
ABSTRACT
Vibrio metschnikovii is an emergent pathogen that causes human infections which may be fatal. However, the phylogenetic characteristics and pathogenicity determinants of V. metschnikovii are poorly understood. Here, the whole-genome features of 103 V. metschnikovii strains isolated from different sources are described. On phylogenetic analysis V. metschnikovii populations could be divided into two major lineages, defined as lineage 1 (L1) and 2 (L2), of which L1 was more likely to be associated with human activity. Meanwhile, we defined 29 V. metschnikovii O-genotypes (VMOg, named VMOg1–VMOg29) by analysis of the O-antigen biosynthesis gene clusters (O-AGCs). Most VMOgs (VMOg1 to VMOg28) were assembled by the Wzx/Wzy pathway, while only VMOg29 used the ABC transporter pathway. Based on the sequence variation of the wzx and wzt genes, an in silico O-genotyping system for V. metschnikovii was developed. Furthermore, nineteen virulence-associated factors involving 161 genes were identified within the V. metschnikovii genomes, including genes encoding motility, adherence, toxins, and secretion systems. In particular, V. metschnikovii was found to promote a high level of cytotoxicity through the synergistic action of the lateral flagella and T6SS. The lateral flagellar-associated flhA gene played an important role in the adhesion and colonization of V. metschnikovii during the early stages of infection. Overall, this study provides an enhanced understanding of the genomic evolution, O-AGCs diversity, and potential pathogenic features of V. metschnikovii.
Introduction
The genus Vibrio consists of a group of bacterial species widely distributed in aquatic habitats. Vibrio species can cause diseases in fish, shellfish, mammals, and humans [Citation1]. Of these, the species Vibrio cholerae, the etiological agent of cholera, is perhaps the best known, with illness associated primarily with strains in O-groups O1 and O139 that carry the genes for cholera toxin (CTX) [Citation2]. “Non-cholera” Vibrio spp., including non-CTX producing V. cholerae within O-groups other than O1 and O139, V. parahaemolyticus, V. vulnificus, V. mimicus, V. fluvialis, and V. furnissii have been associated with mild to moderate gastroenteritis in humans; these species, together with V. alginolyticus, have also been linked with wound infections and, in susceptible hosts, bacteremia and sepsis [Citation3–6]. While markers for virulence have been identified for some of these species/strains (i.e. the presence of genes for CTX in V. cholerae and for the thermostable direct hemolysin [TDH] in V. parahaemolyticus), the factors responsible for virulence in others are poorly understood, making it difficult to clearly identify strains that have an increased likelihood of causing human illness.
V. metschnikovii is an emerging environmental pathogen, capable of colonizing shellfish and causing infections in fish. Humans can become infected by ingesting the organism in fish or shellfish, or by exposure to water containing the organism, with human isolates reported from stool, blood, bile, and wounds [Citation7–11]. In patients who are immunocompromised or have underlying liver disease, the microorganism is capable of causing bacteremia, sepsis, and death, despite what would appear to be appropriate antimicrobial therapy [Citation8, Citation10, Citation12].
In the post-genome era, genome-based phylogeny has become a powerful tool for providing insights into the evolution and spread of Vibrio species [Citation13, Citation14]. However, unlike other pathogens in the genus of Vibrio, the global genomic epidemiology, transmission dynamics, and pathogenicity determinants of V. metschnikovii are poorly understood. In a previous study, whole genome sequence data were obtained from strains of V. metschnikovii isolated from bird and water samples in China [Citation15]. V. metschnikovii has also been isolated from a temperate-climate river basin of central Italy [Citation16] and from surface water in southern and eastern Slovakia [Citation17].
O-antigen analysis is a key element in the serotyping of Gram-negative bacteria. A systematic and large-scale analysis of V. metschnikovii O-antigen biosynthesis gene clusters (O-AGCs) is needed to better understand the diversity of their O-genotypes. In terms of pathogenicity, V. metschnikovii has been found to be pathogenic to freshwater-cultured hybrid sturgeon and pigs, horses, and cattle [Citation7]. Some V. metschnikovii isolates from herring in the Norwegian Sea [Citation18] were positive for cytolysin A (hlyA) and thermolabile hemolysin (tlh) genes, raising questions about the potential role of these and other virulence-associated genes in pathogenicity.
In this study, we investigate the global evolution, population structure, and O-AGCs diversity of the species and explore its pathogenicity and transmission dynamics from aquatic niche to human host. Our findings provided data critical to understanding this underestimated pathogen.
Materials and methods
Bacterial strains, genome sequencing and annotation
A total of 103 V. metschnikovii genomes were analyzed in this study (Table S1). Among them, 30 strains were newly sequenced; this includes 29 isolated from various sources and geographic locations in China during years of 2000–2020, and V. metschnikovii type strain JCM 21189T. Another 73 whole genome sequences were acquired from the GenBank (https://www.ncbi.nlm.nih.gov/genbank/) and SRA (https://www.ncbi.nlm.nih.gov/sra/) database of NCBI (accessed in March 2022). Public genomes with less than 200 contigs were selected.
Genomes were sequenced by using the Illumina HiSeq 2000 platform. The reads data were assembled into contigs using SOAP de novo v2.04 [Citation19]. QUAST v5.0.1 [Citation20] software was applied to evaluate the genome assembly. Open reading frame (ORF) prediction and annotation were done by Prodigal v2.6.3 [Citation21] and Prokka v1.13.3 [Citation22] software, with Rapid Annotation using Subsystem Technology (RAST [Citation23], http://rast.nmpdr.org).
