
ABSTRACT
Vaccination is a crucial prevention and control measure against H9N2 avian influenza viruses (AIVs) that threaten poultry production and public health. However, H9N2 AIVs in China undergo continuous antigenic drift of hemagglutinin (HA) under antibody pressure, leading to the emergence of immune escape variants. In this study, we investigated the molecular basis of the current widespread antigenic drift of H9N2 AIVs. Specifically, the most prevalent h9.4.2.5-lineage in China was divided into two antigenic branches based on monoclonal antibody (mAb) hemagglutination inhibition (HI) profiling analysis, and 12 antibody escape residues were identified as molecular markers of these two branches. The 12 escape residues were mapped to antigenic sites A, B, and E (H3 was used as the reference). Among these, eight residues primarily increased 3`SLN preference and contributed to antigenicity drift, and four of the eight residues at sites A and B were positively selected. Moreover, the analysis of H9N2 strains over time and space has revealed the emergence of a new antigenic branch in China since 2015, which has replaced the previous branch. However, the old antigenic branch recirculated to several regions after 2018. Collectively, this study provides a theoretical basis for understanding the molecular mechanisms of antigenic drift and for developing vaccine candidates that contest with the current antigenicity of H9N2 AIVs.
The H9N2 avian influenza virus (AIV), an RNA virus, was first discovered in Wisconsin in 1966 [Citation1] and was later reported in Guangdong in 1992 [Citation2]. Currently, it is the most widespread low-pathogenicity AIV in China, and co-infection with other pathogens can lead to high mortality in poultry and significant economic losses. The H9N2 AIVs have been identified as sources of internal donor genes for human-infecting AIVs, such as H5N1, H7N9, and H10N8 [Citation3–7]. Several cases of human H9N2 infections have been reported, providing an opportunity to develop potentially pandemic H9N2 strains [Citation8].
H9N2 AIVs can be divided into Eurasian and American lineages based on the hemagglutinin (HA) gene. The Eurasian lineage includes the h9.4.2 sub-lineage, which is composed of six subtypes (h9.4.2.1–h9.4.2.6). The h9.4.2.5-lineage is the most prevalent in China [Citation9–13]. The G57 genotype or S genotype dominates in China based on a genotype analysis of eight gene segments [Citation3,Citation5]. Vaccine immunization is used to prevent and control H9N2 AIVs in China since the first H9N2 subtype avian influenza inactivated vaccine (A/Chicken/Shanghai/F/98 strain) was approved in 2002 [Citation14,Citation15]. However, H9N2 AIVs can easily undergo antigenic drift under vaccine immunization pressure [Citation14,Citation16]. The results of antigenic cartography based on the cross-hemagglutination inhibition (HI) and cross-microneutralization (MN) tests showed that H9N2 isolates and vaccines belonged to different antigenic groups. Challenge experiments also showed that the vaccines provided incomplete protection against some H9N2 AIV isolates [Citation17–20], indicating that the mutated H9N2 AIVs may become a prevailing strain.
Antibody escape mutant screening, identification of molecular markers, and bioinformatics analysis are techniques commonly employed to analyze variations in HA proteins [Citation21–23]. Numerous critical antigenic residues have been identified in the head of the H9N2 HA [Citation22,Citation24–28]. Recently, H9N2 HA genetic analysis has revealed the emergence of new branches [Citation29]. The antigenic properties between subgroup I (emerged in August 1992) and subgroups II (emerged in September 1996), III (emerged in September 2010) in BJ/94-like may associate with 11 residues changes [Citation22]. Additionally, several mutant residues in the H9N2 HA head play a significant role in modifying the antigenicity between h9.4.2.5 and other lineages or some epidemic strains [Citation10,Citation30]. However, the characteristics of antigenic classification and critical antigenic residues in h9.4.2.5 in China remain unclear. In this study, we analyzed the antigenic profile for the h9.4.2.5-lineage of the H9N2 subtype AIVs using 12 monoclonal antibodies (mAbs) and assessed the effect of antibody escape residues on the antigenic drift of H9N2 AIVs.
Materials and methods
Cell and virus
Madin-Darby canine kidney (MDCK) cells were cultured in Minimum Essential Medium (MEM; HyClone, USA) containing 10% fetal bovine serum (FBS), and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; HyClone, USA) containing 10% FBS. The H9N2 AIV strains A/chicken/Shanghai/F/98 (F98) and A/chicken/Taixing/10/2010 (TX) were stored in our lab [Citation31]. Twenty-two H9N2 AIV strains were isolated from poultry in Jiangsu Province, China, from 2017 to 2019, and propagated in 10-d-old specific pathogen-free (SPF) embryonated chicken eggs, 12 of which were purified using viral rescue. The HA genes of these viruses were amplified using Hoffman primers and cloned into the plasmid pHW2000 to generate recombinant viruses [Citation32,Citation33]. The co-transfection plasmids contained the NA gene from the TX strain and internal genes from A/Puerto Rico/8/1934 (PR8). To construct recombinant viruses with HA mutations, mutant HA genes from the A/goose/Jiangyin/JY100405/2018 (100405) and A/chicken/Jiangyin/JY120118/2018 (120118) strains were generated using the Mut Express MultiS Fast Mutagenesis Kit V2 (Vazyme, Nanjing, China). Transfection of plasmids, virus identification, and passaging were performed following the procedures described in a previous study [Citation31,Citation34]. Detailed information on the viruses is provided in Tables S1, 2, and 3, and the primers used for the mutation are shown in Table S4.
