
ABSTRACT
In this study, we conducted a detailed molecular epidemiological analysis of HIV-1 epidemic strains in various sexual risk behaviour groups in a developed area in eastern China based on phylogenetic inference, molecular transmission networks, and Bayesian analyses. A total of 1083 pol sequences (91.23%, 1083/1187) from newly diagnosed HIV-1-positive patients from 2017 to 2020 were successfully obtained and involved thirteen HIV-1 subtypes, in which the major HIV-1 subtypes were CRF07_BC (36.10%, 391/1083) and CRF01_AE (34.63%, 375/1083). 485 individuals (44.78%,485/1083) formed 146 clusters in the network. Of which CRF07_BC showed extensive clustering driven by men who have sex with men (MSM) within larger networks, CRF01_AE and other subtypes were generally driven by small clusters (SCs) and medium clusters (MCs) among various risk groups. Through Sankey diagrams, the MSM group infected with CRF07_BC had a greater impact on the non-commercial heterosexual group among all cross-risk groups. In addition, a higher number of key individuals infected with CRF07_BC (40.22%, 74/127), suggests the crucial role of CRF07_BC-infected individuals as a local epidemic driver in the context of a mixed HIV-1 epidemic. Further Bayesian analysis confirmed that CRF07_BC was introduced into Jiaxing city from other provinces multiple times and spread rapidly among MSM and heterosexual individuals. Overall, our study provided some insights and information to explore the local transmission dynamics of HIV-1 epidemic strains involving various sexual risk groups, and emphasize that it is necessary to conduct in-depth research and precise intervention targeting key clusters/ individuals to effectively block the continued transmission of AIDS.
Introduction
For the past 40 years, human immunodeficiency virus type 1 (HIV-1) has been a serious threat to global public health and disease surveillance systems [Citation1–4]. According to the World Health Organization (https://www.who.int/), the estimated number of persons living with HIV/AIDS (PLWH) globally by the end of 2020 was 37.6 million, with 1.5 million new infections and approximately 690,000 people dying from acquired immunodeficiency syndrome (AIDS)-related illnesses. Even though the Joint United Nations Program on HIV/AIDS (UNAIDS) proposed “90-90-90” prevention and control targets and the vision of “Ending the HIV Epidemic by 2030” [Citation5,Citation6], China is one of the countries with the highest number of PLWH and the most complicated and diversified HIV-1 subtypes and continues to face significant obstacles and pressures.
After the first HIV-1 infection was detected in China in 1985 [Citation7], the number of PLWH rose year after year. By the end of 2020, there were 1,053,000 living PLWH in China, among which 13,1671 PLWH were newly diagnosed in 2020 (https://www.unaids.org/en). The distribution features and epidemiological trends of HIV-1 subtypes in China are also undergoing great changes as the number of PLWH increases rapidly. Novel HIV-1 subtypes and recombinants continue to evolve in China as a result of high genetic diversity and variability, resulting in a national HIV-1 epidemic. China has conducted four National HIV Molecular Epidemiological Surveys over the last 40 years, with the latest survey in 2016 revealing that the types of HIV-1 subtypes have increased rapidly, becoming more complex and diversified [Citation8]. More than twenty HIV-1 subtypes were discovered, among which the most prevalent epidemic subtypes were CRF07_BC, CRF01_AE, CRF08_BC, and B [Citation9]. Since the 1990s, CRF01_AE and CRF07_BC have undergone continuous evolution in China and have caused a widespread epidemic in various risk groups (e.g. men who have sex with men (MSM), heterosexual individuals (those involved in commercial behaviour or not), people who inject drugs (PWID), etc.) [Citation10–15]. Newly reported circulating recombinant form strains (CRFs) (e.g. CRF55_01B, CRF59_01B, CRF65_cpx, CRF67_01B, CRF68_01B, CRF85_BC, etc.) and unique recombinant form strains (URFs) frequently appear in certain groups [Citation16–19]. Furthermore, sexual contact infection has emerged as the primary method of HIV-1 transmission and tends to expand from high-risk groups to the general population, providing significant challenges to AIDS prevention and treatment in China, particularly in economically developed regions [Citation15,Citation20–22].
In the setting of rapid and unbalanced economic development, large-scale migrations of “floating” individuals moving to certain developed regions and urban centres (e.g. Shanghai, Shenzhen, Hangzhou, etc.) in China for increased job opportunities, better educational environments, and the openness and tolerance of large cities have tended to result in diverse HIV-1 subtypes, complex transmission patterns, and increased transmission risks in these regions. Therefore, how to grasp the epidemic characteristics of HIV-1 subtypes in various sexual risk behaviour groups in these areas, understand the emergence and transmission of novel recombinant subtypes, analyse the possible existence of HIV-1 transmission events, and identify potential high-risk groups are still HIV-1 issues that need to be solved urgently for AIDS prevention and treatment. In recent years, combining phylogenetic inference and epidemiological data has been proposed as a feasible approach to ascertain the drivers of HIV-1 prevalence in various groups and regional settings, which plays an increasingly important role in AIDS prevention and treatment [Citation23–27]. Nevertheless, most of these studies have focused on a certain group or single subtype, which was insufficient to understand and disentangle the HIV-1 transmission dynamics involving various risk groups for determining the hotspots or drivers of local HIV-1 prevalence.
Therefore, in this paper, through a high-density sampling strategy of a medium-sized and developed city in eastern China, we report a comprehensive study that describes the molecular genetics and epidemiological characteristics of HIV-1 epidemic strains in various sexual risk behaviour groups, with combined phylogenetic inference, transmission network analysis, and molecular epidemiology data. These findings aim to provide new perspectives for local HIV-1 intervention and prevention through an in-depth analysis of local HIV-1 prevalence and effectively reduce HIV-1 infection rates, which are expected to help inform public health prevention strategies.