Phylogenetic and pangenome analysis of V. metschnikovii
Intergenomic distances were calculated using the GBDP algorithm [Citation24]. A principal co-ordinates analysis (PCoA) was performed using the matrix of V. metschnikovii accessory-genome organized with the gene present or absent. Core genome SNPs (cgSNPs) were called using snippy v4.3.6 software (https://github.com/tseemann/snippy) against the complete genome sequence of V. metschnikovii strain 2012V_1020 (assembly accession: GCA_009665235) as the reference genome. Gubbins v3.2 software (https://github.com/nickjcroucher/gubbins) was used as a recombination-removal tool to mitigate the effect of horizontal sequence transfer on phylogenetic reconstructions [Citation25]. The FastBAPS algorithm was used for rapidly identifying genomic lineages [Citation26]. The maximum-likelihood (ML) tree was constructed based on the non-recombinant sites using IQ-TREE v1.6.12 [Citation27] based on cgSNPs with 1000 bootstrap replicates. The 103 V. metschnikovii genomes were annotated by Prokka v1.13.3 [Citation22] and then all the *.gff files were collected as a local database. The pan-genome matrix was defined using Roary pan-genome pipeline v1.12 (https://sanger-pathogens.github.io/Roary) with a nucleotide identity threshold of 90%.
Identification and annotation of O-AGCs of V. metschnikovii
The sequences of the O-AGCs regions between gmhD and rjg of each V. metschnikovii genome were extracted. The RAST server pipeline was used to predict ORFs and annotate the O-AGCs genes. Web-BLAST (https://blast.ncbi.nlm.nih.gov/Blast) was applied to infer putative functions. Protein domains were found using Pfam program (https://pfam.sanger.ac.uk) with an E value threshold of 0.01 [Citation28]. TMHMM v.2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/) program was used to predict transmembrane domains. Insert sequences (IS) searches were performed using the ISfinder database (https://www-is.biotoul.fr/). In silico O-genotyping system was performed by BLASTn search using the wzx gene (or wzt gene) as a reference gene. Only a perfect match with an identity cutoff of >99% was considered.
Distribution of functional virulence genes and antimicrobial resistance genes
The virulence-associated genes were annotated based on the core dataset of virulence factor database (VFDB [Citation29], http://www.mgc.ac.cn/VFs/). All the protein sequences were searched against the database using BLASTp with a relatively relax cutoff (identity ≥ 60%, length coverage ≥ 30%) and an E-value of 1e-5. The antimicrobial resistance genes were compared by using the comprehensive antibiotic resistance database (CARD, http://arpcard.mcmaster.ca). The BLASTn cutoff values were chosen as follow: an E-value of 1e-5, the identity ≥ 90% and length coverage ≥ 60%. Differences in resistance genes between lineages were statistically compared using the Fisher’s Exact Test.
In vitro cellular infectious, motility and biofilm formation assays
Bacterial infection was performed using the human epidermoid carcinoma cell line Hep-2 (CCC0068; Peking Union Medical College cell resource center) [Citation30]. Cell adhesion and invasion assay was performed in 24-well plates as described previously [Citation31] with some modifications. The lactate dehydrogenase (LDH) released by cells was detected using Cytotox 96 kit (Promega, Madison, USA). The criteria for classifying the cytotoxicity ranks referred to the previous study [Citation30]. All experiments were performed three times in duplicate.
Growth curves of V. metschnikovii strains were determined by measuring the OD600 values after inoculation with fresh medium at ratio of 1/2000 (v/v). Bacterial swimming motility test as well as biofilm formation assays were performed as previously described [Citation32]. Detailed procedures for in vitro cellular infectious, motility and biofilm formation assays are described in the supplementary data.
Construction of flhA mutant and complementary mutant
A V. metschnikovii mutant strain was constructed from clinical strain ZJ20140528 as a wild-type (WT). The suicide plasmid pWM91 [Citation33] was used to construct a flhA mutant (ΔflhA) by homologous recombination. E. coli SM10 λpir served as the donor strain to transfer the recombinant plasmid into the WT by means of conjugative transfer. Incubation culture on LB agar (with 10% sucrose but without NaCl at 22°C for 48 h) was used to select the mutant strain. To construct the complementary mutants (C-ΔflhA), the entire coding region of flhA gene was cloned into plasmid pSRKGm [Citation34]. Then the complementary plasmids were transformed into the above mutant with 0.1% arabinose as an inducer. The primers for the mutation and complementary protocol were described in Tables S2 and S3.
Data availability
The genome sequences of the 30 V. metschnikovi strains sequenced in this study have been deposited at GenBank/DDBJ/ENA under the BioProject ID no. PRJNA900004.
Results
Phylogenetics and population structure of V. metschnikovii
The 103 V. metschnikovii strains were clustered into two distinct lineages (L1 and L2) on the phylogenetic tree ((a)) using the FastBAPS algorithm based on non-recombinant cgSNPs. Similarly, the GBDP-based phylogenomic tree also divided V. metschnikovii populations into two major lineages (Figure S1). Most strains in the L1 were isolated from clinical and food samples, while strains in the L2 were isolated from migratory birds. Using the criteria for defining a clonal group (CG) [Citation35], 17 CGs were identified in this study ((a)). It is noteworthy that the four clinical isolates were nested in L1, which were sourced from three countries (China, the United States, and the United Kingdom), indicating that strains in L1 are more likely to be pathogenic to humans, although the number of clinical strains was limited.
