
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Chikungunya virus is a re-emerging arbovirus that has caused epidemic outbreaks in recent decades. Patients in older age groups with high viral load and severe immunologic response during acute infection are likely to develop chronic arthritis and severe joint pain. Currently, no antiviral drug is available. Previous studies suggested that a flavone derivative, 8-bromobaicalein, was a potential dengue and Zika replication inhibitor in a cell-based system targeting flaviviral polymerase. Here we characterized that 8-bromobaicalein inhibited chikungunya virus replication with EC50 of 0.49 ± 0.11 µM in Vero cells. The molecular target predicted at viral nsP1 methyltransferase using molecular binding and fragment molecular orbital calculation. Additionally, oral administration of 250 mg/kg twice daily treatment alleviated chikungunya-induced musculoskeletal inflammation and reduced viral load in healthy adult mice. Pharmacokinetic analysis indicated that the 250 mg/kg administration maintained the compound level above EC99.9 for 12 h. Therefore, 8-bromobaicalein should be a potential candidate for further development as a pan-arboviral drug.
Introduction
Chikungunya is a mosquito-borne viral disease caused by the chikungunya virus (CHIKV) and primarily affects humans [Citation1]. It is transmitted to humans through the bites of infected Aedes mosquitoes, primarily Aedes aegypti [Citation2]. The virus is endemic in many regions of Africa, Asia, and the Indian subcontinent. However, in recent years, it has spread to new areas, including the Americas and Europe [Citation3–5]. The outbreaks and becoming a global health concern, in which climate change may increase vector abundance [Citation6]. CHIKV infection typically presents with fully resolved and self-limited symptoms such as high fever, joint and muscle pain, headache, rash, and fatigue. However, in some cases, joint pain can be severe and persistent, leading to long-term complications. In fact, persistent arthralgia and CHIKV-induced polyarthritis were observed in 30–40% after acute infections [Citation7]. Furthermore, severe manifestations involving the central nervous system and fulminant hepatitis were documented in the recent La Réunion outbreak [Citation8]. Several vaccines are under development and a live-attenuated Δ5nsP3 (Valneva) is the most advanced vaccine candidate currently in the licensing process [Citation9]. Currently, no specific antiviral treatment is available for CHIKV infection; however, drug discovery efforts are underway to develop effective treatments.
Several potential targets for antiviral drugs have been identified, including viral enzymes and host factors involved in viral replication. CHIKV is a member of the Togaviridae family, the genus Alphavirus. The virus has a single-stranded positive-sense RNA genome encoding early non-structural (nsP1, 2, 3, 4) and late structural (C, E1, E2, E3, and 6 K) polyproteins [Citation5,Citation10]. Nonstructural proteins are primarily responsible for viral replication and modulation of host cell responses, whereas structural proteins play an essential role during entry and exit and genome protection. nsP1 is a capping enzyme with methyltransferase and guanylyltransferase activities [Citation11]. nsP2 is multifunctional as nucleoside triphosphatase (NTPase) [Citation12,Citation13], helicase [Citation14], and RNA 5’triphosphatase (RTPase) [Citation15], and protease [Citation16,Citation17]. nsP3 is a scaffolding protein and nsP4 is an RNA-dependent RNA polymerase (RdRp) [Citation18]. These enzymes, such as viral protease and polymerase, are attractive targets for developing antiviral drugs. In addition, small-molecule inhibitors targeting these viral enzymes have shown promise in nonclinical studies.
Baicalein is a flavonoid compound with potential antiviral properties against mosquito-borne flaviviruses [Citation19,Citation20] and CHIKV[Citation21]. In general, flavonoids were well-established as antivirals against many viral families [Citation22]. Previous reports suggested that baicalein effectively inhibited CHIKV with a 50% inhibitory concentration at 1.891 μg/mL (6.997 μM) in Vero cells [Citation21], and 16.9 μM in Huh-7 cells [Citation23]. Moreover, the mechanisms of action of the drug could be at various steps including attachment, translation/replication, and mainly at the inactivation of the viral particle [Citation21]. Furthermore, the compound modulated pro-inflammatory cytokines, which may help alleviate symptoms associated with CHIKV infection [Citation24]. Baicalein derivatives with modification at the 8 or 4′ position could lead to 5–10 times more potent antivirals than the parent compound [Citation23]. Furthermore, halogenation of flavones could also potentiate antiviral activities [Citation25]. Previous studies suggested that 8-bromobaicalein was a potent inhibitor of the SARS-CoV-2, dengue and Zika viruses [Citation26,Citation27], targeting viral enzymes with affinities and activities stronger than the original baicalein. In this study, we explored the potential of 8-bromobaicalein as an inhibitor of the mouse model with CHIKV-induced musculoskeletal inflammatory disease [Citation28]. Molecular docking, fragment molecular orbital (FMO), and molecular dynamics simulation approaches were used to predict the potential target and characterize the bromine substitution effect. Moreover, this work also included pharmacokinetics and additional drug toxicity analysis to achieve the most efficient and nontoxic administration for the in vivo efficacy test.
Materials and methods
Cells, virus, chemicals, and reagents
LLC/MK2 (ATCC® CCL-7), Vero (ATCC®CCL-81), and C6/36 (ATCC®CRL-1660) cell lines were maintained as previously described [Citation29–33]. A CHIKV virus (Ross isolate; accession number: KM258100) was a courtesy from Professor Padet Siriyasatien. The virus was propagated in the C6 / 36 cell line as previously described in dengue and Zika viruses [Citation29,Citation30]. The 8-bromobaicalein was synthesized, purified, and identified as previously described [Citation27]. The mass spectra of the negative ions of 8-bromobaicalein was demonstrated in supplementary Figure 1.
Cell-based antiviral efficacy
The protocol was adapted from a previous description of flaviviruses [Citation30]. Vero cells were seeded at 5 × 104 cells in each well of 24-well plate and incubated overnight at 37 ° C under 5% CO2. Cells were infected with CHIKV at the multiplicity of infection (M.O.I.) of 0.1 for 1 h. Cells were washed with phosphate buffer saline (PBS) and incubated with 1 mL of MEM medium supplemented with 1% FBS, 100 I.U./mL penicillin, 100 μg/mL streptomycin, and 10 mM HEPES at 37 ° C under 5% CO2. The 8-bromobaicalein was prepared in dimethylsulfoxide (DMSO) at the indicated concentrations before addition to viral infected cells. Supernatants were collected after 3 days of incubation and analyzed by plaque titration [Citation34]. The EC50 values were calculated from nonlinear regression analysis (GraphPad Prism 10 Software, Boston, MA, USA). The results were reported as means and standard error of the the means (SEM) of EC50 values from three independent experiments in which each drug concentration was tested in triplicate.