MATERIALS and methods
Study population and sample collection
In this study, we selected a medium-sized and developed city (Jiaxing city) in eastern China as the study area to conduct a high-density sampling of prospective molecular epidemiological study. Jiaxing city, located in the hinterland of the Yangtze River Delta that borders developed cities such as Hangzhou, Shanghai, and Suzhou, has a permanent population of 5.4 million, among which 1.94 million are migrants (35.87%) and mainly from Yunnan, Sichuan, Anhui, Henan and other provinces. Since 2017, the number of newly diagnosed HIV-1 patients in Jiaxing city had continued to increase, more than 90% were infected by sexual transmission and more than half of all migrants are diagnosed every year.
Blood samples were obtained from 1187 newly confirmed HIV-1-positive patients by the Jiaxing Municipal Center for Disease Control and Prevention (Jiaxing CDC) through the first follow-up between January 2017 and December 2020, representing 93.54% (1187/1269) of the newly diagnosed patients in Jiaxing city in this study. All patients were newly confirmed HIV-1-positive and treatment-naive cases during sampling and had no previous exposure to highly active antiretroviral therapy. As per our previous study [Citation22], demographic and epidemiological data were collected by the staff from the Jiaxing CDC and Zhejiang Provincial CDC through face-to-face investigations as covariates for transmission network analysis. All blood samples were linked to demographic and epidemiological data using anonymous numerical codes. Cases with incomplete information or without the target gene fragment were not included in this study.
Amplification of HIV-1 pol for phylogenetic analyses
According to the manufacturer's instructions, viral RNA was extracted from 140 μl of plasma samples using a QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA, United States). RNA samples were directly subjected to reverse transcription PCR and nested PCR to generate the partial HIV-1 pol coding region (HXB2: 2253-3306), which included the entire protease (PR) and the first 300 codons of the reverse transcriptase (RT) gene. Target PCR products were sent to Hangzhou TsingKe Bio-Tech Co. for purification and sequencing, as previously described [Citation22]. Every nucleotide position was sequenced at least once on each strand.
Subtype identification and maximum-likelihood phylogenetic analysis
The trimming and assembly of sequences were performed using Sequencher® v5.4.6 (Gene Codes, Ann Arbor, MI) [Citation28]. Ambiguous bases were considered when the secondary peak was more than 30% of the main peak. As previously described, sequences with ≥ 5% ambiguous bases were excluded from the study [Citation29]. Sequences were subjected to multiple alignments using MAFFT v7.487 [Citation30] and trimmed to identical lengths (≥ 1000 bp, HXB2: 2253–3265 nt). Reference sequences were obtained from the Los Alamos National Laboratory (LANL) HIV sequence database (https://www.hiv.lanl.gov), which covers the major HIV-1 subtypes. HIV-1 subtypes were defined using a set of methods, including the online automated HIV-1 subtyping tool COMET (COntext-based Modeling for Expeditious Typing) v2.2 [Citation31], phylogenetic analyses, the jumping profile Hidden Markov Model (jpHMM) [Citation32], and Simplot v3.5.1 [Citation33]. The maximum-likelihood (ML) phylogenetic trees with different HIV-1 subtypes were constructed with RAxML v8.2.12 under the GTR + G + I nucleotide substitution model [Citation34] and then were visually edited by FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and the web-based tool Interactive Tree of Life (iTOL) v6.0 [Citation35] for further analysis involved geographic location, phylogenetic cluster, and risk groups. Local support values were computed with a Shimodaira-Hasegawa (SH) test [Citation36]. If ML tree nodes had SH-like support values ≥ 0.90, the viral sequences were identified as the subtypes of reference sequences.
Identification and analysis of the HIV-1 transmission network
All sequences were aligned, and pairwise genetic distances were computed using HIV Trace based on the Tamura-Nei 93 (TN93) method [Citation37]. To construct an effective transmission network for identifying potential transmission clusters using the visualization software Cytoscape v3.9.1 [Citation22], an optimal genetic distance threshold was selected from 0.001 substitutions/site to 0.02 substitutions/site (the optimal threshold was established by the maximum number of clusters below this threshold). Graphically, molecular networks consist of two basic elements: nodes and links. Each node represents an individual pol sequence, and transmission linkages are represented by lines drawn between both nodes according to a specific threshold. An independent connected group with at least two nodes in a molecular network is labelled a molecular cluster (cluster) [Citation38]. In this study, the formed clusters in the network were classified according to their size, namely, large cluster (LC, ≥10 nodes), medium cluster (MC, 4–9 nodes), and small cluster (SC, 2–3 nodes). In addition, individuals with ≥4 links were identified as key individuals for further analysis [Citation39].
Bayesian evolution and spatiotemporal geographic analysis of key clusters
Temporal Exploration of Sequences and Trees (TempEst) v1.5.3 [Citation40] was used to perform a regression analysis of root-to-tip genetic divergence obtained from the ML phylogeny against sampling year for the key cluster sequences and reference sequences, which eliminated the sequences that significantly deviated for subsequent Bayesian analysis. The best-fitting evolution model according to the Akaike information criterion (AIC) was confirmed using jModelTest v2.0.1 [Citation41]. Then, a series of Bayesian analyses were performed using Bayesian evolutionary analysis by sampling trees (BEAST) v1.10.4 [Citation42], which involved the calculations of the time of the most recent common ancestors (tMRCAs) and rates of evolution (in units of nucleotide substitutions/site/year) and Bayesian skyline plot (BSP) analysis. The BEAUti programme operating parameters were modified, and the two independent chains, both adjusted to 200 million steps with sampling every 20,000 steps, were run under the strict clock and relaxed clock models, assuming a constant substitution rate as estimated from the dataset. The sample collection dates were included as temporal data. In general, the marginal likelihood data were the largest, effective sample size values monitored for consistency using Tracer v1.7.1 [Citation43] exceeded 200, and the model was considered an optimal clock model. Maximum clade credibility (MCC) trees were generated using the Bayesian TreeAnnotator and visually edited in FigTree v1.4.4. Visual Bayesian phylogenetic analysis was performed on the generated KML files using spatial phylogenetic reconstruction of evolutionary dynamics (SPREAD) v1.0.7 [Citation44].
Statistical analyses
EpiData 3.0 software was used to establish the database, and Statistical Product and Service Solutions (SPSS) 20.0 software was used to organize and analyse the data. A Chisquared test was used for comparisons among different groups. P values <0.05 indicated statistical significance.
Patient consent statement
This study was reviewed and approved by the Medical Ethics Committee of the National Center for AIDS/STD Control and Prevention (Approval ID: X140617334), and written informed consent was obtained from the study participants. All experiments were performed by following the approved guidelines and regulations.
Results
Patient demographics in jiaxing city
In this prospective study, 1083 HIV sequences (91.23%, 1083/1187) were successfully obtained from 1187 newly diagnosed HIV-positive patients in Jiaxing city from 2017 to 2020. Of the 1083 successfully sequenced patients, most were male (81.63%, 884/1083), and the median age was 43 years (ranging from 5 to 86 years). A total of 94.92% were of Han ethnicity (1028/1083), and the proportion of married individuals was 46.63% (505/1083). Only 417 (38.50%) of these cases were born in Jiaxing city, while a majority (58.45%, 633/1083) were domestic migrants. MSM transmission was the main method of sexual contact (39.52%, 428/1083), followed by non-commercial heterosexual contact (30.19%, 327/1083) and commercial heterosexual contact (22.35%, 242/1083). The majority of the individuals had a junior high school education or below (69.81%, 756/1083). Other detailed demographic characteristics of the study participants are summarized in . Moreover, a demographic comparison of two groups (individuals in the networks (Clusters; links ≥1) and individuals not in the networks (No cluster; no link)) is shown in .
Table 1. Sociodemographic characteristics of the participants in Jiaxing city from 2017 to 2020.
HIV-1 subtype distribution and characteristics in jiaxing city
The HIV-1 epidemic in Jiaxing city was driven by different HIV-1 subtypes, and thirteen HIV-1 subtypes and a proportion of URFs were identified from the 1083 pol sequences obtained successfully. CRF07_BC (36.10%, 391/1083) and CRF01_AE (34.63%, 375/1083) were the major subtypes, and other subtypes showed small-scale sporadic characteristics, among which were CRF08_BC (10.99%, 119/1083), CRF85_BC (4.99%, 54/1083), CRF55_01B (3.88%, 42/1083), B (2.49%, 27/1083), CRF57_BC (0.18%, 2/1083), CRF59_01B (1.11%, 12/1083), CRF63_02A (0.18%, 2/1083), CRF65_cpx (0.18%, 2/1083), CRF67_01B (0.74%, 8/1083), CRF68_01B (0.28%, 3/1083), URF (01_AE/07_BC) (12.31%, 25/1083), URF (01_AE/B) (0.09%, 1/1083) and URF (B/C) (1.39%, 15/1083).
Constructing the maximum-likelihood phylogenetic trees of different HIV-1 subtypes (), we found that CRF01_AE in Jiaxing city was highly homologous to several CRF01_AE epidemic clusters in China. The CRF01_AE-infected individuals of two clusters, g4a and g1, accounted for the highest proportions, 15.47% (58/375) and 12.00% (45/375), respectively. Cluster g4a was dominated by MSM (94.83%, 55/58), and cluster g1 was dominated by heterosexual individuals (66.67%, 30/45). Among the other CRF01_AE clusters, g2, g2a, g3, and g7 were dominated by MSM, and g5 was dominated by heterosexual individuals. Other CRF01_AE-infected individuals who were not concentrated among the CRF01_AE clusters above were mainly scattered in small clusters, indicating that the prevalence of CRF01_AE in Jiaxing city has a long history of transmission and has evolved along with different evolutionary aspects.
Figure 1. Maximum-likelihood phylogenetic trees of patients infected with different HIV-1 subtypes in Jiaxing city from 2017 to 2020. The phylogenetic trees based on the pol region of different HIV-1 subtypes were constructed by an approximate maximum-likelihood method with RAxML v8.2.12 under the GTR + G + I nucleotide substitution model. The SH-like node support value (≥0.90) was indicated and considered credible, which was defined as the same subtype or subcluster. Clades of different colours represent different HIV-1 subtypes and subclusters. The circle shows the risk group (the outer circle; ▪ represents heterosexual individuals; □ represents MSM). (A) Maximum-likelihood phylogenetic tree of CRF01_AE. (B) Maximum-likelihood phylogenetic tree of CRF07_BC. (C) Maximum-likelihood phylogenetic tree of other HIV-1 subtypes.
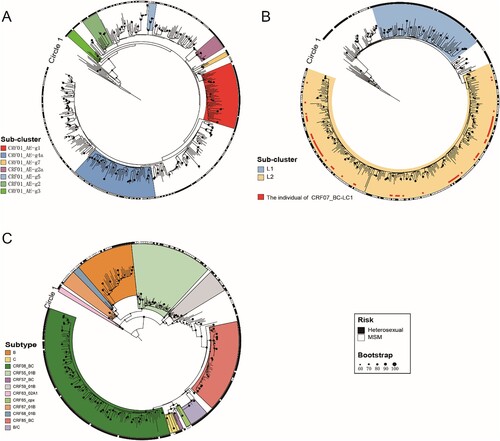
CRF07_BC mainly presented two distinct evolutionary branches, among which CRF07_BC-L1 was present mostly in heterosexual individuals (82.65%, 81/98), while CRF07_BC detected in Jiaxing city was mostly concentrated among people with CRF07_BC-L2 (65.98%, 258/391), mainly driven by the MSM population (63.95%, 165/258), indicating that the MSM population played an important role in driving the prevalence of CRF07_BC in Jiaxing city.
Besides the two major HIV-1 subtypes, the other HIV-1 subtypes (CRF08_BC, CRF85_BC, B, C, and URF (B/C)) were concentrated among most heterosexual individuals, in which 15 cases of URF (B/C) had high homology (99.9%−100.00%), indicating that the URF (B/C) detected in Jiaxing city had a high transmission correlation. And the other HIV-1 subtypes (CRF55_01B, CRF59_01B, CRF67_01B, and CRF68_01B) were mainly driven by MSM individuals.
Distribution characteristics of different HIV-1 subtypes in the network
To clearly understand the relationship between cluster and demographic characteristics among different HIV-1 subtypes in Jiaxing city, we plotted the molecular transmission networks based on pol sequences using the optimal genetic distance of 0.01 substitutions/site ( and ).
Figure 2. Molecular transmission network analysis of patients infected with different HIV-1 subtypes in Jiaxing city from 2017 to 2020. HIV-1 transmission cluster diagrams illustrating the structure and demographics of the putative transmission clusters among newly diagnosed individuals from 2017 to 2020. All edges represent a genetic distance between nodes of less than 0.01 substitutions/site, and the colour of the node indicates the different HIV-1 subtypes (A) and the different sexual contact risk groups (B). Different shapes denote sex, and the node size indicates the number of associated links.
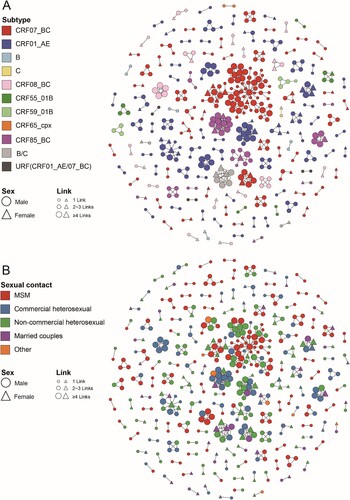
Table 2. Molecular network clustering of different HIV-1 subtypes in Jiaxing city from 2017 to 2020
The results showed that the distribution of different HIV-1 subtypes had obvious diversity in the network based on this threshold. In this study, 485 individuals (44.78%, 485/1083) formed 146 clusters (cluster sizes ranging from 2 to 74, median cluster size 2), which included ten subtypes. Of these, the CRF01_AE network had the largest number of clusters and was largely driven by small clusters. By the end of 2020, 55 clusters (cluster sizes ranging from 2 to 11) had been formed, including 42 SCs, 11 MCs, and 2 LCs. In addition, 168 CRF01_AE-infected individuals (44.80%, 168/375) were included in the network, including 75 (44.64%) MSM, 68 (40.48%) heterosexual males, and 25 (14.88%) heterosexual females. Among the 55, 16 clusters (29.09%, cluster sizes ranging from 2 to 13) involved only heterosexual individuals, 13 clusters (23.64%, cluster sizes ranging from 2 to 3) involved only MSM, and 26 clusters (47.27%, cluster sizes ranging from 2 to 6) involved multiple risk groups. The two LCs (CRF01_AE-LC1 and LC2) contained 11 and 10 individuals, respectively, and both were clusters of elderly heterosexual individuals (all were over 50 years old, with the oldest being 85 years old) and showed obvious regional aggregation. Besides, the other CRF01_AE clusters were SCs and MCs (cluster sizes ranging from 2 to 6) and included mostly heterosexual individuals, indicating that CRF01_AE showed a small epidemic spread in Jiaxing city.
In stark contrast, the CRF07_BC network had the largest span per cluster (cluster sizes ranging from 2 to 74), forming 45 clusters (37 SCs, 7 MCs, and 1 LC). By the end of 2020, 184 CRF07_BC-infected individuals (47.06%, 184/391) were included in the network, including 78 MSM (42.39%), followed by 71 heterosexual males (38.59%) and 33 heterosexual females (17.93%). Among the 45, 23 clusters (51.11%, cluster sizes ranging from 2 to 6) involved heterosexual individuals, 12 clusters (26.67%, cluster sizes ranging from 2 to 4) involved only MSM, and the other 10 clusters (22.22%, cluster sizes ranging from 2 to 74) involved multiple risk groups. An anomalously significant large cluster (CRF07_BC-LC1) involved 74 CRF07_BC-infected individuals and accounted for 40.22% of CRF07_BC-infected individuals in the network. The cluster was detected in 2017, included 6 individuals (3 MSM and 3 heterosexual males), and then rapidly expanded to 74 individuals (41 MSM, 29 heterosexual males, and 5 heterosexual females) by the end of 2020. Except for CRF07_BC-LC1, the other 44 clusters were largely driven by SCs and MCs (37 SCs and 7 MCs, cluster sizes ranging from 2 to 6) and dominated by heterosexual individuals.
Among the 317 individuals infected with other HIV-1 subtypes, 133 individuals were included in the network, forming 46 clusters (cluster sizes ranging from 2 to 13) involving eight subtypes, which were mainly small scale (37 SCs, 8 MCs, and 1 LCs) and were largely driven by heterosexual individuals (78.20%, 104/133).
Risk group characteristics of different HIV-1 subtypes in the network
The links connected to various sexual risk behaviour groups in the network reflect the relationship and risk among these groups. Hence, according to the characteristics of sexual contact, we classified the heterosexual group into commercial heterosexual contact group (those involved in commercial behaviour), non-commercial heterosexual contact group (those not involved in commercial behaviour), and heterosexual contact within spouses (those involved in spousal transmission) to further explore the correlation characteristics of different HIV-1 subtypes among the various risk groups in the network.
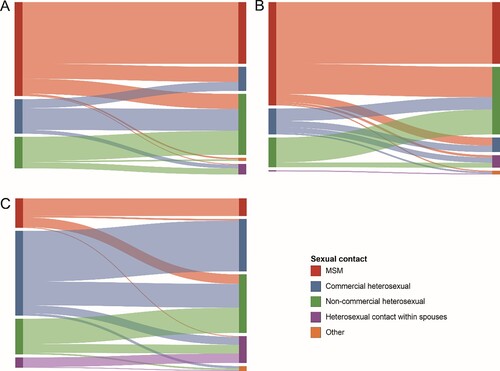
We drew Sankey diagrams based on the links of different HIV-1 subtypes among the various risk groups in the network (). In the CRF01_AE network (which included 213 links), the correlation between MSM in the same sexual risk behaviour group was the highest, accounting for 38.50% (82/213); the sexual risk behaviour groups of heterosexual individuals involved in commercial and non-commercial behaviour accounted for 15.02% (32/213) and 5.63% (12/213), respectively. The correlation between the commercial heterosexual and non-commercial heterosexual groups among all cross-risk groups was the highest, accounting for 13.62% (29/213). In the CRF07_BC network (which included 296 links), the correlation between MSM in the same risk group was the highest, accounting for 38.51% (114/296), followed by non-commercial heterosexual and commercial heterosexual groups, accounting for 15.54% (46/296) and 4.05% (12/296), respectively. Among all cross-risk groups, MSM had the highest correlation with the non-commercial heterosexual group, accounting for 18.92% (56/296). In the other subtype network (which included 202 links), the correlation between the commercial heterosexual group in the same risk groups was the highest, accounting for 31.68% (64/202), followed by non-commercial and MSM groups, accounting for 15.84% (32/202) and 10.89% (22/202), respectively. The correlation between the commercial heterosexual and non-commercial heterosexual groups among all cross-risk groups was the highest, accounting for 14.85% (30/202).