Preparation of mAbs and screening of antibody escape mutants
To generate hybridoma cell lines secreting mAbs, BALB/c mice were immunized subcutaneously with the inactivated and purified virus every 2 weeks. Mice were first immunized with Freund’s complete adjuvant, followed by subsequent immunization with Freund’s incomplete adjuvant. Three days before fusion, mice were intraperitoneally immunized with the inactivated purified virus. The mAbs were screened using the HI assay and mAb escape mutants were selected using a previously described protocol [Citation27].
Total RNA extraction and sequencing
Total viral RNA was extracted using TRIzol Reagent (Vazyme, Nanjing, China), and reverse transcription-polymerase chain reaction (RT–PCR) was performed using universal primers [Citation32]. Amplified PCR products were sequenced by Sangon Biotech Company (Shanghai, China).
Three-dimensional (3D) structure rendering
HA monomers H9N2 and H3N2 (PDB ID: 1JSD and 3VUN, respectively) were aligned using PyMOL to compare their 3D structures. Five distinct antigenic sites (A, B, C, D, and E) of H9N2 AIVs were mapped based on the H3N2 HA structure [Citation35,Citation36].
Serological assays
For antigenic profiling, HI assays were performed using standard methods [Citation37]. The mAbs were obtained from the mouse ascites fluid, and polyclonal antibodies were pooled from the sera of the vaccinated chickens. Heatmap analysis was performed using the OmicStudio tool (https://www.omicstudio.cn/tool). Antigenic distances were calculated using antigenic cartography (http://www.antigenic-cartography.org). The MN assay was performed on MDCK cells as described previously study [Citation27,Citation38]. The 50% protective dose (PD50) was calculated using the Reed-Muench assay.
Solid-phase direct binding and receptor-destroying enzyme-based avidity assay
Solid-phase binding assays using 3′-Sialyl-N-acetyllactosamine-biotin (3`SLN, Dextra, UK), 6′-Sialyl-N-acetyllactosamine-biotin (6`SLN, Dextra, UK) and 32 HA units viruses per well were conducted as previously study [Citation39]. A receptor-destroying enzyme-based avidity assay was performed as described previously [Citation21,Citation40]. The chicken erythrocytes were treated with different concentrations (200–0.05 U/0.2 mL) of the receptor-destroying enzyme (RDE) α2-3,6,8 Neuraminidase (NEB, USA) and the data are expressed as the maximal concentration of RDE that allowed full agglutination.
Phylogenic and selection pressure analyses
6342 HA gene sequences of avian H9N2 AIVs isolated in China between 1996 and 2021 were downloaded from the Global Initiative on Sharing Avian Influenza Data (GISAID; https://platform.epicov.org/). Redundant sequences with >95% identity were removed using BioAider software (V1.334). The neighbor-joining (NJ) tree of the HA nucleotide sequences (open reading frame) was analyzed using MEGA (version 7.0.14) software with 1000 bootstrap replicates. The Interactive Tree of Life (ITOL; https://itol.embl.de/) software was used to manage and optimally visualize the tree [Citation41]. The mean genetic distance was analyzed using MEGA (version 7.0.14) software based on HA nucleotide sequences (open reading frame). Sequences with >95% identity in each branch were re-identified using TBtools (V1.098769) [Citation42] based on the classification record generated in BioAider (V1.334). The Mixed Effects Model of Evolution (MEME), Fast Unconstrained Bayesian AppRoximation (FUBAR), Fixed Effects Likelihood (FEL), and Single-Likelihood Ancestor Counting (SLAC) tools were used to estimate selection pressure using the Datamonkey web server (http://www.datamonkey.org/analyses) [Citation43]. Positive selection sites were identified using at least three out of four methods with P-value <0.1 (MEME, FEL and SLAC) or posterior probability >0.9 (FUBAR) [Citation44].
Statistical analysis
Statistical significance was analyzed using one-way analysis of variance (ANOVA) with GraphPad Prism 7 software. P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001). Excel was used to calculate the amino acid mutation rates and the proportion of branches. A heatmap and bar graph were generated using the OmicStudio tool (https://www.omicstudio.cn/tool).