Time of addition and time of removal assays
The protocol was adapted from a previous description [Citation35]. Vero cells were seeded at 5 × 104 cells in each well of 24-well plate and incubated overnight at 37 ° C under 5% CO2. Cells were infected with CHIKV at the multiplicity of infection (M.O.I.) of 0.5 for 1 h. Cells were washed with phosphate buffer saline (PBS) and incubated with 1 mL of MEM medium supplemented with 1% FBS, 100 I.U./mL penicillin, 100 μg/mL streptomycin, and 10 mM HEPES at 37 ° C under 5% CO2. In time of addition assay, the 10 µM 8-bromobaicalein was prepared in dimethylsulfoxide (DMSO) and added to virus-infected cells at designated time points after infection. Supernatants were collected after 2 days of incubation and analyzed by plaque titration [Citation34]. In time of removal assay, the 10 µM 8-bromobaicalein was added to virus-infected cells after 1 h of infection and removed at designated time points. The infected cells were then maintained in MEM medium supplemented with 1% FBS, 100 I.U./mL penicillin, 100 μg/mL streptomycin, and 10 mM HEPES at 37 ° C under 5% CO2 until 2 days of incubation. The supernatants collected from the infected cells at 2 days of incubation (time of removal) and the supernatants collected from the removals at designated time points were taken for plaque titration. The results were reported as means and standard error of the means (SEM) tested in triplicate (supplementary Figure 2) (GraphPad Prism 10 Software, Boston, MA, USA).
Mice
The Institutional Animal Care and Use Committee of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand, approved the Animal Care and Use Protocol, based on the criteria of the National Institutes of Health, USA, for the use and treatment of laboratory animals (certificate number: 015/2565). C57BL/6 mice purchased from the National Laboratory Animal Center, Nakhornpathom, Thailand, were used in the experiment.
Animal toxicity test
In a single-dose oral administration, ten 8-week-old C57BL/6 mice were divided equally into treatment and control groups. The 8-bromobaicalein was prepared in DMSO and added to the vehicle (35% polyethylene glycol 400, 2% ethanol and 63% deionized water) at the final concentration of 6.25 mg/mL in 10% DMSO [Citation32]. The compound was administered using a 18-gauge rounded tipped gavage to the treatment group (n = 5) and the body temperature, activity score, foot width and length were monitored daily. The control group (n = 5) was administered orally with 10% DMSO in the vehicle. Blood samples were collected on the 1st, 3rd, 7th day after the administration and the sera were separated by centrifugation at 5000 rpm for 10 min for analysis of alanine transaminase (ALT) (EnzyChrom, EALT-100, BioAssay, Hayward, CA, USA), and serum creatinine (Cr) (QuantiChromTM, DICT-500, BioAssay, Hayward, CA, USA) [Citation26].
In a multiple-dose oral administration, the sera from the drug-treated and vehicle-treated groups (n = 5/group) of in vivo efficacy experiment (Figure 3A) were analyzed for ALT and Cr (GraphPad Prism 10 Software, Boston, MA, USA). The results were compared with the previous report [Citation27].
Sample preparation
Analytical conditions to determine the level of 8-bromobaicalein in mice plasma were adapted from the pharmacokinetic study of baicalein in monkeys [Citation36]. Briefly, the 20 μL aliquot of plasma sample was vortex mixed with 80 μl HPLC-grade acetonitrile (Merck, Darmstadt, Germany) for 1 min before centrifugation at 10,000 rpm at 20 °C for 5 min. The supernatants were transferred to the autosampler and injected into the LC–MS/MS system for analysis.
LC–MS/MS conditions
The compound concentrations (ng/mL) were analyzed using LC-MS/MS (Shimadzu, Japan), consisting of a solvent delivery module (LC-20AD), auto sampler (SIL-20ACXR), Column oven (CTO-20AC), degasser (DGU-20A5R), Reservoir switching valve (FCV-11AL) and triple quadrupole tandem mass spectrometer (LCMS-8040) with an Electrospray ionisation (ESI). The mass spectrometer was operated in negative ionization mode. Quantifications were performed using the Multiple Reaction Monitoring (MRM) mode to detect precursor and product ion. Chromatographic conditions: (1) column: ACE Excel 5 Super C18, 150 × 2.1 mm; (2) Guard column: Phenomenex C18, 4.0 × 2 mm. The product ion transitions at m/z 346.90 ffb 239.00 (target ion) was identified as the 8-bromobaicalein (supplementary Figure 1). Column temperature was maintained at 40 ° C and auto-sampler at 20 °C. The mobile phase consisted of 0.1% formic acid-acetonitrile (25: 75, v/v) with a flow rate of 0.4 ml/min. The total run time was 5 min. The chromatogram was shown in supplementary Figures 3 and 4.
Validation for analytical method
The specificity was evaluated by comparing the response of blank mice plasma with the corresponding spiked plasma to exclude interference at the retention time of the analyte. Linearity was assessed by analyzing eight concentration levels of calibration samples to generate a calibration curve in the range of 5–10,000 ng/mL. The calibration curve was plotted by linear least-squares regression analysis (weighting factor = 1/C2) through the compound measurement of the peak area ratio. The back-calculated concentrations of the calibration standards must be within ±15% of the nominal concentration, except for the lowest limit of quantification (LLOQ), where it was ±20%. In addition, LLOQ was defined as the amount that could be detected with a signal-to-noise ratio at least five times the signal of a blank sample.
Pharmacokinetic experiment and data analysis
Twenty 8-week-old C57BL/6 mice were divided into four groups (n = 5/group); one group for intravenous (IV, 10 mg/kg) and three groups for oral administrations (PO, 50, 150, and 250 mg/kg) [Citation37]. Blood samples were collected through tail vein nicking at 15, 30 min, 1, 2, 3, and 6 h after IV administration and at 1, 2, 4, 6, 8, 12, and 24 h after PO administration. The blood volume was limited to less than 50 µl per draw (less than 2% body weight) to prevent hemodynamic instability. All blood samples were separated by centrifugation at 5,000 rpm for 10 min and the sera were kept at –80 °C until analysis.
The pharmacokinetic parameters were analyzed with PhoenixTM 64 version 8.3 (Certara, Princeton, NJ) using the non-compartmental model. Maximum plasma concentration (Cmax), time to Cmax (Tmax), area under the plasma concentration-time curve from 0 to t (AUC0–t), area under the plasma concentration-time curve from 0 to infinity (AUC0–inf), volume of distribution (Vd), half-life (t1/2) and drug clearance (CL) were reported as mean ± standard deviation (SD).
In vivo anti-CHIKV efficacy
Thirty mice were divided into three groups; the 250 mg/kg compound-treated (n = 10), vehicle-treated (n = 10), the mock infection (n = 5) and the group before treatment (n = 5) [Citation38,Citation39]. The mock infection group (n = 5) was injected into their Lt hind footpad with maintenance medium or minimal essential medium (MEM) (Gibco®, Langley, USA) supplemented with 1% fetal bovine serum (Gibco®, Langley, USA), 100 I.U./mL penicillin (Bio Basic Canada, Ontario, Canada) and 100 μg/mL streptomycin (Bio Basic Canada, Ontario, Canada), and 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (Sigma Aldrich, St. Louis, USA). The group before treatment (n = 5) was injected into their Lt footpad with CHIKV (5 × 106 pfu/mouse) and monitored for clinical signs, activity scores, and foot width and length on day 1 after infection. Footpad tissues were collected for viral quantification and histopathological study. Similarly, the compound-treated groups (n = 10) and vehicle-treated (n = 10) were injected with CHIKV (5 × 106 pfu/mouse) and daily monitored for tissue inflammation before administrations of 5 consecutive doses twice daily of 250 mg/kg 8-bromobaicalein or DMSO vehicle, respectively. The rationale for administering the treatment after infecting the mice is that it is clinically relevant. The patient should seek medical interventionaafter the infection is well-established. Parameters such as body temperature [Citation40], activity score [Citation41,Citation42], and foot width and length were monitored daily. Height and width of the Lt hind feet were measured by using Mitutoya C/N 530-104 vernier callipers (Kanagawa, Japan). At two and twelve hours after the last 8-bromobaicalein dose administered, blood samples were taken for analysis of Cmax and Ctrough levels, respectively. Footpad tissues were collected for viral quantification and histopathological study. The indicated tissues (∼5 mg) were collected in lysis buffer (RNeasy kit, QIAGEN, Hilden, Germany) on ice and stored at −80 ° C within 15 min after collection. The samples were thawed and macerated using the bead and vortex method, followed by a 30-s spin with a microcentrifuge at top speed. The RNA extraction process was performed according to the manufacturer’s protocol (RNeasy minikit, QIAGEN, Hilden, Germany). RNA products were quantified at 100 ng/sample prior to addition to RT-qPCR (Power SYBR® Green RNA-to Ct, Applied Biosystem, Waltham, MA, USA). The primers for the CHIKV envelope protein and mouse β-actin were listed in previous descriptions [Citation43,Citation44]. The remaining footpads were sent for histopathological study at the Department of Pathology, Faculty of Medicine, Chulalongkorn University. The staining method was Hematoxylin & Eosin, conducted on the paw preserved in neutral buffer formalin with a slice thickness of 3 μm. The chemicals for histological stain process included haematoxylin solution, eosin solution (C.V. Laboratories Company Ltd., Bangkok, Thailand), Xylene (Merck, Darmstadt, Germany), Ethanol (Merck, Darmstadt, Germany). The histological scoring system followed the previous description [Citation45]. Briefly, the total score (0–8) were for (1) the severity of inflammatory cell influx (0–4, none to severe), (2) synovial hyperplasia (0–3), and (3) bone loss (0–1). Scores were added to obtain an arthritis index (ranging from 0 to 8), representing three investigators’ means and standard errors.
Molecular docking and FMO calculation
The 3D crystal structures of envelope protein, nsP2, and 3 were obtained from the Protein Data Bank (PDB) with PDB ID 3N42 [Citation46], 4ZTB [Citation47], and, 6VUQ [Citation48], respectively. To predict the 3D structure of nsP1 and 4, a templated homology model was generated using SwissModel [Citation49], based on the cryo-EM structure of CHIKV-nsP1 (PDB ID: 6Z0V [Citation50]) and the crystal structure of alphavirus-nsP4 (PDB ID: 7F0S [Citation51]). The charge parameter and the topology of baicalein and 8-bromobaicalein optimized by the B3LYP/6-31G(d) theory of the base set using Gaussian 16 were obtained from our previous study [Citation52]. Native inhibitors including sinefungin (nsP1) [Citation53], D160d (nsP2) [Citation54], ADP-ribose (nsP3) [Citation48], and ribavirin (nsP4) [Citation55] were prepared into 3D structures as previously described [Citation52]. The binding energy and the pose were predicted using Autodock VinaXB program [Citation56], and the lowest binding affinity for each ligand-nsP1 complex was chosen as the initial complex structure for the calculation of the molecular orbital (FMO) calculation [Citation57–59]. The complexes were then minimized and prepared using a standard procedure, in which the protein within 7 of the ligand was considered as a QM region for the calculation of the FMO. Pair interaction energy (PIE) and decomposition analysis (PIEDA) were calculated with RIMP2/6-31G* under PCM solution () [Citation60,Citation61]. The energy contribution obtained from PIEDA included electronic properties, such as electrostatic (
), dispersion (
), charge transfer with higher order mixed terms (CT + mix) energies (
), and charge exchange (
) which was conducted in the GAMESS software [Citation57,Citation60]. The ligand-binding pose was visualized using UCSF Chimera software [Citation62].
Results
In vitro antiviral efficacy of baicalein and its derivatives
8-bromobaicalein was previously evaluated and the results did not show any apparent cytotoxicity (CC50 > 100 µM) against various cell lines, including Vero cells [Citation27]. In this work, the compound was tested against CHIKV-infected Vero cells, and the effective concentration of 8-bromobaicalein was 0.49 ± 0.11 µM (A). The EC50 of the parent compound, baicalein, was 0.52 ± 0.06 μM (A); therefore, 8-bromobaicalein was slightly more potent than the parent compound. Furthermore, the effective concentration of 8-bromobaicalein against DENV1-4 and ZIKV was previously reported to be 0.66–0.88 µM, and slightly more potent than baicalein [Citation27]. Additional time-of-addition assay suggested that the 8-bromobaicalein inhibited the translation/replication most effectively at early time points after infection (supplementary Figure 2A). Moreover, time-of-removal confirmed that the compound inhibition remained effective after removal out of the system (supplementary Figure 2B). Supernatants removed at indicated time points were also analyzed by plaque titration and the 8-bromobaicalein indeed inhibited the virion progeny compared to DMSO treatment. Therefore, this baicalein derivative was further analyzed for CHIKV antiviral efficacy in vivo.
Figure 1. Comprehensive analysis of 8-bromobaicalein and baicalein, (A) antiviral efficacy in CHIKV (M.O.I. of 0.1) infected Vero cells for 72 h. Supernatants were analyzed by plaque titration. (B) Molecular docking-based prediction of CHIKV protein targets. (C-D) Binding conformation of 8-Bromobaicalein and baicalein compared to GTP at nsP1. (E) Fragment molecular orbital calculation-based assessment of energy contribution and key interacted residues for 8-bromobaicalein and baicalein.
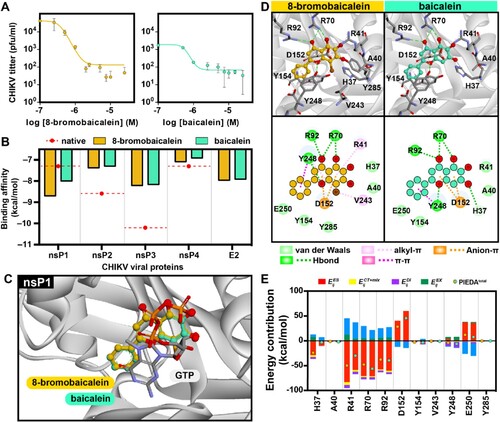
To predict the potential target of 8-bromobaicalein, molecular docking was performed on various CHIKV viral targets, including nsPs and the E2 protein, in comparison to the native inhibitor for each vital target. However, it should be noted that there is no reported native inhibitor for the E2 glycoprotein. By considering the binding affinity for each viral target, the results showed that 8-bromobaicalein demonstrated comparable affinity to baicalein across all viral targets, except for nsP1. Notably, 8-bromobaicalein exhibited a stronger binding affinity (–8.70 kcal/mol) for nsP1 compared to baicalein (–8.00 kcal/mol), as determined by the vinaXB scoring function (B). Furthermore, the binding affinity of these compounds to nsP1 surpassed that of the native inhibitor of nsP1 protein, sinefungin (–7.30 kcal/mol). These compounds bind to the GTP binding pocket located in the capping domain of nsP1. The tip of strand β4, loop β4-αD, and helix αZ defined this binding pocket. Furthermore, the study showed that the loops αC-β4 and β4-αD interact with the adjacent SAM site as the guanosine base moiety of GTP [Citation63]. The interaction between the compounds and nsP1 involves key amino acid residues, including D152, Y248, and E250 (C and D). Fragment molecular orbital (FMO) calculations were carried out using the B3LYP/6-31g* density functional theory with PCM solvation to assess the energy contribution of key interacting residues and the influence of bromine substitution (E). The chromone moiety of the compounds was found to insert between the charged residue D152 and the aromatic amino acid Y248, forming an anion-π interaction. However, the FMO calculation revealed an electrostatic repulsion () resulting from the negative charge of D152, which was lower in 8-bromobaicalein (∼20 kcal/mol). Y248, R70, and R92 participated in hydrogen bonding interactions with the hydroxyl group and oxygen in the chromone, contributing to a similar energy range between the compounds (E). Additionally, Y248 formed a π-π interaction with the benzene ring. Furthermore, an alkyl-π interaction was observed between substituted bromine and two residues, V243 and Y248. In particular, 8-bromobaicalein exhibited stronger electrostatic (
) and hydrophobic interactions (
) with H37 and R41. This enhancement might be attributed to the presence of the bromine atom, resulting in a greater total PIEDA for 8-bromobaicalein. Consequently, the presence of bromine atom could strengthen the interaction by (i) increasing hydrophobic interactions with V243 and Y248 and (ii) influencing the arrangement of the ligand at the binding site involving R41 and Y285, as shown in D.
Animal toxicity of intravenous and oral administrations
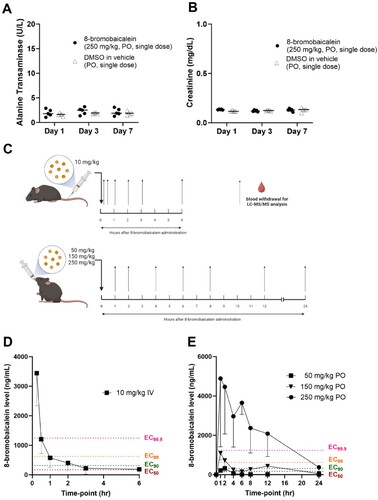
A previous toxicity study [Citation26] was performed using a 10 mg/kg 8-bromobaicalein diluted in 10% DMSO in a standard saline solution, single dose, intravenously through the tail vein (n ≥ 5/group) of 8-week-old C57BL/6 mice. Blood samples were collected at 1, 3, and 8 days after administration to evaluate liver (ALT) and kidney (Cr) functions. The results showed a slight but insignificant elevation of ALT on the first day after administration. Moreover, another study [Citation27] performed seven consecutive doses of 10 mg/kg 8-bromobaicalein diluted in 10% DMSO in vehicle (35% polyethylene glycol 400, 2% ethanol and 63% deionized water), intraperitoneally of adult (8-week-old) and infant C57BL/6 mice. ALT and Cr levels on the day after treatment were insignificantly different in the treated and control groups. Therefore, the single dose, 10 mg/kg 8-bromobaicalein in 10% DMSO in vehicle, was chosen for the pharmacokinetic study.
In addition to intravenous administration, the toxicity in oral administration was also evaluated for further pharmacokinetic analysis. Baicalein was previously administered at 500 mg/kg in monkeys [Citation36] and no apparent toxicity was observed throughout the pharmacokinetic study. However, the maximal solubility concentration of 8-bromobaicalein in the vehicle preparation is 6.25 mg/mL and the maximum oral dose volume was 5 mL/kg (bolus). Therefore, the maximum dose to be given orally was around 250 mg/kg. In this study, mice were orally administered with 250 mg/kg 8-bromobaicalein and blood samples were taken for the analysis of ALT and Cr at 1, 3, and 7 days after the drug administration (A and B). All mice were healthy throughout the experiment, and ALT and Cr levels were insignificantly different between the compound-treated and vehicle-treated groups. Therefore, we concluded that 250 mg/kg 8-bromobaicalein was safe for further pharmacokinetic analysis.
Figure 2. Hepatorenal toxicity of 8-bromobaicalein (250 mg/kg, per oral (PO), single dose, n = 5) measured by (A) alanine transaminase and (B) creatinine levels on day 1, 3, and 7 after administration. (C) Scheme of 8-bromobaicalein pharmacokinetic analysis and plasma concentration-time profiles (created by Biorender) (D) intravenous (IV) and (E) PO routes of administrations (n = 5). Effective concentrations at 50, 90, 99, and 99.9 were plotted to demonstrate the level of drug in systemic circulation. Data represented means and standard deviation of the tested samples.
Pharmacokinetic study
In method validation for the LC–MS/MS analysis, 8-bromobaicalein was completely separated without any significant interferences observed. The retention time of 8-bromobaicalein was approximately 1.4 min. The calibration curve was constructed from the concentration ranges of 5–10,000 ng/mL. The regression equation was as follows: y = 122.894x − 353.148 (R2 = 0.9959). The LLOQ was 5 ng/mL with the accuracy at 118.66% and the back calculated concentrations of all calibration standards ranged from 88.17% to 118.66% accuracy. Therefore, the method was sensitive and valid for the quantification of 8-bromobaicalein in mouse plasma.
An 8-bromobaicalein was administered in a single dose IV (10 mg/kg) or PO (50, 150, and 250 mg/kg). Blood samples were collected at multiple time points for LC-MS/MS analysis (C). Plasma concentration–time profiles and pharmacokinetic parameters were described with the levels of effective concentrations (EC99.9, EC99, EC90, EC50) against CHIKV-infected cells (D–E, ). In the IV administration group, the 8-bromobaicalein level rapidly declined within 2 h after administration, similar to the baicalein profile in monkey (IV, 10 mg/kg) [Citation36,Citation64]. Moreover, the peak plasma concentration and time (Cmax and Tmax) of 8-bromobaicalein was 3,442.8 ± 1,102 ng/mL at 15 min after administration (D, ). The compound approximately reached EC99.9, EC99, EC90, EC50 levels at 30 min, 1.0, 2.0, and 6.0 h, respectively. Therefore, IV administration might not be the most appropriate route of administration.
Table 1. Pharmacokinetic parameters of 8-bromobaicalein.
In PO administration, plasma concentration–time profiles of three doses with effective concentration levels (EC99.9, EC99, EC90, EC50) was reported (E). The time to maximum plasma concentration (Tmax) was 2.0, 1.0, and 1.6 h after administering 50, 150, and 250 mg/kg, respectively, similar to previous descriptions of the parent compound [Citation36,Citation64]. The Tmax results of baicalein in rats (PO, 18 mg/kg) and monkeys (PO, 50, 150, and 500 mg/kg) were 1.1, 1.6, 1.4, and 2.3 h [Citation36,Citation64], respectively. The first pass metabolism of 8-bromobaicalein was also suspected to be the same as that of the parent compound in other mammals [Citation36,Citation64]. The t1/2 were 4.6 ± 0.1, 6.4 ± 1.9, and 5.5 ± 1.9 h after administering 50, 150, and 250 mg/kg, respectively. Moreover, the secondary peaks were observed at 6 and 12 h in 150 mg/kg, and 12 h in 250 mg/kg, respectively, suggesting an enterohepatic recirculation. Only the 250 mg/kg 8-bromobaicalein maintained the plasma level above EC99.9 for 12 h after a single dose administration. Therefore, the oral dose of 250 mg/kg, twice daily, was chosen for further in vivo efficacy analysis.
In vivo efficacy of 8-bromobaicalein
CHIKV-induced musculoskeletal inflammation was demonstrated in an immunocompetent mouse model [Citation28,Citation65]. However, the infection is locally confined to the site of injection and viremia could rarely be detected. This experiment was designed as a proof-of-concept that the 250 mg/kg, PO, BID, 8-bromobaicalein effectively alleviated the CHIKV-induced inflammation in adult immunocompetent mice (A). On day 1 after infection, the CHIKV-inoculated mice became inactive with increasing activity scores, body temperature (B–C), compared to mock infection. In addition, swellings at the site of injection (D–E, 4A) increased to 101–106% of the measurement day 0. Histopathology showed marked infiltration of polymorphonuclear cells (PMN) and other leukocytes in connective tissues (B-C). The viral load was 1.89-fold higher than the β-actin (D). All measurements on day 1 were performed before the first dose of compound administration.
Figure 3. In vivo efficacy of 8-bromobaicalein against CHIKV-induced musculoskeletal inflammation (A) Scheme of experimental procedure, and clinical parameters (created by Biorender) (B) activity score, (C) body temperature, (D) foot width change, and (E) foot length change. Data represented means and standard deviation (SD) and *, ** represented the significant difference at p < 0.05, p < 0.01, respectively using one-way ANOVA. Ns represented non-significant difference.
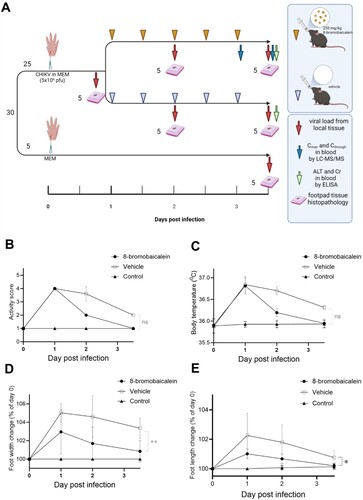
Figure 4. Pathology of the In vivo efficacy experiment. The experimental procedure was shown in A. (A) Gross examination and (B) histopathology by H & E staining of the (i–vi) Lt footpad tissues under the following conditions; (i) CHIKV-infection before treatment (day 1), (ii) no infection control (day 3.5), (iii) 8-bromobaicalein (day 2), and (iv) vehicle-treated (day 2), (v) 8-bromobaicalein (day 3.5), and (vi) vehicle-treated (day 3.5). The Rt footpad tissues were shown in (vii) 8-bromobaicalein (day 3.5), and (viii) vehicle-treated (day 3.5). The slice thickness was 0.3 mm, stained by H&E, and the scale bar (100 µm) was indicated. (C) Histopathological scores, (D) CHIKV RNA level in tissue samples were demonstrated in before treatment (day 1), 8-bromobaicalein-treated (day 2–3.5), and vehicle-treated (day 2–3.5) groups. PO represented per oral, and BID represented twice a day administration, respectively. (E) ALT and Cr levels of drug-treated, and vehicle-treated groups (day 3.5) were evaluated for potential hepatorenal toxicity. An unpaired t-test was used and ** indicated a significant difference at p < 0.01.
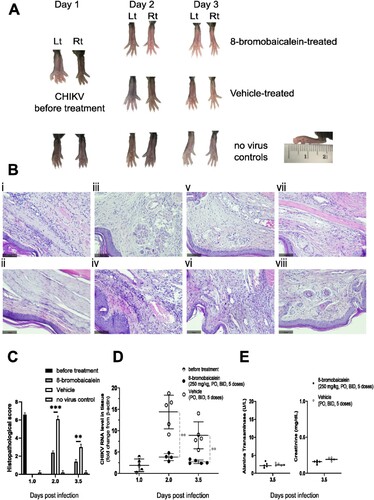
On days 2 and 3 after infection, the compound-treated mice improved clinically with reduced local swelling compared to vehicle-treated mice (B–E). Histology showed reduced inflammation and infiltration of leukocytes on day 2, or after two treatment doses, whereas the inflammation was still not fully resolved in the vehicle-treated group (A–B). The mean peak viremia on days 2 and 3 was significantly different in the compound-, and vehicle-treated groups with p < 0.01 (C). Moreover, the Cmax and Ctrough of 8-bromobaicalein taken 2 and 12 h after the 5th dose were 10,043.19 ± 5416.78 ng/mL, and 2,194.65 ± 1,444.01 ng/mL, respectively (Supplementary Figures 3 and 4). Interestingly, the 8-bromobaicalein level at 12 h after a single oral administration was 2,084.82 ± 1,157.54 ng/mL (E), similar to the Ctrough level. The finding suggested that the compound should be extensively metabolized and excreted so that accumulation of the original compound was not observed. Furthermore, ALT and Cr in both groups were within normal limits (D). In summary, the treatment with 250 mg/kg 8-bromobaicalein per oral, twice daily, for 5 doses can alleviate the clinical burden and reduce the CHIKV-induced inflammation without any detectable hepatorenal toxicity in adult mice.
Discussion
An 8-bromobaicalein is one of the baicalein derivatives reported with anti-flaviviral activities [Citation27]. The efficacy (EC50) against CHIKV-infected Vero cells was 0.49 ± 0.11 µM (A), which was slightly more potent than the EC50 against dengue and Zika viruses [Citation27]. The compound inhibited CHIKV-infected cells when added earlier than 12 h post-infection but the inhibitory effect remained even after removal for 48 h (supplementary Figure 2). This finding indicated that the target could located at viral translation/replication. The in silico pan-docking and FMO suggested nsP1 as a target because the compounds bound more stable than the native inhibitor (C and E). A bromine atom could increase the stability of the interactions and arrangement of 8-bromobaicalein at the GTP-binding site of nsP1. However, other nsPs and E2 could not be excluded as they may contribute to additional interactions. Further MTase activity-based investigation should confirm nsP1 as the target of the 8-bromobaicalein and baicalein.
Baicalein derivatives were potential inhibitors of dengue, Zika, and chikungunya viruses in cell-based assays [Citation23,Citation66,Citation67]. However, this work is the first to describe the in vivo anti-CHIKV efficacies of a flavone derivative. In a single dose experiment, the ALT or Cr did not increase despite 8-bromobaicalein being administered at a high dose of 250 mg/kg (A and B). Previous finding showed a slight increase of ALT on day 1 after 10 mg/kg intravenous (IV) administration [Citation26], or seven doses of 10 mg/kg intraperitoneal (IP) administration [Citation27]. This finding suggested that the oral administration did not show hepatorenal toxicity as ALT or Cr were within normal range. Moreover, the ALT and Cr levels were also within normal range after 5 doses of 250 mg/kg, PO, BID in the in vivo efficacy test (E). One of the potential explanation that the oral administration did show any acute hepatic toxicity could be from the low absorption rate. The compound could be slowly absorbed and mainly excreted via large intestine. However, multiple rounds of enterohepatic recirculation maintained the baicalein level in blood for 12 h. Acute drug-induced hepatotoxicity usually occurred by heavy metabolism of the drug causing apoptosis and necrosis of the hepatocytes [Citation68]. It is likely that the elelvated ALT on IV and IP routes could be caused by the exhaustion of hepatocytes after the abrupt surge of baicalein in blood. Therefore, we concluded that the acute hepatorenal toxicity was not observed after oral administration with the therapeutic dose in mice.
In pharmacokinetic study, the single dose IV and three doses PO were administered and the therapeutic range (EC99.9) was reached and maintained for 12 h only in 250 mg/kg PO (E). The pharmacokinetic profiles of the original baicalein have been extensively studied in rats, monkeys, and humans [Citation36,Citation64,Citation69] but not in mice. However, similar profiles between baicalein in various mammals and 8-bromobaicalein in mice have been reported, especially with their rapid metabolism [Citation36,Citation64,Citation69]. In brief, liver is mainly responsible for glucuronidation, sulfation, and methylation of baicalein conjugates, in which will subsequently undergo bile excretion [Citation70]. However, enterohepatic recirculation were noted in monkey [Citation36] and in our experiment (E, 150 and 250 mg/kg). Therefore, it is likely that the pharmacokinetics of 8-bromobaicalein would be similar to those of baicalein in humans.
CHIKV-induced inflammation in C57BL/6 immunocompetent mice has a limitation. In human, the pathogenesis of CHIKV starts with a local infection in the mosquito bite, followed by systemic infection, and eventually, a local infection at multiple joints. The mice footpads were directly injected with CHIKV imitating the local infection at the joints. Systemic infection cannot be evaluated in our setting as the viremia was undetectable (Ct >35, data not shown). This observation was previously described in mice up to 12 weeks old [Citation71]. However, the viral strains, titers, or the age of C57BL/6 mice could establish divergent outcomes observed in each of them [Citation72]. In addition, baicalein and its derivatives have been well-recognized for their immunomodulatory effects [Citation73,Citation74]. In this work, the treatment group was clinically resolved after two doses, corresponding to a reduction in histopathology and tissue viral load. The rapid response could be a combination of the antiviral and anti-inflammatory effects of 8-bromobaicalein.
Previous reports with in vivo efficacies include suramin [Citation75] and berberin [Citation76]. Suramin is an anti-parasitic drug for Trypanosomiasis where the mechanism of action is still unclear. Suramin targeted CHIKV entry and replication [Citation77], and could reduce the inflammation-induced arthritis [Citation78]. Similarly, berberine is an alkaloid used as a nutritional supplement in diabetes and other metabolic syndrome [Citation79]. Berberine inhibited CKIKV-induced MAPK signaling and alleviate the local inflammation [Citation76]. Therefore, the potential anti-CHIKV drugs shared common combinatorial characters of viral replication inhibitor and anti-inflammation.
In summary, 8-bromobaicalein inhibited CHIKV-infected cells at the EC50 of 0.49 ± 0.11 µM and reduced the clinical burden and local inflammation of immunocompetent mice. Furture investigation of this compound as anti-arboviral inhibitor would be the safety, and efficacy in secondary animal species, potentially a nonhuman primate. In fact, a nonhuman primate is an appropriate disease model for dengue efficacy test. Moreover, the drug formulation could be improved for better compliance in further nonclinical and clinical trials.
Limitations of the study
The number of blood samples for pharmacokinetic analysis was limited by the total blood volume of the mice. Furthermore, the chosen model was a direct injection of CHIKV into immunocompetent mice, causing local inflammation to the joints and surrounding connective tissues. Therefore, the systemic infection cannot be evaluated.
Author contributions
Conceptualization, S.B.; methodology, V.C., N.L., K.H., N.W., T.K.; validation, S.B., S.W., Y.S., W.C.; formal analysis, V.C., K.H., N.P., S.B.; investigation, V.C., N.L., R.H., N.P.; data curation, V.C., K.H., N.P., S.B.; writing – original draft preparation, V.C.; writing – review and editing, K.H., N.W., T.K., S.B.; project administration, V.C.; funding acquisition, S.B., K.H., T.K. All authors have read and agreed to the published version of the manuscript.
Competing interests statement
The authors declare no conflict of interest.
Supplemental Material
Download MS Word (183 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Silva JJ, Ludwig-Begall LF, Oliveira-Filho EF, et al. A scoping review of chikungunya virus infection: epidemiology, clinical characteristics, viral co-circulation complications, and control. Acta Trop. 2018 Dec;188:213–224.
- Lounibos LP, Kramer LD. Invasiveness of aedes aegypti and aedes albopictus and vectorial capacity for chikungunya virus. J Infect Dis. 2016 Dec 15;214(suppl 5):S453–s458.
- Rougeron V, Sam IC, Caron M, et al. Chikungunya, a paradigm of neglected tropical disease that emerged to be a new health global risk. J Clin Virol. 2015 Mar;64:144–152.
- Khongwichit S, Chansaenroj J, Chirathaworn C, et al. Chikungunya virus infection: molecular biology, clinical characteristics, and epidemiology in Asian countries. J Biomed Sci. 2021 Dec 2;28(1):84.
- Cavalcanti TYV dL, Pereira MR, de Paula SO, et al. A review on chikungunya virus epidemiology, pathogenesis and current vaccine development. Viruses. 2022 May 5;14(5).
- Iwamura T, Guzman-Holst A, Murray KA. Accelerating invasion potential of disease vector aedes aegypti under climate change. Nat Commun. 2020;11(1):2130.
- Murillo-Zamora E, Mendoza-Cano O, Trujillo-Hernandez B, et al. Persistent arthralgia and related risks factors in laboratory-confirmed cases of chikungunya virus infection in Mexico. Rev Panam Salud Publica. 2017 Jun 8;41(e72.).
- Mazaud R, Salaün JJ, Montabone H, et al. [Acute neurologic and sensorial disorders in dengue and chikungunya fever]. Bull Soc Pathol Exot Filiales. 1971 Jan-Feb;64(1):22–30.
- Roongaraya P, Boonyasuppayakorn S. Chikungunya vaccines: An update in 2023. Asian Pac J Allergy Immunol. 2023 Mar;41(1):1–11.
- Toivanen A. Alphaviruses: an emerging cause of arthritis? Curr Opin Rheumatol. 2008 Jul;20(4):486–490.
- Cross RK. Identification of a unique guanine-7-methyltransferase in semliki forest virus (SFV) infected cell extracts. Virology. 1983 Oct 30;130(2):452–463.
- Karpe YA, Aher PP, Lole KS. NTPase and 5'-RNA triphosphatase activities of chikungunya virus nsP2 protein. PLoS One. 2011;6(7):e22336.
- Rikkonen M, Peranen J, Kaariainen L. ATPase and GTPase activities associated with semliki forest virus nonstructural protein nsP2. J Virol. 1994 Sep;68(9):5804–5810.
- de Cedron M G, Ehsani N, Mikkola ML, et al. RNA helicase activity of semliki forest virus replicase protein NSP2. FEBS Lett. 1999 Apr 1;448(1):19–22.
- Vasiljeva L, Merits A, Auvinen P, et al. Identification of a novel function of the alphavirus capping apparatus. RNA 5'-triphosphatase activity of Nsp2. J Biol Chem. 2000 Jun 9;275(23):17281–7.
- Hardy WR, Strauss JH. Processing the nonstructural polyproteins of sindbis virus: nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. J Virol. 1989 Nov;63(11):4653–4664.
- Vasiljeva L, Valmu L, Kaariainen L, et al. Site-specific protease activity of the carboxyl-terminal domain of semliki forest virus replicase protein nsP2. J Biol Chem. 2001 Aug 17;276(33):30786–30793.
- Tomar S, Hardy RW, Smith JL, et al. Catalytic core of alphavirus nonstructural protein nsP4 possesses terminal adenylyltransferase activity. J Virol. 2006 Oct;80(20):9962–9969.
- Zandi K, Teoh BT, Sam SS, et al. Novel antiviral activity of baicalein against dengue virus. BMC Complement Altern Med. 2012 Nov 9;12:214.
- Oo A, Rausalu K, Merits A, et al. Deciphering the potential of baicalin as an antiviral agent for chikungunya virus infection. Antiviral Res. 2018 Feb;150:101–111.
- Lani R, Hassandarvish P, Shu MH, et al. Antiviral activity of selected flavonoids against chikungunya virus. Antiviral Res. 2016 Sep;133:50–61.
- Badshah SL, Faisal S, Muhammad A, et al. Antiviral activities of flavonoids. Biomed Pharmacother. 2021 Aug;140(111596.
- Qian X-J, Zhou H-Y, Liu Y, et al. Synthesis of baicalein derivatives and evaluation of their antiviral activity against arboviruses. Bioorg Med Chem Lett. 2022;72:128863.
- Liao H, Ye J, Gao L, et al. The main bioactive compounds of scutellaria baicalensis georgi. for alleviation of inflammatory cytokines: A comprehensive review. Biomed Pharmacother. 2021;133:110917.
- Puranik NV, Rani R, Singh VA, et al. Evaluation of the antiviral potential of halogenated dihydrorugosaflavonoids and molecular modeling with nsP3 protein of chikungunya virus (CHIKV). ACS Omega. 2019 Dec 3;4(23):20335–20345.
- Hengphasatporn K, Wilasluck P, Deetanya P, et al. Halogenated baicalein as a promising antiviral agent toward SARS-CoV-2 main protease. J Chem Inf Model. 2022 Mar 28;62(6):1498–1509.
- Boonyasuppayakorn S, Saelee T, Huynh TNT, et al. The 8-bromobaicalein inhibited the replication of dengue, and Zika viruses and targeted the dengue polymerase. Sci Rep. 2023 2023/03/25;13(1):4891.
- Morrison TE, Oko L, Montgomery SA, et al. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol. 2011 Jan;178(1):32–40.
- Srivarangkul P, Yuttithamnon W, Suroengrit A, et al. A novel flavanone derivative inhibits dengue virus fusion and infectivity. Antiviral Res. 2018 Mar;151:27–38.
- Suroengrit A, Yuttithamnon W, Srivarangkul P, et al. Halogenated chrysins inhibit dengue and Zika virus infectivity. Sci Rep. 2017 Oct 20;7(1):13696.
- Boonyasuppayakorn S, Saelee T, Visitchanakun P, et al. Dibromopinocembrin and dibromopinostrobin are potential anti-dengue leads with mild animal toxicity. Molecules. 2020 Sep 11;25(18).
- Phumee A, Chompoosri J, Intayot P, et al. Vertical transmission of Zika virus in culex quinquefasciatus Say and aedes aegypti (L.) mosquitoes. Sci Rep. 2019 Mar 27;9(1):5257.
- Boonyasuppayakorn S, Reichert ED, Manzano M, et al. Amodiaquine, an antimalarial drug, inhibits dengue virus type 2 replication and infectivity. Antiviral Res. 2014 Jun;106:125–134.
- Boonyasuppayakorn S, Suroengrit A, Srivarangkul P, et al. Simplified dengue virus microwell plaque assay using an automated quantification program. J Virol Methods. 2016 Nov;237:25–31.
- Loeanurit N, Tuong TL, Nguyen V-K, et al. Lichen-derived diffractaic acid inhibited dengue virus replication in a cell-based system. Molecules. 2023;28(3):974.
- Tian S, He G, Song J, et al. Pharmacokinetic study of baicalein after oral administration in monkeys. Fitoterapia. 2012 Apr;83(3):532–540.
- Bei D, An G. Pharmacokinetics and tissue distribution of 5,7-dimethoxyflavone in mice following single dose oral administration. J Pharm Biomed Anal. 2016 Feb 5;119:65–70.
- Patil P, Agrawal M, Almelkar S, et al. In vitro and in vivo studies reveal alpha-mangostin, a xanthonoid from garcinia mangostana, as a promising natural antiviral compound against chikungunya virus. Virol J. 2021 Feb 28;18(1):47.
- Chang AY, Tritsch SR, Porzucek AJ, et al. A mouse model for studying post-acute arthritis of chikungunya. Microorganisms. 2021 Sep 21;9(9).
- Kawakami Y, Sielski R, Kawakami T. Mouse body temperature measurement using infrared thermometer during passive systemic anaphylaxis and food allergy evaluation. J Vis Exp. 2018 Sep 14;139.
- van Griensven M, Dahlweid FM, Giannoudis PV, et al. Dehydroepiandrosterone (DHEA) modulates the activity and the expression of lymphocyte subpopulations induced by cecal ligation and puncture. Shock. 2002 Nov;18(5):445–449.
- Schuerholz T, Doemming S, Hornef M, et al. The anti-inflammatory effect of the synthetic antimicrobial peptide 19-2.5 in a murine sepsis model: a prospective randomized study. Crit Care. 2013 Jan 9;17(1):R3.
- Chen H, Parimelalagan M, Lai YL, et al. Development and evaluation of a SYBR green-based real-time multiplex RT-PCR assay for simultaneous detection and serotyping of dengue and chikungunya viruses. J Mol Diagn. 2015 Nov;17(6):722–728.
- Dang CP, Leelahavanichkul A. Over-expression of miR-223 induces M2 macrophage through glycolysis alteration and attenuates LPS-induced sepsis mouse model, the cell-based therapy in sepsis. PLoS One. 2020;15(7):e0236038.
- Schneider AH, Machado CC, Veras FP, et al. Neutrophil extracellular traps mediate joint hyperalgesia induced by immune inflammation. Rheumatology. 2020;60(7):3461–3473.
- Voss JE, Vaney MC, Duquerroy S, et al. Glycoprotein organization of chikungunya virus particles revealed by X-ray crystallography. Nature. 2010 Dec 2;468(7324):709–712.
- Narwal M, Singh H, Pratap S, et al. Crystal structure of chikungunya virus nsP2 cysteine protease reveals a putative flexible loop blocking its active site. Int J Biol Macromol. 2018 Sep;116:451–462.
- Zhang S, Garzan A, Haese N, et al. Pyrimidone inhibitors targeting chikungunya virus nsP3 macrodomain by fragment-based drug design. PLoS One. 2021;16(1):e0245013.
- Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303.
- Jones R, Bragagnolo G, