a target for the anticancer diterpene oridonin in Jurkat cells. J Proteomics. 2013;82:14–26.
- Chen L, Lv D, Chen X, et al. Biosensor-based active ingredients recognition system for screening STAT3 ligands from medical herbs. Anal Chem. 2018;90(15):8936–8945.
- He Z, Zhu X, Wen W, et al. Dengue virus subverts host Innate immunity by targeting adaptor protein MAVS. J Virol. 2016;90(16):7219–7230.
- Kleinert RDV, Montoya-Diaz E, Khera T, et al. Yellow fever: integrating current knowledge with technological innovations to identify strategies for controlling a re-emerging virus. Viruses. 2019;11(10):960.
- Yau C, Gan ES, Kwek SS, et al. Live vaccine infection burden elicits adaptive humoral and cellular immunity required to prevent Zika virus infection. EBioMedicine. 2020;61:103028.
- Chen LH, Wilson ME. Yellow fever control: current epidemiology and vaccination strategies. Trop Dis Travel Med Vaccines. 2020;6:1.
- Garriga D, Headey S, Accurso C, et al. Structural basis for the inhibition of poxvirus assembly by the antibiotic rifampicin. Proc Natl Acad Sci U S A. 2018;115(33):8424–8429.
- Green M, Bragdon J, Rankin A. 3-Cyclic amine derivatives of rifamycin: strong inhibitors of the DNA polymerase activity of RNA tumor viruses. Proc Natl Acad Sci U S A. 1972;69(5):1294–1298.
- Szabo C, Bissell MJ, Calvin M. Inhibition of infectious Rous sarcoma virus production by rifamycin derivative. J Virol. 1976;18(2):445–453.
- Perera-Lecoin M, Meertens L, Carnec X, et al. Flavivirus entry receptors: an update. Viruses. 2013;6(1):69–88.
- Selisko B, Papageorgiou N, Ferron F, et al. Structural and functional basis of the fidelity of nucleotide selection by flavivirus RNA-dependent RNA polymerases. Viruses. 2018;10(2):59.
- Dubankova A, Boura E. Structure of the yellow fever NS5 protein reveals conserved drug targets shared among flaviviruses. Antiviral Res. 2019;169:104536.