Results
H9N2 AIV isolates from 2017 to 2019 exhibited antigen variation
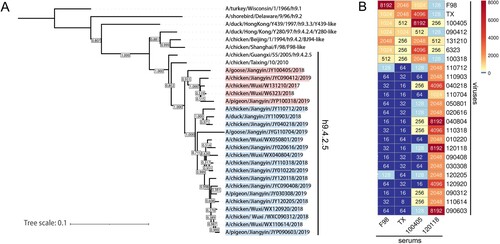
Phylogenetic analysis indicated that the HA genes of the H9N2 AIV isolates from 2017 to 2019 belonged to the h9.4.2.5-lineage (A). The HI assay revealed that the antisera from 100405 vaccinated chickens displayed higher cross-HI titres (HI titres ≥512) against isolates 090412, 131210, 6323, and 100318, and previous strains F98 and TX, but lower cross-HI titres (HI titres ≤256) against the other 17 strains. The antisera from 120118 vaccinated chickens displayed lower cross-HI titres (most HI titres ≤256) against isolates 100405, 090412, 131210, 6323, and 100318, and strains F98 and TX, but higher cross-HI titres (HI titres >512) against the other 17 H9N2 isolates (B). These data indicate that the H9N2 AIV isolates from 2017 to 2019 could be classified into two groups according to their antigenicity. Since strains 100405 and 120118 shared 94.2%−95.3% and 94.6%−98.8% nucleotide identity and good cross-reactivity with other strains of their respective groups, the two strains were chosen as the parental viruses.
Figure 1. Phylogenetic evolutionary and cross-HI assays. (A) NJ tree of HA gene of several representative strains and 22 H9N2 viruses isolated from 2017 to 2019. (B) Cross-HI results between F98, TX, and 22 H9N2 viruses to the serum of F98, TX, 100405, and 120118.
MAb escape residues were located on antigenic sites A, B, and E of HA of H9N2 viruses
The HI assay identified two mAbs against F98, one against TX, and four against 120118. These seven mAbs and five other mAbs against F98 and TX [Citation27] were used to select mAb escape variants. All 12 mAbs showed high HI titres (HI titres ≥28) and MN titres (PD50 ≥ 101.8) against the parent strains (). The results of the HI profiling analysis revealed that the mAbs prepared from F98 and TX reacted well with group 1 viruses (most HI titres >512) and exhibited low reactivity with group 2 viruses (most HI titres<256). However, the mAbs against the 120118 isolate showed lower HI titres (most HI titres <256) to group 1 viruses and higher HI titres (most HI titres ≥ 512) to group 2 viruses (A). The HI titre in the escape strain was reduced by at least 5log2 (m5D3, m3F6, m10G8, m1C9, m1C11, m5E5, m4D7, and m6C2) or completely abolished (m3E6, m2F7, m3F9, and m2D6) compared to the parent strain (). The escape residues of the mAbs against strains F98 and TX were R92K, S145N, D153E, N166D, N167D, A168D/T, T197A, T200I, P247S, and I386M, whereas those of the mAbs against strain 120118 were D145N, G153D, T198K, and S201R. In alignment with the above and other reported antigenic residues in H9N2 HA [Citation45], we found that residues 92, 247 and 386 lacked consistent mutations across groups. Residues 90, 149, and 164 showed consistent mutations (B). Therefore, the selected 12 mutant residues, including G90E, S145D, K149T/N, D153G, Q164R, N166D, N167G, A/V168N/E, T197D/N, V/A198T, T200R, and N201S/R/G, were mutated in groups 1 and 2, and were more conserved in group 1 (B). All residues except for residue 90 were located close to the receptor-binding site (RBS). Residue 90 of HA in H9N2 was located close to antigenic site E (H3 was used as a reference); residues 145, 149, and 153 were located in antigenic site A; and residues 164, 166, 167, 168, 197, 198, 200, and 201 were located in antigenic site B (C). Compared to Site I, Overlapping area, Site II defined by Kaverin [Citation24] and H9-A, H9-B defined by Peacock [Citation28], the H3 modelling antigenic sites encompassed more of the 12 mutated residues we selected (Table S5).
Figure 2. HI profiling analysis and screening of critical antigenic sites in H9N2 viruses. (A) Cross-HI of H9N2 viruses with mAbs. (B) HA amino acids alignment (H9 number with signal peptide). (C) Structural modelling of antigenic sites in HA. RBS is represented by six conserved sites, 109, 110, 148, 161, 191, and 202 (H9 number with signal peptide). H3 antigenic sites A, B, C, D, and E have been described in previous studies [Citation35,Citation36].
![Figure 2. HI profiling analysis and screening of critical antigenic sites in H9N2 viruses. (A) Cross-HI of H9N2 viruses with mAbs. (B) HA amino acids alignment (H9 number with signal peptide). (C) Structural modelling of antigenic sites in HA. RBS is represented by six conserved sites, 109, 110, 148, 161, 191, and 202 (H9 number with signal peptide). H3 antigenic sites A, B, C, D, and E have been described in previous studies [Citation35,Citation36].](/cms/asset/eaf5b716-4dc8-46bb-a34e-edf6226b3fcd/temi_a_2246582_f0002_oc.jpg)
Table 1. Biological properties of mAbs and mutations in the HA of mAb escape mutants.
The 12 mutant residues affected receptor-binding avidity and caused the antigenic drift
Strains 100405 and 120118 showed receptor-binding avidity for both 3`SLN and 6`SLN in the solid-phase binding assay, but 100405 exhibited a stronger binding specifically to 6`SLN. Mutations 405-T200R, 405-AE and 405-AB1E highly increased binding avidity towards 3`SLN, while mutations 405-G90E, 405-S145D, 405-Q164G(W160R), 405-201S, 405-B1, 405-B2, 405-AE, 405-AB1E substantially reduced binding to 6`SLN (). Notably, the 405-D153G displayed elevated receptor binding to both 3`SLN and 6`SLN, and exhibited significantly increased binding avidities in RDE assay. In addition, mutations 405-A168N, 405-V198T(N191T), and 405-T200R also showed significantly increased binding avidities in the RDE assay (Fig S1). For the strain 120118, mutations 118-D197 T, 118-B1, 118-B2, 118-AB1E, and 118-AB1B2E revealed increased receptor binding to both 3`SLN and 6`SLN, while 118-145S, 118-T149 K, 118-D166N, 118-T198V, 118-S201N, and 118-A exhibited a stronger binding to 6`SLN (). Moreover, mutations 118-T149K, 118-D197T, 118-T198V, and 118-AB1B2E also displayed significantly increased binding avidities in the RDE assay (Fig S1).
Figure 3. Receptor preference of 100405 recombinant viruses in the solid-phase binding assay. In multi-site mutants of 100405, the mutations A, B1, B2 and E represent SKD145 149 153DTG, QNNQ164 166 167 168RDGN, TVTN197 198 200 201DTRS and G90E mutations, respectively. 405-T197D and 405-A were excluded from solid-phase direct binding assays due to the low HA titres.
Figure 4. Receptor preference of 120118 recombinant viruses in the solid-phase binding assay. In multi-site mutants of 120118, the mutations A, B1, B2 and E represent DTG145 149 153SKD, RDGN164 166 167 168 QNNQ, DTRS197 198 200 201TVTN and E90G mutations, respectively.
The antigenicity of the recombinant viruses was assessed by HI testing of their reactivity with homologous and heterologous sera. Antigenic cartography revealed that 405-D153G, 405-Q164G (W160R), 405-AE, 405-AB1E, 118-D197T, 118-A, 118-B1, 118-B2, 118-AE, 118-AB1E, and 118-AB1B2E caused at least 2.18 AU antigenic drifts (3.23, 3.05, 2.18, 4.10, 2.51, 4.16, 4.71, 4.07, 4.11, 5.95, and 7.45 AU, respectively), compared to the parental viruses (A).
Figure 5. The antigenic drift of recombinant 100405 and 120118 viruses. (A) Antigen cartography based on the cross-HI assay. One unit (grid) represents a 2-fold change in the HI assay. 100405 and its single-site mutants are indicated in blue, and its multi-site mutants are indicated in cyan. 120118 and its single-site mutants are indicated in red and its multi-site mutants are indicated in orange. 100405-A did not stably agglutinate chicken erythrocytes but had an available TCID50 titre in MDCK cells. Therefore, it was only analyzed using the MN assay. (B and C) PD50 titres of serums against recombinant 100405 viruses in MN assay. (D and E) PD50 titres of serums against recombinant 120118 viruses in MN assay. a: one set of results were below the test line. b: two sets of results were below the test line.
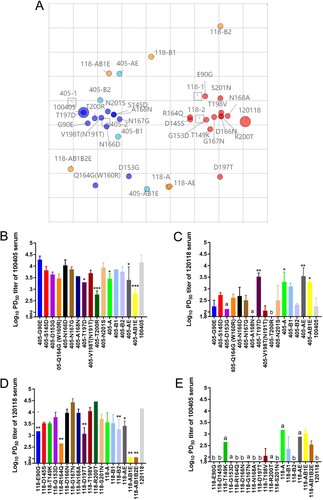
The MN assay showed that, compared to the parental virus 100405, mutant strains 405-T197D, 405-T200R, 405-A, 405-AE, and 405-AB1E exhibited a significantly decreased PD50 of 100405 antisera, whereas the mutant strains 405-T197D, 405-A, 405-AE, and 405-AB1E showed a significant increased PD50 against 120118 antisera (B and C). In contrast, when compared to the parental virus 120118, mutant strains 118-E90G, 118-R164Q, 118-D197T, 118-B2, 118-AE, 118-AB1E, and 118-AB1B2E showed a significantly decreased PD50 against 120118 antisera, whereas mutant strains 118-AB1E and 118-AB1B2E showed an increased PD50 against 100405 antisera (D and E). These data indicate that residues 90, 145, 149, 153, 164, 166, 167, and 168 play critical roles in antigen drift between these two groups.