Figure 3. Linkages analysis of different sexual risk behaviour groups with the main HIV-1 subtypes in the network. The colour indicates the different sexual contact risk groups. (A) The Sankey diagram of CRF01_AE. (B) The Sankey diagram of CRF07_BC. (C) The Sankey diagram of other HIV-1 subtypes.
Risk factor and dynamic analysis of key individuals in jiaxing city
In addition to the network characteristics of different HIV-1 subtypes in various risk groups in the network, a total of 127 key individuals with more links (≥4 links, ranging from 4 to 31 links) were detected (), among which the newly confirmed patients in 2018 accounted for the highest proportion (38.58%, 49/127); the majority had CRF07_BC (58.27%, 74/127), most were male (86.61%, 110/127), most of them had a junior high school or below education (73.22%, 93/127), and most of them were MSM (36.22%, 46/127). The 127 key individuals were mainly distributed in 15 clusters (cluster sizes ranging from 5 to 74), of which 37 individuals (29.13%, 37/127) were concentrated in the cluster CRF07_BC-LC1, mainly MSM, and the remaining individuals were among distributed in small and medium clusters.
Through the above analysis, we observed that CRF07_BC-infected individuals played an important role in driving the HIV-1 epidemic in Jiaxing city. Therefore, we further conducted a dynamic Bayesian analysis based on CRF07_BC in Jiaxing city to better understand the evolution process and transmission path of CRF07_BC. We downloaded 2969 available CRF07_BC reference sequences from the Los Alamos National Laboratory (LANL) HIV Sequence Database (https://www.hiv.lanl.gov) and excluded repetitive sequences. Finally, 58 CRF07_BC reference sequences from other regions in China and 122 CRF07_BC sequences from various risk groups in Jiaxing city were selected based on homology (>98.00%) to be included in the dynamic analysis.
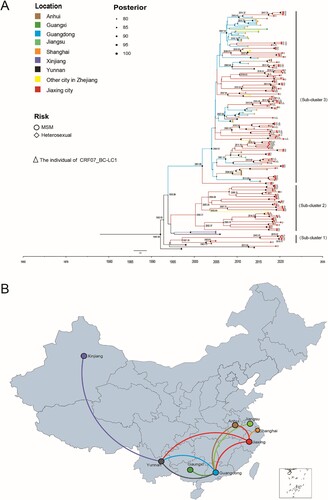
In this study, TempEst v1.5.3 was used to test the molecular clock hypothesis of the selected 180 CRF07_BC sequences, and the R2 value was calculated. The R2 value was generally between 0.1 and 1.0. The larger the value was, the better the fit of the molecular clock model. The results showed that the calculated R2 was 0.9954, indicating that the 180 CRF07_BC sequences were generally in line with the molecular clock hypothesis. The origin time of the most recent common ancestor of CRF07_BC was estimated by the lognormal relaxed molecular clock model, the general time-reversible model (GTR) + G + I nucleotide substitution, and the Bayesian skyline demographic growth model. The Bayesian skyline demographic growth model analysis showed that CRF07_BC had an exponential growth trend from 2006 to 2011, stabilized from 2011 to 2017, and declined in 2018. By Bayesian and spatiotemporal pathway analysis, the average evolution rate of the CRF07_BC strains was 1.567×10−3 nucleotide substitutions/site/year (95% HPD: 1.050×10−3-2.343×10−3). reveals the origin time of the different evolutionary branches of CRF07_BC in Jiaxing city. With the support of high posterior probability, combining the average evolution rate and pairwise genetic distance to estimate the evolution time, we found that CRF07_BC in Jiaxing city gradually formed three independent evolutionary clades during the evolution process, among which Subcluster 1 (including four heterosexual individuals in Jiaxing city) and Subcluster 2 (including 35 heterosexual individuals in Jiaxing city) showed a stable local epidemic with no evidence of an expanding trend, and both subclusters included mainly heterosexual individuals. In contrast, Subcluster 3 contained 79 CRF07_BC-infected individuals in Jiaxing city, of which 47 were MSM and 26 were heterosexual individuals, and all 37 key individuals with CRF07_BC-LC1 were in this subcluster. In addition, based on the close geographical relationship, Subcluster 3 showed that CRF07_BC in Jiaxing city was introduced from Yunnan, Guangdong, and Anhui Provinces in China and further transmitted locally, revealing that CRF07_BC maybe play a key role in driving the local HIV-1 epidemic.
Figure 4. Maximum clade credibility (MCC) tree and map of geographic location transition based on the CRF07_BC sequences. Bayesian analyses were performed using BEAST v1.10.4 (see Methods for details). In the MCC tree (A), sequences from 180 CRF07_BC sequences (122 CRF07_BC reference sequences and 58 CRF07_BC sequences in Jiaxing city) form three unique phylogenetic clades, labelled Subclusters 1–3; the branch lengths reflect the evolutionary time, and nodes labelled with evolutionary time were supported by a high posterior probability (≥0.90). The corresponding time scale was marked at the bottom of the MCC tree; different colours of branches indicate that the formed clades contain reference sequences from different provinces/cities in China. In the geographic location transition map (B), points are colour-coded by the geographic location of origin. Lines are colour-coded by the geographic location of the destination.
Discussion
In this study, we conducted high-density sampling of newly diagnosed patients in a developed city (Jiaxing city) in eastern China from 2017 to 2020 to achieve larger and proportional sample coverage across all risk groups and then performed a detailed molecular epidemiological study involving comprehensive phylogenetic, transmission characteristics, and risk factor analyses. This study is unique in that it is the first to apply a detailed molecular epidemiological approach and high-density sampling to better explore the local transmission characteristics and epidemic patterns of HIV-1 epidemic strains involved in various sexual risk behaviour groups.
The four National HIV-1 Molecular Epidemiological Surveys revealed that the characteristics of different HIV-1 subtypes in China are diverse and involve geographical distributions, transmission patterns, and risk groups, in which CRF07_BC and CRF01_AE have become the major HIV-1 subtypes and are widespread epidemic subtypes in most parts of the country, creating a considerable challenge for intervention [Citation8,Citation9]. In this study, through the phylogenetic analysis of 1083 samples successfully collected from Jiaxing city from 2017 to 2020, the results revealed that the HIV-1 subtypes in Jiaxing city were relatively diverse, and a total of thirteen HIV-1 subtypes and a proportion of URFs were identified (). The major HIV-1 subtypes were CRF07_BC (36.10%, 391/1083) and CRF01_AE (34.63%, 375/1083), and there were certain differences in the distribution of different HIV-1 subtypes among various risk groups. CRF01_AE and other subtypes were mainly distributed in small and medium-sized clusters with various risk groups. In stark contrast, CRF07_BC had little clustering in the heterosexual group but demonstrated extensive clustering in the MSM group. Such diversity and difference suggested that the HIV-1 epidemic in Jiaxing city may be related to large-scale migrations and certain types of human mobility and led to the introduction of HIV-1 epidemic subtypes in other regions, resulting in the continuous spread of the local HIV-1 epidemic among various risk groups, which further thwarted AIDS prevention and control in this region.
The HIV-1 molecular network has been proposed as a feasible approach to ascertain the correlation among individuals involved in various risk groups [Citation3,Citation15,Citation28,Citation38,Citation45,Citation46]. The accuracy of the network is closely related to the coverage rate of sample collection. According to the published guidelines on HIV-1 Transmission Network Monitoring and Intervention [Citation47], the sampling rate should be no less than 60% of the number of newly diagnosed patients in the testing area. In this study, the sample coverage rate of newly confirmed HIV-1 patients in Jiaxing city from 2017 to 2020 exceeded 90% per year, and subtypes were identified per patient, which could relatively comprehensively reflect the local molecular transmission network. We thoroughly analysed the molecular transmission networks of different HIV-1 subtypes among various risk groups below the optimal genetic distance of 0.01 substitutions/site. The molecular network of different HIV-1 subtypes showed significantly different clustering conditions (). CRF01_AE and other subtypes had more clusters in the network, in which there were few LCs and which was generally driven by SCs and MCs. In stark contrast, CRF07_BC showed extensive clustering within larger networks, suggesting that the close transmission linkages of CRF07_BC-infected individuals in Jiaxing city may be related to social networks and behavioural patterns among the various sexual risk groups in the network. The distinctly large cluster (CRF07_BC-LC1), which contained 74 cases, was formed in 2017 (containing 6 cases) and rapidly expanded to 74 cases in 2020, a 12-fold increase, indicating ongoing expansion and highlighting the need for enhanced monitoring of key clusters in the region to disrupt spread chains promptly.
In the molecular network, possible transmission linkages among various risk groups were estimated based on the genetic sequence associations among clusters [Citation9,Citation19,Citation48,Citation49]. Although the molecular network cannot determine whether it is a direct transmission event, the transmission linkages among different risk groups can reflect the relationship of these groups to a certain extent. These linkages across different risk groups can provide clues and directions for identifying the “source” of the hotspot populations through different risk pathways[Citation45,Citation50]. In this study, through Sankey diagrams based on the links of different HIV-1 subtypes among the various risk groups (), the transmission linkages among various risk groups showed significant differences. In the CRF07_BC network, the correlation among MSM was the highest, while in CRF01_AE and other subtypes, heterosexual individuals were more correlated, which suggested that the “sources” of the different HIV-1 subtypes in different risk groups were diverse. Among all cross-risk groups, MSM infected with CRF07_BC had a greater impact on heterosexual individuals who were not involved in commercial behaviour, while heterosexual individuals involved in commercial behaviour infected with CRF01_AE and other subtypes (especially heterosexual males involved in commercial behaviour) had a greater impact on heterosexual individuals not involved in commercial behaviour. These findings indicated that there may be more bridge populations between MSM and heterosexual individuals not involved with commercial behaviour infected with CRF07_BC in Jiaxing City, while there may be more crossover between heterosexual individuals involved in commercial behaviour and heterosexual individuals not involved in commercial behaviour infected with CRF01_AE and other subtypes. In addition, due to the bridge populations between MSM and non-commercial heterosexual groups, MSM has a great impact on the non-commercial heterosexual individuals compared with the other risk groups, which further drives the formation of a large-scale transmission network, as evidenced by the large cluster of CRF07_BC-LC1, reflecting the complex local epidemic pattern. It is necessary to conduct further in-depth analysis of the epidemiological characteristics of different subtypes to determine the key populations and implement targeted prevention and control measures.
In addition, 127 key individuals with more links (≥4 links, ranging from 4 to 31 links) were detected in the network. CRF07_BC-infected individuals were significantly more likely to be high-risk communicators than those infected with CRF01_AE and other subtypes and were mostly concentrated in the large cluster (CRF07_BC-LC1). These key individuals generally had a lower education level, relatively limited knowledge of AIDS, and may be more prone to high-risk behaviours. Therefore, strengthening real-time monitoring of key molecular clusters where such key individuals are clustered, targeted publicity and education, and the development of easy-to-understand publicity materials for people with low education levels may help reduce high-risk behaviours in this population.
Through the above analysis, our study highlights the crucial role of CRF07_BC-infected individuals as a local epidemic driver in the context of a mixed HIV-1 epidemic. We further performed a molecular evolutionary dynamic analysis based on CRF07_BC (). The results showed that CRF07_BC was introduced into Jiaxing city from other provinces (Yunnan, Guangdong, and Anhui Provinces) multiple times, and then infections spread rapidly among MSM and heterosexual individuals who may share practices with MSM. These findings showed the important position of key individuals in the network and revealed the mutual transmission relationship between different risk groups and different regions. Meanwhile, our findings also confirmed that the path of geographic location transitions for HIV-1 CRF07_BC largely conforms to the characteristics of population migration in China and mainly tends to flow within and between economically developed areas, such as Shanghai city, Hangzhou city, and Guangzhou city [Citation51]. Jiaxing city is located in the hinterland of the Yangtze River Delta region in China, bordering large cities such as Hangzhou city, Shanghai city, and Suzhou city, and attracts a large number of migrant workers from Yunnan, Sichuan, and Anhui provinces every year. Furthermore, with the rapid development of transportation and internet applications, MSM population seeks sexual partners with more convenience and secrecy to avoid stigmatization and discrimination, providing favourable conditions for the cross-regional and -group transmission of HIV-1 [Citation52,Citation53]. Therefore, capturing these potential suspected high-risk groups using molecular surveillance data for targeted publicity and education, enhancing their awareness of using protection during sexual encounters, and strengthening treatment will help reduce the transmission risk of these groups.
Our study provided some insights and information to explore the local transmission characteristics and epidemic patterns of HIV-1 epidemic strains involving various sexual risk behaviour groups. However, this study has certain limitations. First, although we included as many newly diagnosed patients as possible in our study, with a sampling density of more than 90%, the molecular network we deduced based on HIV-1 pol sequences is only a part of the overall local risk behaviour network, and many unsequenced and undiagnosed individuals were not included in the molecular network analysis. Second, molecular network analysis was not able to determine whether there was a direct transmission relationship between two associated patients. Patients infected by different sexual contact methods were connected, and it can be speculated that between these sexual contact methods, at a certain time node in the past, cross-groups transmission occurred. Third, the epidemiological data, including the sexual contact methods, were based on individual reports, and there may be some information bias. Due to the limited time of the study, more epidemiological characteristics of the survey subjects were not collected, which requires further in-depth research and exploration in the future. However, the wealth of information found in this study will lay a good foundation for further research.
Conclusions
Our study is the first to apply a detailed molecular epidemiological approach to better explore the local transmission characteristics and epidemic pattern of HIV-1 epidemic strains involved in various sexual risk behaviour groups in developed areas (Jiaxing city) in eastern China. Our findings emphasize that it is necessary to conduct in-depth research and precise intervention targeting key clusters/individuals, explore new models of precise intervention based on the HIV-1 molecular transmission network, and implement measures including early AIDS detection and exposure prevention to effectively block the continued transmission of AIDS and reduce the rate of new infections. Understanding these epidemic dynamics in real time is of increasing public health importance in terms of guiding prevention efforts.
Author contributions
Xiaohong Pan and Chengliang Chai conceived and designed the study. Qin Fan, Jiafeng Zhang, Xiaobei Ding, Yan Xia, Zhihong Guo, Rui Ge, and Yong Yan performed experiments. Yi Feng and Ping Zhong provided guidance. Qin Fan, Jiafeng Zhang, and Mingyu Luo analyzed the data. Qin Fan wrote the main manuscript and prepared all figures. All authors have reviewed the manuscript.
Acknowledgments
We appreciate all the volunteer patients enrolled in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hemelaar J. The origin and diversity of the HIV-1 pandemic. Trends Mol Med. 2012;18(3):182–192.
- Bbosa N, Kaleebu P, Ssemwanga D. HIV subtype diversity worldwide. Curr Opin HIV AIDS. 2019;14(3):153–160.
- Sivay MV, Totmenin AV, Zyryanova DP, et al. Characterization of HIV-1 epidemic in Kyrgyzstan. Front Microbiol. 2021;12:753675.
- Rashid A, Li K, Feng Y, et al. HIV-1 genetic diversity a challenge for AIDS vaccine development: a retrospective bibliometric analysis. Hum Vaccin Immunother. 2022;18(1):2014733.
- McMahon JH, Medland N. 90-90-90: how do we get there? The Lancet HIV. 2014;1(1):e10–e11.
- Fauci AS, Redfield RR, Sigounas G, et al. Ending the HIV epidemic: a plan for the United States. JAMA. 2019;321(9):844–845.
- Zeng Y, Fan J, Zhang Q, et al. Detection of antibody to LAV/HTLV-III in sera from hemophiliacs in China. Aids Res. 1986;2(suppl 1):S147–S149.
- Zhong P. Progress in research and practice of molecular epidemiology of HIV-1. Electronic Journal of Emerging Infectious Diseases. 2019;4(3):137–144.
- Vrancken B, Zhao B, Li X, et al. Comparative circulation dynamics of the five main HIV types in China. J Virol. 2020;94(23):e00683-20.
- Xin R, He X, Xing H, et al. Genetic and temporal dynamics of human immunodeficiency virus type 1 CRF07_BC in Xinjiang, China. J Gen Virol. 2009;90(Pt 7):1757–1761.
- Feng Y, He X, Hsi JH, et al. The rapidly expanding CRF01_AE epidemic in China is driven by multiple lineages of HIV-1 viruses introduced in the 1990s. AIDS. 2013;27(11):1793–1802.
- Chen Y, Chen S, Kang J, et al. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the Southwest border of China. PLoS One. 2014;9(9):e107578.
- Yan H, Ding Y, Wong FY, et al. Epidemiological and molecular characteristics of HIV infection among money boys and general men who have sex with men in Shanghai, China. Infect Genet Evol. 2015;31:135–41.
- Zhang W, Chen J, Pan X, et al. Trends of HIV-1 subtypes among young people in Hangzhou, China. AIDS Res Hum Retroviruses. 2017;33(3):219–227.
- Zheng S, Wu J, Hu Z, et al. Epidemiology and molecular transmission characteristics of HIV in the capital city of Anhui Province in China. Pathogens. 2021;10(12):1554.
- Zai J, Liu H, Lu Z, et al. Tracing the transmission dynamics of HIV-1 CRF55_01B. Sci Rep. 2020;10(1):5098.
- Fan Q, Zhang WJ, Yang JZ, et al. Characteristic analysis of molecular subtypes and recombinant structure of HIV-1 infection in Zhejiang Province, 2015. Chin J Prev Med. 2018;52(4):409.
- Su L, Feng Y, Liang S, et al. The origin and spread of CRF85_BC, driven by heterosexual trasmission among older people in Sichuan, China. BMC Infect Dis. 2020;20(1):772.
- Chen X, Qin C, Chen R, et al. Epidemiological profile and molecular genetic characterization of HIV-1 among female sex workers and elderly male clients in Guangxi, China. Emerg Microbes Infect. 2021;10(1):384–395.
- Zhou PP, Yu G, Kuang YQ, et al. Rapid and complicated HIV genotype expansion among high-risk groups in Guangdong Province, China. BMC Infect Dis. 2019;19(1):185.
- Zhang Z, Dai L, Jiang Y, et al. Transmission network characteristics based on env and gag sequences from MSM during acute HIV-1 infection in Beijing, China. Arch Virol. 2017;162(11):3329–3338.
- Fan Q, Zhang J, Luo M, et al. Analysis of the driving factors of active and rapid growth clusters among CRF07_BC-infected patients in a developed area in Eastern China. Open Forum Infect Dis. 2021;8(3):ofab051.
- Hansson D, Leung KY, Britton T, et al. A dynamic network model to disentangle the roles of steady and casual partners for HIV transmission among MSM. Epidemics. 2019;27:66–76.
- Alexiev I EMC, Knyazev S, et al. Molecular Epidemiology of the HIV-1 Subtype B Sub-Epidemic in Bulgaria. Viruses. 2020;12(4):441.
- Gil H, Delgado E, Benito S, et al. Transmission clusters, predominantly associated with men who have sex with men, play a main role in the propagation of HIV-1 in Northern Spain (2013-2018). Front Microbiol. 2022;13:782609.
- Nduva GM, Otieno F, Kimani J, et al. Phylogeographic assessment reveals geographic sources of HIV-1 dissemination among men who have sex with men in Kenya. Front Microbiol. 2022;13:843330.
- Kwan TH, Wong NS, Lui GCY, et al. Incorporation of information diffusion model for enhancing analyses in HIV molecular surveillance. Emerg Microbes Infect. 2020;9(1):256–262.
- Zhang J, Yao J, Jiang J, et al. Migration interacts with the local transmission of HIV in developed trade areas: a molecular transmission network analysis in China. Infect Genet Evol. 2020;84:104376.
- Jiang J, Fan Q, Zhang J, et al. A geographic hotspot and emerging transmission cluster of the HIV-1 epidemic among older adults in a rural area of Eastern China. AIDS Res Hum Retrovir. 2020;36(9):712–720.
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780.
- Daniel S, Glenn L, Anne-Marie T, et al. COMET: adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014;18:e144–e144.
- Anne-Kathrin S, Ming Z, Ingo B, et al. jpHMM: improving the reliability of recombination prediction in HIV-1. Nucleic Acids Res. 2009;37(Web Server issue):W647–51.
- Fan Q, Zhang WJ, Yang JZ, et al. Characteristic analysis of molecular subtypes and recombinant structure of HIV-1 infection in Zhejiang Province, 2015. Zhonghua yu fang yi xue za zhi Chinese journal of preventive medicine. 2018;52(4):409.
- Alexandros S. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;9:1312–1313.
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296.
- Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol.biol.evol. 1999;8:1114–1116.
- Mi YA, Wertheim JO, Kim WJ, et al. HIV-1 transmission networks across South Korea. Aids Research & Human Retroviruses. 2017;33(8):827–831.
- Han X, Zhao B, An M, et al. Molecular network-based intervention brings us closer to ending the HIV pandemic. Front Med. 2020;14(2):136–148.
- Oster AM, Wertheim JO, Hernandez AL, et al. Using molecular HIV surveillance data to understand transmission between subpopulations in the United States. JAIDS J Acquir Immune Defic Syndr. 2015;70(4):444–51.
- Andrew R, Lam TT, Luiz MC, et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;1:vew007.
- arriba DD, Taboada GL, Doallo R, et al. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
- Suchard MA, Philippe L, Guy B, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(1):vey016.
- Rambaut A, Drummond AJ, Xie D, et al. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst Biol. 2018;67(5):901–904.
- Bielejec F, Baele G, Vrancken B, et al. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol Biol Evol. 2016;33(8):2167–2169.
- Ragonnet-Cronin M, Hayford C, D'Aquila R, et al. Forecasting HIV-1 genetic cluster growth in Illinois,United States. JAIDS J Acquir Immune Defic Syndr. 2022;89(1):49–55.
- Stecher M, Hoenigl M, Eis-Hubinger AM, et al. Hotspots of transmission driving the local human immunodeficiency virus epidemic in the Cologne-Bonn Region, Germany. Clin Infect Dis. 2019;68(9):1539–1546.
- Prevention Detecting and Responding to HIV Transmission Clusters: A Guide for Health Department. 2018.
- Brenner BG. Phylogenetic insights on patterns of HIV-1 spread and the design of epidemic control measures. Viruses. 2022;14(2):332.
- Nduva GM, Nazziwa J, Hassan AS, et al. The role of phylogenetics in discerning HIV-1 mixing among vulnerable populations and geographic regions in Sub-Saharan Africa: A systematic review. Viruses. 2021;13(6):1174.
- Oster AM, France AM, Panneer N, et al. Identifying clusters of recent and rapid HIV transmission through analysis of molecular surveillance data. JAIDS J Acquir Immune Defic Syndr. 2018;79(5):543–550.
- Li X, Li Y, Liu H, et al. The emergence and transmission dynamics of HIV-1 CRF07_BC in Mainland China. Virus Evol. 2022;8(1):veac014.
- Zhang J, Guo Z, Pan X, et al. Highlighting the crucial role of Hangzhou in HIV-1 transmission among men who have sex with men in Zhejiang, China. Sci Rep. 2017;1:13892.
- Fan Q, Yao J, Luo M, et al. Molecular transmission characteristics of human immunodeficiency virus type 1 in Northern Zhejiang Province. Chin J Infect Dis. 2021;39(2):74–79.