Figure 1. Population structure and phylogenetic analyses of V. metschnikovi. (a) Phylogenetic ML tree based on cgSNPs with 1000 bootstrap replicates. The red and blue branches represented Lineage 1 (L1) and Lineage 2 (L2), respectively. The purple dots on the top of the branches represented the 17 identified clonal groups (CGs). The inner ring showed different isolation sources, and the outer ring showed different isolation regions. (b) Correlation analysis between genetic distances and geographical distances within L1 strains. (c) Correlation analysis between genetic distances and geographical distances within L2 strains. (d) Splits-tree of phylogenetic networks based on the single-copy genes. (e) Heatmap of paired-SNP variants among 103 V. metschnikovii genomes. The colours from blue to red indicated the increase of SNP number. (f) Comparison of the ratios of six mutagenesis types between L1 and L2. (g) Chord diagram between drug resistance genes (DRG) and V. metschnikovii strains. A total of 17 DRG-positive isolates were identified, including 13 in L1 and 4 in L2.
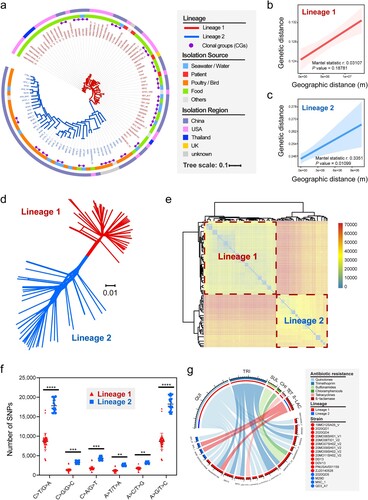
We then assessed whether L1 of V. metschnikovii was preferentially associated with human activity using all 98 strains with well-defined isolation sites (including 59 strains of L1 and 39 strains of L2). By calculating the genetic distances and the actual geographical distances among V. metschnikovii isolates, we found that strains in L1 (including strains isolated from the global human community, i.e. food sources and clinical samples) were more affected by the factor of human activity (Mantel statistic r: 0.1325, P = 0.18781, (b)) than strains in L2 (Mantel statistic r: 0.3351, P = 0.010989, (c)), which were mainly isolated from migratory birds [Citation15].
We re-examined all orthologous sequences of single-copy genes, resulting in a splits-tree of phylogenetic networks ((d)). The two lineages were clearly distinguished in the spatial networks diagram. The heatmap of paired-SNP variants among the 103 genomes also confirmed the phylogenetic differences between the two major lineages ((e)). More transitions than transversions were found in both lineages, and strains in L2 had a higher quantity of mutations than those in L1 (P < 0.001, (f)), especially in the following two types of mutations (shift to C > T/G > A and shift to A > G/T > C).
The distribution of antibiotic resistance genes of V. metschnikovii also strongly indicated a clear evolutionary difference between L1 and L2. Nine kinds of antimicrobial resistance genes belonged to six categories were identified ((g)). Gene qnrVC6 associated with quinolone resistance accounted for the highest positive rate at 12.6% (13/103), followed by trimethoprim resistance gene (dfrA6) and ß-lactamase resistance gene (blaCARB-4), accounting for 5.8% (6/103) and 2.9% (3/103), respectively. Regardless of the category or quantity of drug resistance genes, the positive strains in L1 were significantly more than that in L2 (P < 0.05, (g)), suggesting again that L1 strains is more closely related to human activities, as compared with L2 strains.
O-antigen biosynthesis gene clusters (O-AGCs) of V. metschnikovii
Due to the draft genomes, only 49 contiguous O-AGCs (located in the same contig) were obtained in this study. The 49 O-AGCs were grouped into 29 clearly distinguishable O-antigen genotypes based on gene organization (), defined as VMOg1-VMOg29 respectively, including two sub-groups within VMOg10 (VMOg10-a and VMOg10-b) and four sub-groups within VMOg29 (VMOg29-a, VMOg29-b, VMOg29-c, and VMOg29-d). These four VMO29 subgroups had one or two hypothetical protein gains or losses between the orf19 to orf22 and the orf29 to orf35. Of all VMOgs including six sub-groups, 39.4% of them belonged to L1 (13/33), and the rest were in L2 (n = 20, 60.6%) (). The putative functions of the identified ORFs in these V. metschnikovii VMOgs were listed in Tables S4.1–S4.29.
Figure 2. Genetic structures of the 29 V. metschnikovii O-genotypes identified from the V. metschnikovii reference strains. The different colourful arrows indicated the function, location and direction of gene transcripts in the O-AGCs. For putative glycosyltransferases, in green, types of domains families (family 1, 2, 4, UT, untypable) were indicated instead of gene names.

O-AGCs exhibited a high diversity due to variations in their arrangement and organization, but they mostly contained three major classes of genes, namely nucleotide sugar biosynthesis genes, glycosyltransferase genes, and O-unit processing genes. For nucleotide sugar biosynthesis genes, as an example, rmlBADC genes which are involved in converting glucopyranose-1-phosphate to dTDP-Rha in the deoxythymidine diphosphate (dTDP) sugar pathway, were present in VMOg5, VMOg7, VMOg16 and VMOg29. While eight genotypes (VMOg2, VMOg3, VMOg4, VMOg11, VMOg15, VMOg17, VMOg23, and VMOg24) only contained rmlBA genes, more than half (17/29) contained no nucleotide sugar biosynthesis genes or only a limited number of these genes. For putative glycosyltransferase genes, each genotype contained two to six genes encoding glycosyltransferases, with genes in families 2 and 4 being widely distributed. For O-unit processing genes, wzx/wzy in the Wzx/Wzy pathway or wzm/wzt in the ABC transporter pathway, were identified in V. metschnikovii O-AGCs. Most O-AGCs (97%, 28/29) were assembled by the Wzx/Wzy pathway (Figure S2a, S2b), while only VMOg29-a/b/c/d used the ABC transporter pathway to assemble the O-unit (Figure S2c, S2d). We also developed an in silico O-genotyping system for V. metschnikovii according to the sequence variation of the wzx gene (or wzt gene). Table S5 showed the VMOg distribution of V. metschnikovii with known O-AGCs sequences.
Carriage and distribution of virulence-associated genes in V. metschnikovii
Based on the annotations and predictions of the VFDB database, 161 putative virulence-associated genes involving 19 virulence factors in 9 virulence factor classes were found within the 103 V. metschnikovii genomes, including genes associated with adherence, chemotaxis, motility, secretion system, and toxins ((a), Table S6). Almost all V. metschnikovii isolates contained virulence factors like flagella, cytolysin (hlyA), thermolabile hemolysin (tlh), and mannose sensitive hemagglutinin (MSHA) type IV pili. The type VI secretion system (T6SS) also appears to have evolved in V. metschnikovii populations. More than half of the strains in L1 carry T6SS-related genes. Interestingly, nine strains in L1 which belonged to VMOg29 and formed into two distinct evolutionary branches contained four genes related to the lateral flagella. In contrast, these nine strains of VMOg29 all appear to lack the ugd gene coincidentally, which is related to core tetrasaccharide synthesis in the process of capsular polysaccharide biosynthesis ((a)). The RTX clusters (rtxA, rtxB, rtxC, rtxD), an important virulence factor in V. cholerae encoding the RTX toxin, were only present in four strains in L2. Also, only three strains in L1 carried the T3SS gene cluster ((a)). Putative virulence-associated genes in V. metschnikovii were found to have homologs in other 23 foreign species (or genera). Among these species, V. cholerae, Haemophilus influenzae, Aeromonas hydrophila, Francisella tularensis, and Legionella pneumophila were the top five species sharing homologous virulence genes (Figure S3).
Figure 3. Cytotoxic and genomic-based analyses of the pathogenicity factors in V. metschnikovii. (a) Distribution of virulence-associated genes, O-genotypes, and cytotoxicity ranks. The colours from blue to red represented the identity of the gene sequences. The detail of the different cytotoxicity ranks and O-genotypes are listed on the right. (b) Cytotoxicity value of 30 V. metschnikovii strains. Low cytotoxicity (below 0.3), medium cytotoxicity (between 0.3 and 0.5) and high cytotoxicity (above 0.5). Vibrio cholerae strain N16961 (O1 biovar El Tor) and MO45 (O139) served as cytotoxicity positive controls. (c) Principal component analysis (PCA) of virulence genes for the 30 V. metschnikovii strains. The colours from blue to red represented the values of cos2, and the sizes of the circles represented the contributions of each strain sample. Gene lfhA (accession: VF0474) was described in the VFDB database as a gene encoding lateral flagellar biosynthesis protein in Aeromonas genus. The nomenclature of the flhA gene in V. parahaemolyticus was used for the sake of uniform description [Citation55]. (d) Relationships among cytotoxicity levels, O-genotypes, and virulence factors, according to the binary logistic regression model used in this study. Virulence factors associated with the lateral flagella and the T6SS are marked with red dashed boxes.
![Figure 3. Cytotoxic and genomic-based analyses of the pathogenicity factors in V. metschnikovii. (a) Distribution of virulence-associated genes, O-genotypes, and cytotoxicity ranks. The colours from blue to red represented the identity of the gene sequences. The detail of the different cytotoxicity ranks and O-genotypes are listed on the right. (b) Cytotoxicity value of 30 V. metschnikovii strains. Low cytotoxicity (below 0.3), medium cytotoxicity (between 0.3 and 0.5) and high cytotoxicity (above 0.5). Vibrio cholerae strain N16961 (O1 biovar El Tor) and MO45 (O139) served as cytotoxicity positive controls. (c) Principal component analysis (PCA) of virulence genes for the 30 V. metschnikovii strains. The colours from blue to red represented the values of cos2, and the sizes of the circles represented the contributions of each strain sample. Gene lfhA (accession: VF0474) was described in the VFDB database as a gene encoding lateral flagellar biosynthesis protein in Aeromonas genus. The nomenclature of the flhA gene in V. parahaemolyticus was used for the sake of uniform description [Citation55]. (d) Relationships among cytotoxicity levels, O-genotypes, and virulence factors, according to the binary logistic regression model used in this study. Virulence factors associated with the lateral flagella and the T6SS are marked with red dashed boxes.](/cms/asset/e1163ab6-cafd-4365-a6d4-461638a21900/temi_a_2252522_f0003_oc.jpg)
Cytotoxicity features among V. metschnikovii strains
We performed cytotoxicity assays on a selected set of 30 Chinese V. metschnikovii strains collected as part of this study. By cytotoxicity, the 30 V. metschnikovii strains were divided into three groups, i.e. high-, median- and low- cytotoxicity ((b)). Most strains were classified as median- and low- cytotoxicity (MC and LC). Seven strains were identified as high toxicity (HC), including strain ZJ20140528 isolated from a patient with diarrhea.
We further investigated the relationships between virulence genes, population structure, cytotoxicity levels as well as O-genotypes. By the principal component analysis (PCA), four lateral flagella genes (i.e. lfgG, lfiQ, flhA, lfiN) were the variables that contributed the most to the observed variance in the PCA, followed by gene vgrG and hcp (related to T6SS, (c)).
Using the cytotoxicity data of the 30 V. metschnikovii strains, a Binary Logistic Regression (HC vs. non-HC) was performed to predict correlation between virulence genes and cytotoxicity. Lateral flagella genes were found to be an independent risk factor for HC (Odds ratio = 11.244; 95% CI = 2.725–30.647), together with the prevalence of the complete T6SS gene clusters (including hcp and vgrG, Odds ratio = 15.833; 95% CI = 1.175–28.843). A relatively good accuracy (sensitivity 85.7%, specificity 95.6%) was achieved in identifying whether a given V. metschnikovii strain belonging to HC using this model. Sankey diagram was then used to depict the correspondence of virulence factors and O-genotypes with cytotoxicity ranks ((d)). Only three sub-groups within VMOg29 (VMOg29-b, VMOg29-c, and VMOg29-d) showed an increasing susceptibility to HC group, while the rest of the O-genotypes were more likely to be related to non-HC groups.
Lateral flagellar biosynthesis gene (flhA) is essential for the pathogenicity of V. metschnikovii
The flhA gene was detected in 9.7% (9/103) of V. metschnikovii strains, which all belonged to VMOg29 groups (Table S7). Early studies on lateral flagella were investigated in Aeromonas [Citation36], and the flhA gene cluster of V. metschnikovii was similar to that of Aeromonas hydrophila ((a)). On analysis of a flhA mutant (ΔflhA) constructed as part of this study, together with and a complemented flhA mutant (C-ΔflhA), ΔflhA was found to be significantly impaired in swimming motility, while C-ΔflhA reverted to similar swimming motility as the WT ((b)). Phenotypic validation of four representative strains (belonging to LC, MC, HC, and control) revealed that the deletion of the flhA gene affected swimming motility ((c,d)) and also weakened the biofilm formation ability of the V. metschnikovii strains ((e,f)). Next, we examined the ability of V. metschnikovii to adhere to Hep-2 cells and cause infection. ΔflhA exhibited little cell infections relative to the WT and the C-ΔflhA strains, for example, a reduction in cytotoxicity ((g)), cell adhesion ((h)) and invasion ((j)).
Figure 4. Genetic and functional analyses of lateral flagellar related flhA gene. (a) Gene cluster organization of a 15.8 kb fragment surrounding the flhA gene in the V. metschnikovii chromosome. (b) Swimming motility patterns of the ZJ20140528 (WT), the flhA mutant (ΔflhA), the complemented flhA mutant (C-ΔflhA) and control (2020QD6) strains. (c) Swimming motility of four representative V. metschnikovii strains for 12 h on soft agar (0.35%) and (d) statistical analysis. Strain ZJ20140528 for high cytotoxicity, VMOg29-d and flhA positive; strain ZJ060925 for median cytotoxicity, VMOg29-c and flhA positive; strain 2020QD3 for low cytotoxicity, VMOg-undefined and flhA negative; strain 2020QD6 for control, low cytotoxicity and flhA negative. (e) Crystal violet staining for 48 h and (f) statistical analysis. (g) Cytotoxicity values of five V. metschnikovii strains. (h) Adhesion ability in Hep-2 cells. (i) Invasion ability in Hep-2 cells. (j) Growth curves. The four time points (T1 to T4) for subsequent RT-qPCR were marked with black arrows. (k) Transcription levels of flhA gene at T2. The 16S rRNA gene was used as an internal control. All primer sequences were shown in Table S8. (l) Transcription levels of flhA gene at T3. Those groups with significant differences were marked with * (P < 0.05), ** (P < 0.01), *** (P < 0.001), **** (P < 0.0001).
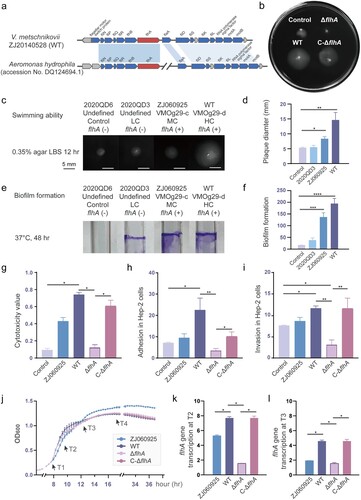
It should be emphasized that the positive phenotypes observed in the strain ZJ060925 (flhA+) were weaker than that in WT ((c–i)). Four different time points (T1 to T4) were selected based on the growth curves ((j)), and then reverse transcription quantitative PCR (RT-qPCR) was performed to estimate the transcript levels of flhA gene in different strains at the same growth state. During the mid and late stage of the exponential phase (T2 and T3), the transcription of flhA in the strain ZJ060925 was higher than that of the ΔflhA but remarkably less than that of the WT and C-ΔflhA strains (P < 0.05, (k–l)). The higher the expression of flhA, the stronger the swimming and cell infection activity of V. metschnikovii strains. The transcription of the flhA gene at the four time points is shown in the Figure S4. These data indicate that the lateral flagella are essential for the initial infection stage of chemotaxis and adhesion to the cell in the cell infection model.
Discussion
V. metschnikovii is a rare human pathogen. Previous studies have shown that V. metschnikovii belongs to the Cincinnatiensis clade [Citation37]. Our comparative genome analysis of V. metschnikovii identified two major lineages with a high genetic diversity of O-genotypes. Our finding also revealed a pathogenic link among virulence-related factors, O-genotypes, and cellular infection models. This study provides an important basis for further study on the evolution and potential pathogenicity of V. metschnikovii.
We defined two major lineages of V. metschnikovii. The strains we collected in China were isolated from different regions in different years. The Mantel test and phylogenetic analyses strongly suggested that the spread of strains from L1 was likely to be closely related to human activities, while strains from L2 tended to be locally endemic as clonal complex groups. A range of important evidence was found: (1) L1 strains were sourced from food and clinical settings, while the origin of L2 strains was not closely related to humans. The sources of food often involve multiple epidemiological factors of human activity, such as food supply, packaging, transportation, and retail trading; (2) By analyzing six different mutation patterns, we found that both C > T/G > A and A > G/T > C were more frequent in L2 strains relative to L1 strains. The cumulative effect of C > T/G > A and A > G/T > C mutations is a continuous process that shapes bacterial ecological-niche adaptation to host immune environments [Citation38]; (3) L1 strains had more drug resistance genes compared to L2, both in terms of category and quantity of resistance genes. A growing trend over antibiotic resistance in human-pathogenic bacteria was observed under environmental selective pressures [Citation39], and part of the antibiotic-resistance rate in L1 strains can be attributed to the potential impact of frequent antibiotic utilization or even drug abuse in human communities. Of course, the real resistance of strains to those antibiotics is related to the presence or absence of resistance genes in addition to a variety of other factors, such as the capsule. Ernst et al. reported the global evolution of pathogenicity in carbapenem-resistant K. pneumoniae, and they demonstrated that mutations disrupting capsule biosynthesis genes impair capsule production, which enhances drug-tolerant K. pneumoniae to persist to yield potentially untreatable, persistent infection [Citation40].
The O-AGCs based Vibrio serotyping became the “gold standard” for the clinical detection and epidemiological surveillance of human Vibrio pathogens [Citation41]. We defined 29 O-genotypes, which were named VMOg1–VMOg29, respectively. But one concern is that only 61% (63/103) of the strains were matched in the in silico O-genotyping, leaving the VMOgs of other 40 strains undefined. Repeats has always been the source of most mis-assemblies, although the vast majority of genes in the bacterial genome are single-copy. Assembly errors from short reads or variations resulting from different assembly methods deserve our attention [Citation42, Citation43]. And a potential mechanism for recombination and deletion events could contribute to the antigenic diversity [Citation44]. Our collection of V. metschnikovii has a considerable O-antigen diversity, suggesting that the genetic differences of these strains are also representative. On the other hand, nearly 40% of the strains still have O-genotypes to be identified in future work. We hypothesize that the real O-antigen diversity of V. metschnikovii that exists in nature is far greater than we currently know. It would be interesting to know much more about the O-AGCs diversity of V. metschnikovii.
The pathogenicity of V. metschnikovii to humans is often overlooked. In the present work, the pathogenicity of V. metschnikovii may be the outcome of multiple factors like some opportunistic pathogenic vibrios [Citation45]. Virulence genes (e.g. hlyA, tlh and MSHA) were commonly present, which have been proved to cause infection in other vibrios [Citation46, Citation47]. Some strains carried virulence genes associated with chemotaxis and motility, secretion systems, and toxins. The contribution of lateral flagella (the first principal component, P1) to the cell infections can explain more observed variance. The absence of flhA reduces the swimming activity and biofilm formation capacity of the V. metschnikovii. And their motility and biofilm-forming capacity in the early stages of infection [Citation48] is associated with the transcript levels of flhA. The mixed flagellation system was initially described in V. metschnikovii in the early 1950s [Citation49]. V. parahaemolyticus and some other members of Vibrios also contain elaborate lateral flagella that are used for rapid movement on surfaces [Citation50]. A previous study has suggested that the transition from the motility phase to the biofilm formation phase is one of the crucial events in infection [Citation51]. Inconsistent with previous studies [Citation18, Citation52], virulence genes related to cytolysin (or hemolysin) were not found to be statistically significant in differentiating cytotoxicity ranks.
Our data also suggested a link among evolutionary lineage, O-genotypes, and virulence factors. To begin with, the evolutionary tree based on cgSNPs and heatmap ((a)) revealed virulence heterogeneity between the two lineages. The lateral flagella, T6SS and T3SS were mainly found in the L1 strains. While several V. metschnikovii strains in L2 carried the RTX toxin, which can play a pathogenic role in cell adhesion and/or pathogenesis [Citation53] in V. cholerae [Citation54]. Next, V. metschnikovii strains carrying different virulence genes exhibited different ranks in cytotoxicity and pathogenic phenotypes. These observations suggest that pathogenicity in humans can be linked with the presence of specific virulence-associated genes; however, further studies, with additional human isolates, are needed to confirm this hypothesis. Thirdly, we found that the cytotoxic rank was related to the O-genotype of V. metschnikovii. The HC group of V. metschnikovii contained VMOg29 specific virulence-associated genes, for example, the lateral flagella, whereas it was absent in the non- VMOg29 (MC or LC O-genotypes) strains. In this study, we propose a potential pathogenic mechanism for V. metschnikovii in the early stages of infection: the lateral flagella may play a fundamental and necessary role in the chemotaxis and host adhesion; other virulence factors could also promote pathogenicity, acting synergistically.
Supplemental Material
Download MS Word (1.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Baker-Austin C, Oliver JD, Alam M, et al. Vibrio spp. infections. Nat Rev Dis Primers. 2018;4(1):8.