The h9.4.2.5-lineage of H9N2 AIVs was divided into two branches
A phylogenetic tree was generated to analyze the evolution of the HA gene of H9N2 AIVs isolated in China from 1996 to 2021. The h9.4.2.5-lineage was divided into two branches, with groups 1 and 2 viruses distributed in branches 1 and 2, respectively. Most of the 12 residues were mutated between branches 1 and 2 (). The amino acid mutation rates at 12 mutant residues of all avian H9N2 AIVs isolated in China from 1996 to 2021 were calculated. The 90G/E, 145S, 149K, 153D, 164Q, 166N/D, 167N, 168A/N, 197 T, 198A/V/T, 200 T, and 201N were dominant in h9.4.2-lineage (not including h9.4.2.5), whereas 90G, 145S, 149K, 153D, 164Q, 166N, 167N, 168A, 197T, 198A/V, 200T, and 201N were dominant in branch 1 (Table S6). Therefore, the 12 mutant residues of group 1 were consistent with those of branch 1 and similar to those of the h9.4.2-lineage without h9.4.2.5. In contrast, branch 2 contained 90E, 145D, 149T/N, 153G, 164R, 166N/D, 167G, 168N/D/E, 197D/T, 198 T, 200R, and 201N/D/G as the dominant residues. The 12 mutant residues in group 2 were similar to those in branch 2, except for 166N (68.32%) and 201N (33.89%) (Table S6). These data indicate that these 12 antigenic residues can serve as molecular markers for branch classification.
Figure 6. HA gene phylogenetic evolutionary and 12 critical antigenic residues alignment of avian H9N2 AIV from 1996 to 2021 in China.
Genetic distance analysis revealed that the h9.4.2.5-lineage of H9N2 AIVs exhibited the largest genetic distance within the lineage (8.77%) and between the lineages (12.93–15.32%) among the six tertiary branches of h9.4.2. In contrast to the distances between the other tertiary branches and branch 1 of the h9.4.2.5-lineage (9.85-13.40%), branch 2 displayed greater genetic distance (9.85-16.21%). Furthermore, there was a large genetic distance (9.85%) between branch 1 and branch 2 (Table S7).
Positive selection analysis revealed 8, 11, and 55 positively selected sites in h9.4.2 without h9.4.2.5, branch 1, and branch 2, respectively. Furthermore, residue 198 was under positive selection in h9.4.2 (not excluding h9.4.2.5). Residues 145, 168, 197 and 198 were under positive selection in branch 1, and antigenic residues 145, 149, 164, 168, 200, and 201 were under positive selection in branch 2 (Table S8). These findings suggest greater evolutionary pressure in branch 2 and that the antigenic sites screened were indeed subject to positive selection in their natural state.
A new antigenic branch in h9.4.2.5-lineage became circulating
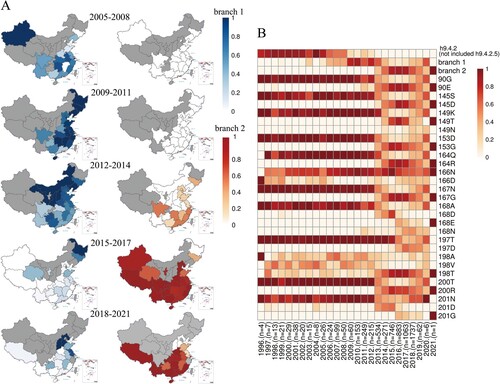
Before 2005, h9.4.2 (not excluding h9.4.2.5) was dominant in China. Branch 1 viruses of h9.4.2.5 have been predominantly found in Xinjiang and several southern regions since 2005 and have gradually extended to the northeast, eventually becoming the major circulating viruses in most areas of China from 2009 to 2011. However, from 2012 to 2014, the prevalence of branch 1 viruses decreased slightly in several southern regions, where branch 2 of h9.4.2.5 viruses emerged. From 2015 to 2017, branch 2 viruses spread rapidly throughout China, replacing branch 1 viruses with the major circulating viruses. Notably, the prevalence of branch 2 viruses decreased slightly, whereas that of branch 1 viruses increased in several northern regions from 2018 to 2020 (A and B, and Table S9). The prevalence of 12 amino acid residues was similar to that of branch 1, branch 2, and the h9.4.2-lineage without h9.4.2.5 from 1996 to 2021 (B).
Figure 7. Isolation information of H9N2 AIVs in China from 1996 to 2021.(A) The percentages of branches 1 and 2 in each region from 2005 to 2021. The period during which no H9N2 viruses were counted is indicated in grey. (B) The annual isolation rates of h9.4.2 lineage without h9.4.2.5, branch 1, and branch 2, and annual natural mutation rates at the 12 antigenic residues. “n” below the picture refers to the total number of H9N2 AIV samples for that year.
Discussion
In China, the HA genes of most H9N2 isolates currently belong to the h9.4.2.5 lineage. The viruses isolated from 2011 to 2012 shared 92–93.5% nucleotide identity with the HA gene of classical vaccine strains, whereas the viruses collected from 2013 to 2015 shared 90–92% nucleotide identity with the vaccine strains [Citation12]. In this study, 22 H9N2 AIVs isolated from 2017 to 2019 were divided into two antigenic groups according to the cross-HI assay of chicken antisera and mAbs. The two groups contained 12 residue mutations based on mAb escape sites and sequence analysis. The h9.4.2.5-lineage of H9N2 AIVs in China could be divided into two branches, and mutant sites on the HA genes of the two branches were consistent with the grouping of the mAb HI profiling analysis (). The genetic distance between the branch 1 and branch 2 viruses was 9.85%, reaching the genetic distance between the tertiary branches (Table S7). Branch 1 viruses (group 1) showed HI reactivity similar to that of the classical vaccine strain F98. However, branch 2 (group 2) exhibited a difference in HI reactivity between branch 1 and F98-like viruses (B and 2A).
It has been reported that R92K was responsible for the decreased binding of 1C9 [Citation27]. However, 1C9 exhibited low reactivity against nine group 2 viruses harbouring 92R. The escape residues could not explain the reactivity of 2D6 (D153E) to 110712 (153D), 3F6 (T200I) to TX (200T), and 6C2 (S201R) to 110704 (201N) (A and B).
H9N2 AIVs with a single mutation at the reported escape residues had small HI titre changes against parental antisera, indicating that mouse mAbs do not consistently recognize the same epitopes as chicken serum, or that there may be differences in immunodominance [Citation21]. Evidence suggests that residue substitutions in the HA antigenic site in H3N2 and H2N2 cause different antigenic cluster transitions, with seven residues at antigenic sites A or B being the primary determinants [Citation36,Citation46–48]. Simultaneous mutation of 12 sites in 120118 was successfully rescued, whereas simultaneous mutation of 12 sites in 100405 was not. Therefore, we used the simultaneous mutation of eight sites in 100405 for HI and MN assays. Our study showed that the simultaneous mutation of eight antigenic sites (90, 145, 149, 153, 164, 166, 167, and 168) on the HA of H9N2 AIVs could transform the antigenicity from group 1 viruses to group 2 viruses, and vice versa, indicating that the eight antigenic sites located at antigenic sites A, B, and E can be molecular markers for antigenic drift and branch classification (C and 5).
Amino acid mutations can increase the receptor-binding avidity to both avian-like α2,3-linked sialic acids and human-like α2,6-linked sialic acids, which may reduce antibody binding [Citation40,Citation49,Citation50]. The mutants 405-D153G, 405-T200R, 405-AE, 405-AB1E, 118-D197T, 118-A, 118-B1, 118-B2, 118-AE, 118-AB1E, and 118-ABE1 showed increased viral binding avidity towards 3`SLN ( and 4), which resulted in decreased antibody titres in HI and/or MN assays (). Therefore, the increased 3`SLN preference of H9N2 AIVs appears a positive correlation with antigenic drift.
Positively selected codons in H3 are associated with immune escape and are useful in predicting future epidemics [Citation23]. Both the total number of positively selected sites and the number of positively selected antigenic sites in branch 2 were higher than those in h9.4.2 (not excluding h9.4.2.5), except for h9.4.2.5, and branch 1 (Table S8). As branch 2 has only been prevalent since 2013, these sites are more likely to undergo rapid evolution and antigenic drift in branch 2 viruses, especially the 12 antigenic sites screened in this study.
In China, 28 H9N2 vaccines had been approved by 2021, all of which were isolated before 2012 [Citation14]. However, since 2015, the H9N2 strain has undergone significant antigenic drift. We hypothesize that branch 2, prevalent from 2015 to 2021, may have arisen under vaccine pressure. Notably, the old branch 1 was not completely replaced from 2015 to 2017 and has become prevalent again in several regions from 2018 to 2021 (). Considering the co-prevalence of branches 1 and 2 in several regions after 2018, an appropriate immunization strategy based on these two antigenic branches is required.
In summary, our study demonstrated that 12 antigenic residues were molecular markers of the two antigenic branches in h9.4.2.5. Most of these residues were located in the immunodominant antigenic sites A and B and were mutated under serum pressure, leading to an antigenicity transition. Therefore, an immunization strategy should be developed based on the prevalence of these two antigenic branches in China. This study provides a theoretical basis for analyzing the molecular mechanisms of antigenic drift and selecting vaccine candidates that contest with the current antigenicity of H9N2 AIVs.
Compliance and ethics
All experiments were approved by the Jiangsu Laboratory Animal Welfare guidelines and the Ethicals of the Jiangsu Administrative Committee of Laboratory Animals (Permit number SYXKSU-2017-0044).
Supplemental Material
Download JPEG Image (349.3 KB){kind=link}
Supplemental Material
Download MS Word (257.7 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Homme PJ, Easterday BC. Avian influenza virus infections. I. Characteristics of influenza A-turkey-Wisconsin-1966 virus. Avian Dis. 1970 Feb;14(1):66–74. doi:10.2307/1588557
- Sun Y, Liu J. H9n2 influenza virus in China: a cause of concern. Protein Cell. 2015 Jan;6(1):18–25. doi:10.1007/s13238-014-0111-7
- Pu J, Wang S, Yin Y, et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc Natl Acad Sci U S A. 2015 Jan 13;112(2):548–553. doi:10.1073/pnas.1422456112
- Guan Y, Shortridge KF, Krauss S, et al. Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci U S A. 1999 Aug 3;96(16):9363–9367. doi:10.1073/pnas.96.16.9363
- Gu M, Chen H, Li Q, et al. Enzootic genotype S of H9N2 avian influenza viruses donates internal genes to emerging zoonotic influenza viruses in China. Vet Microbiol. 2014 Dec 5;174(3-4):309–315. doi:10.1016/j.vetmic.2014.09.029
- Liu D, Shi W, Gao GF. Poultry carrying H9N2 act as incubators for novel human avian influenza viruses. Lancet. 2014 Mar 8;383(9920):869. doi:10.1016/S0140-6736(14)60386-X
- Arai Y, Ibrahim MS, Elgendy EM, et al. Genetic compatibility of reassortants between avian H5N1 and H9N2 influenza viruses with higher pathogenicity in mammals. J Virol. 2019 Feb 15;93(4):e01969–18. doi:10.1128/JVI.01969-18
- Xu J, Li S, Yang Y, et al. Human infection with a further evolved avian H9N2 influenza A virus in sichuan, China. Sci China Life Sci. 2018 May;61(5):604–606. doi:10.1007/s11427-017-9150-8
- Jiang W, Liu S, Hou G, et al. Chinese and global distribution of H9 subtype avian influenza viruses. PloS one. 2012;7(12):e52671. doi:10.1371/journal.pone.0052671
- Liu Q, Zhao L, Guo Y, et al. Antigenic evolution characteristics and immunological evaluation of H9N2 avian influenza viruses from 1994-2019 in China. Viruses. 2022 Mar 30;14(4):726. doi:10.3390/v14040726
- Liu S, Ji K, Chen J, et al. Panorama phylogenetic diversity and distribution of type A influenza virus. PloS one. 2009;4(3):e5022. doi:10.1371/journal.pone.0005022
- Zhu R, Xu D, Yang X, et al. Genetic and biological characterization of H9N2 avian influenza viruses isolated in China from 2011 to 2014. PloS one. 2018;13(7):e0199260.
- Xia J, Cui JQ, He X, et al. Genetic and antigenic evolution of H9N2 subtype avian influenza virus in domestic chickens in southwestern China, 2013-2016. PloS one. 2017;12(2):e0171564.
- Cao Y, Liu H, Liu D, et al. Hemagglutinin gene variation rate of H9N2 avian influenza virus by vaccine intervention in China. Viruses. 2022 May 13;14(5):1043. doi:10.3390/v14051043
- Gu M, Xu L, Wang X, et al. Current situation of H9N2 subtype avian influenza in China. Vet Res. 2017 Sep 15;48(1):49. doi:10.1186/s13567-017-0453-2
- Zhang P, Tang Y, Liu X, et al. Characterization of H9N2 influenza viruses isolated from vaccinated flocks in an integrated broiler chicken operation in eastern China during a 5 year period (1998-2002). J Gen Virol. 2008 Dec;89(Pt 12):3102–3112. doi:10.1099/vir.0.2008/005652-0
- Belser JA, Sun X, Brock N, et al. Genetically and antigenically divergent influenza A(H9N2) viruses exhibit differential replication and transmission phenotypes in mammalian models. J Virol. 2020 Aug 17;94(17):e00451–20. doi:10.1128/JVI.00451-20
- Xu C, Ye H, Qiu W, et al. Phylogenetic classification of hemagglutinin gene of H9N2 avian influenza viruses isolated in China during 2012-2016 and evaluation of selected candidate vaccine strains. Poult Sci. 2018 Sep 1;97(9):3023–3030. doi:10.3382/ps/pey154
- Wei Y, Xu G, Zhang G, et al. Antigenic evolution of H9N2 chicken influenza viruses isolated in China during 2009-2013 and selection of a candidate vaccine strain with broad cross-reactivity. Vet Microbiol. 2016;182:1–7. doi:10.1016/j.vetmic.2015.10.031
- Sun Y, Pu J, Jiang Z, et al. Genotypic evolution and antigenic drift of H9N2 influenza viruses in China from 1994 to 2008. Vet Microbiol. 2010;146(3-4):215–225. doi:10.1016/j.vetmic.2010.05.010
- Peacock TP, Harvey WT, Sadeyen JR, et al. The molecular basis of antigenic variation among A(H9N2) avian influenza viruses. Emerg Microbes Infect. 2018 Nov 7;7(1):176. doi:10.1038/s41426-018-0178-y
- Yan W, Cui H, Engelsma M, et al. Molecular and antigenic characterization of avian H9N2 viruses in Southern China. Microbiol Spectr. 2022 Feb 23;10(1):e0082221.
- Bush RM, Fitch WM, Bender CA, et al. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol. 1999 Nov;16(11):1457–1465. doi:10.1093/oxfordjournals.molbev.a026057
- Kaverin NV, Rudneva IA, Ilyushina NA, et al. Structural differences among hemagglutinins of influenza A virus subtypes are reflected in their antigenic architecture: analysis of H9 escape mutants. J Virol. 2004 Jan;78(1):240–249. doi:10.1128/JVI.78.1.240-249.2004
- Okamatsu M, Sakoda Y, Kishida N, et al. Antigenic structure of the hemagglutinin of H9N2 influenza viruses. Arch Virol. 2008;153(12):2189–2195. doi:10.1007/s00705-008-0243-2
- Wan Z, Ye J, Xu L, et al. Antigenic mapping of the hemagglutinin of an H9N2 avian influenza virus reveals novel critical amino acid positions in antigenic sites. J Virol. 2014 Apr;88(7):3898–3901. doi:10.1128/JVI.03440-13
- Zhu Y, Yang D, Ren Q, et al. Identification and characterization of a novel antigenic epitope in the hemagglutinin of the escape mutants of H9N2 avian influenza viruses. Vet Microbiol. 2015 Jul 9;178(1-2):144–149. doi:10.1016/j.vetmic.2015.04.012
- Peacock T, Reddy K, James J, et al. Antigenic mapping of an H9N2 avian influenza virus reveals two discrete antigenic sites and a novel mechanism of immune escape. Sci Rep. 2016 Jan 7;6:18745.
- Dong J, Zhou Y, Pu J, et al. Status and challenges for vaccination against avian H9N2 influenza virus in China. Life. 2022 Aug 27;12(9):1326.
- Zheng Y, Guo Y, Li Y, et al. The molecular determinants of antigenic drift in a novel avian influenza A (H9N2) variant virus. Virol J. 2022 Feb 5;19(1):26. doi:10.1186/s12985-022-01755-9
- Chen S, Zhu Y, Yang D, et al. Efficacy of live-attenuated H9N2 influenza vaccine candidates containing NS1 truncations against H9N2 avian influenza viruses. Front Microbiol. 2017;8:1086. doi:10.3389/fmicb.2017.01086
- Hoffmann E, Stech J, Guan Y, et al. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146(12):2275–2289. doi:10.1007/s007050170002
- Hoffmann E, Neumann G, Kawaoka Y, et al. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A. 2000 May 23;97(11):6108–6113. doi:10.1073/pnas.100133697
- Qin T, Chen Y, Huangfu D, et al. PA-X protein of H1N1 subtype influenza virus disables the nasal mucosal dendritic cells for strengthening virulence. Virulence. 2022 Dec;13(1):1928–1942. doi:10.1080/21505594.2022.2139474
- Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981 Jan 29;289(5796):373–378. doi:10.1038/289373a0
- Smith DJ, Lapedes AS, de Jong JC, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004 Jul 16;305(5682):371–376. doi:10.1126/science.1097211
- Spackman E. Animal influenza virus: methods and protocols. New York (NY): Humana Press; 2020.
- Steel J, Lowen AC, Wang TT, et al. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. mBio. 2010 May 18;1(1):e00018–10. doi:10.1128/mBio.00018-10
- Gao R, Gu M, Liu K, et al. T160a mutation-induced deglycosylation at site 158 in hemagglutinin is a critical determinant of the dual receptor binding properties of clade 2.3.4.4 H5NX subtype avian influenza viruses. Vet Microbiol. 2018 Apr;217:158–166. doi:10.1016/j.vetmic.2018.03.018
- Hensley SE, Das SR, Bailey AL, et al. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science. 2009 Oct 30;326(5953):734–736. doi:10.1126/science.1178258
- Letunic I, Bork P. Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021 Jul 2;49(W1):W293–W296. doi:10.1093/nar/gkab301
- Chen C, Chen H, Zhang Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020 Aug 3;13(8):1194–1202. doi:10.1016/j.molp.2020.06.009
- Weaver S, Shank SD, Spielman SJ, et al. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 2018 Mar 1;35(3):773–777. doi:10.1093/molbev/msx335
- Grinev A, Chancey C, Volkova E, et al. Genetic variability of west Nile virus in U.S. blood donors from the 2012 epidemic season. PLoS Negl Trop Dis. 2016 May;10(5):e0004717. doi:10.1371/journal.pntd.0004717
- Carnaccini S, Perez DR. H9 influenza viruses: an emerging challenge. Cold Spring Harbor Perspect Med. 2020 Jun 1;10(6):a038588. doi:10.1101/cshperspect.a038588
- Koel BF, Burke DF, Bestebroer TM, et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013 Nov 22;342(6161):976–979. doi:10.1126/science.1244730
- Linster M, Schrauwen EJA, van der Vliet S, et al. The molecular basis for antigenic drift of human A/H2N2 influenza viruses. J Virol. 2019 Apr 15;93(8):e01907–18. doi:10.1128/JVI.01907-18
- Zost SJ, Wu NC, Hensley SE, et al. Immunodominance and antigenic variation of influenza virus hemagglutinin: implications for design of universal vaccine immunogens. J Infect Dis. 2019 Apr 8;219(Suppl_1):S38–s45. doi:10.1093/infdis/jiy696
- Sealy JE, Peacock TP, Sadeyen JR, et al. Adsorptive mutation and N-linked glycosylation modulate influenza virus antigenicity and fitness. Emerg Microbes Infect. 2020 Dec;9(1):2622–2631. doi:10.1080/22221751.2020.1850180
- Sealy JE, Yaqub T, Peacock TP, et al. Association of increased receptor-binding avidity of influenza A(H9N2) viruses with escape from antibody-based immunity and enhanced zoonotic potential. Emerging Infect Dis. 2018;25(1):63–72. doi:10.3201/eid2501.180616