- Tamplin ML, Carrillo Parodi C. Environmental spread of Vibrio cholerae in Peru. Lancet. 1991;338(8776):1216–1217.
- Hooper WL, Barrow GI, McNab DJ. Vibrio parahaemolyticus food-poisoning in Britain. Lancet. 1974;1(7866):1100–1102.
- Plotkin BJ, Konaklieva MI. Surface properties of Vibrio vulnificus. Lett Appl Microbiol. 2007;44(4):426–430. doi:10.1111/j.1472-765X.2006.02083.x
- Jacobs Slifka KM, Newton AE, Mahon BE. Vibrio alginolyticus infections in the USA, 1988–2012. Epidemiol Infect. 2017;145(7):1491–1499. doi:10.1017/S0950268817000140
- Morris JG. Cholera and other types of vibriosis: a story of human pandemics and oysters on the half shell. Clin Infect Dis. 2003;37(2):272–280. doi:10.1086/375600
- Konechnyi Y, Khorkavyi Y, Ivanchuk K, et al. Vibrio metschnikovii: current state of knowledge and discussion of recently identified clinical case. Clin Case Rep. 2021;9(4):2236–2244. doi:10.1002/ccr3.3999
- Hansen W, Freney J, Benyagoub H, et al. Severe human infections caused by Vibrio metschnikovii. J Clin Microbiol. 1993;31(9):2529–2530. doi:10.1128/jcm.31.9.2529-2530.1993
- Pariente Martín M, Escribano Garaizábal E, Liria Sánchez PJ, et al. Vibrio metschnikovii from a human infected leg ulcer. Rev Inst Med Trop Sao Paulo. 2008;50(5):311–312. doi:10.1590/S0036-46652008000500012
- Jensen J, Jellinge ME. Severe septic shock and cardiac arrest in a patient with Vibrio metschnikovii: a case report. J Med Case Rep. 2014;8:348. doi:10.1186/1752-1947-8-348
- Linde HJ, Kobuch R, Jayasinghe S, et al. Vibrio metschnikovii, a rare cause of wound infection. J Clin Microbiol. 2004;42(10):4909–4911. doi:10.1128/JCM.42.10.4909-4911.2004
- Wallet F, Tachon M, Nseir S, et al. Vibrio metschnikovii pneumonia. Emerg Infect Dis. 2005;11(10):1641–1642. doi:10.3201/eid1110.050177
- Materna AC, Friedman J, Bauer C, et al. Shape and evolution of the fundamental niche in marine Vibrio. ISME J. 2012;6(12):2168–2177. doi:10.1038/ismej.2012.65
- Ke HM, Liu D, Ogura Y, et al. Tracing genomic divergence of vibrio bacteria in the Harveyi clade. J Bacteriol. 2018;200:15.
- Zheng L, Zhu LW, Jing J, et al. Pan-genome analysis of Vibrio cholerae and Vibrio metschnikovii strains isolated from migratory birds at Dali Nouer Lake in Chifeng, China. Front Vet Sci. 2021;8:638820. doi:10.3389/fvets.2021.638820
- Caldini G, Neri A, Cresti S, et al. High prevalence of Vibrio cholerae non-O1 carrying heat-stable-enterotoxin-encoding genes among vibrio isolates from a temperate-climate river basin of central Italy. Appl Environ Microbiol. 1997;63(7):2934–2939. doi:10.1128/aem.63.7.2934-2939.1997
- Valáriková J, Korcová J, Ziburová J, et al. Potential pathogenicity and antibiotic resistance of aquatic Vibrio isolates from freshwater in Slovakia. Folia Microbiol. 2020;65(3):545–555. doi:10.1007/s12223-019-00760-w
- Håkonsholm F, Lunestad BT, Aguirre Sánchez JR, et al. Vibrios from the Norwegian marine environment: characterization of associated antibiotic resistance and virulence genes. Microbiologyopen. 2020;9(9):e1093.
- Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8(1):61–65. doi:10.1038/nmeth.1527
- Gurevich A, Saveliev V, Vyahhi N, et al. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–1075. doi:10.1093/bioinformatics/btt086
- Hyatt D, Chen GL, Locascio PF, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi:10.1186/1471-2105-11-119
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
- Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genom. 2008;9:75. doi:10.1186/1471-2164-9-75
- Meier-Kolthoff JP, Auch AF, Klenk HP, et al. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013;14:60. doi:10.1186/1471-2105-14-60
- Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucl Acids Res. 2015;43(3):e15. doi:10.1093/nar/gku1196
- Tonkin-Hill G, Lees JA, Bentley SD, et al. Fast hierarchical Bayesian analysis of population structure. Nucl Acids Res. 2019;47(11):5539–5549. doi:10.1093/nar/gkz361
- Nguyen LT, Schmidt HA, von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274. doi:10.1093/molbev/msu300
- Mistry J, Chuguransky S, Williams L, et al. Pfam: the protein families database in 2021. Nucl Acids Res. 2021;49(D1):D412–D419. doi:10.1093/nar/gkaa913
- Liu B, Zheng D, Jin Q, et al. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucl Acids Res. 2019;47(D1):D687–d692. doi:10.1093/nar/gky1080
- Liu L, Chen D, Liu L, et al. Genetic diversity, multidrug resistance, and virulence of Citrobacter freundii from diarrheal patients and healthy individuals. Front Cell Infect Microbiol. 2018;8:233. doi:10.3389/fcimb.2018.00233
- Guo P, Zhu B, Liang H, et al. Comparison of pathogenicity of invasive and carried Meningococcal isolates of ST-4821 complex in China. Infect Immun. 2019;87:12.
- Gu D, Liu H, Yang Z, et al. Chromatin immunoprecipitation sequencing technology reveals global regulatory roles of low-cell-density quorum-sensing regulator AphA in the pathogen Vibrio alginolyticus. J Bacteriol. 2016;198(21):2985–2999. doi:10.1128/JB.00520-16
- Metcalf WW, Jiang W, Daniels LL, et al. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35(1):1–13. doi:10.1006/plas.1996.0001
- Kamal NM, Zamri-Saad M, Masarudin MJ, et al. Interaction between Pasteurella multocida B:2 and its derivatives with bovine aortic endothelial cell (BAEC). BMC Vet Res. 2017;13(1):186. doi:10.1186/s12917-017-1109-1
- Yang C, Zhang X, Fan H, et al. Genetic diversity, virulence factors and farm-to-table spread pattern of Vibrio parahaemolyticus food-associated isolates. Food Microbiol. 2019;84:103270. doi:10.1016/j.fm.2019.103270
- Canals R, Altarriba M, Vilches S, et al. Analysis of the lateral flagellar gene system of Aeromonas hydrophila AH-3. J Bacteriol. 2006;188(3):852–862. doi:10.1128/JB.188.3.852-862.2006
- Fang Y, Chen A, Dai H, et al. Vibrio fujianensis sp. nov., isolated from aquaculture water. Int J Syst Evol Microbiol. 2018;68(4):1146–1152. doi:10.1099/ijsem.0.002642
- Liu Q, Liu H, Shi L, et al. Local adaptation of Mycobacterium tuberculosis on the Tibetan Plateau. Proc Natl Acad Sci U S A. 2021;118:17.
- Peng M, Salaheen S, Buchanan RL, et al. Alterations of Salmonella enterica Serovar Typhimurium antibiotic resistance under environmental pressure. Appl Environ Microbiol. 2018;84:19.
- Ernst CM, Braxton JR, Rodriguez-Osorio CA, et al. Adaptive evolution of virulence and persistence in carbapenem-resistant Klebsiella pneumoniae. Nat Med. 2020;26(5):705–711. doi:10.1038/s41591-020-0825-4
- Li Z, Lu X, Wang D, et al. Genomic comparison of serogroups O159 and O170 with other Vibrio cholerae serogroups. BMC Genom. 2019;20(1):241), doi:10.1186/s12864-019-5603-7
- Salzberg SL, Yorke JA. Beware of mis-assembled genomes. Bioinformatics. 2005;21(24):4320–4321. doi:10.1093/bioinformatics/bti769
- Collins A. The challenge of genome sequence assembly. Open Bioinform J. 2018;11:1.
- Drummelsmith J, Amor PA, Whitfield C. Polymorphism, duplication, and IS1-mediated rearrangement in the chromosomal his-RFB-GND region of Escherichia coli strains with group IA and capsular K antigens. J Bacteriol. 1997;179(10):3232–3238. doi:10.1128/jb.179.10.3232-3238.1997
- Li L, Meng H, Gu D, et al. Molecular mechanisms of Vibrio parahaemolyticus pathogenesis. Microbiol Res. 2019;222:43–51. doi:10.1016/j.micres.2019.03.003
- Zhang W, Luo M, Feng C, et al. Crash landing of Vibrio cholerae by MSHA pili-assisted braking and anchoring in a viscoelastic environment. Elife. 2021;10:e60655. doi:10.7554/eLife.60655
- Sathyamoorthy V, Huntley JS, Hall AC, et al. Biochemical and physiological characteristics of HlyA, a pore-forming cytolysin of Vibrio cholerae serogroup O1. Toxicon. 1997;35(4):515–527. doi:10.1016/S0041-0101(96)00163-8
- Gavín R, Rabaan AA, Merino S, et al. Lateral flagella of Aeromonas species are essential for epithelial cell adherence and biofilm formation. Mol Microbiol. 2002;43(2):383–397. doi:10.1046/j.1365-2958.2002.02750.x
- Chu J, Liu J, Hoover TR. Phylogenetic distribution, ultrastructure, and function of bacterial flagellar sheaths. Biomolecules. 2020;10:3.
- McCarter LL. Polar flagellar motility of the Vibrionaceae. Microbiol Mol Biol Rev. 2001;65(3):445–462. doi:10.1128/MMBR.65.3.445-462.2001
- Khan F, Tabassum N, Anand R, et al. Motility of Vibrio spp.: regulation and controlling strategies. Appl Microbiol Biotechnol. 2020;104(19):8187–8208. doi:10.1007/s00253-020-10794-7
- Miyake M, Honda T, Miwatani T. Purification and characterization of Vibrio metschnikovii cytolysin. Infect Immun. 1988;56(4):954–960. doi:10.1128/iai.56.4.961-965.1988
- Chatterjee R, Nag S, Chaudhuri K. Identification of a new RTX-like gene cluster in Vibrio cholerae. FEMS Microbiol Lett. 2008;284(2):165–171. doi:10.1111/j.1574-6968.2008.01199.x
- Sheahan KL, Cordero CL, Satchell KJ. Autoprocessing of the Vibrio cholerae RTX toxin by the cysteine protease domain. Embo J. 2007;26(10):2552–2561. doi:10.1038/sj.emboj.7601700
- Kirov SM. Bacteria that express lateral flagella enable dissection of the multifunctional roles of flagella in pathogenesis. FEMS Microbiol Lett. 2003;224(2):151–159. doi:10.1016/S0378-1097(03)00445-2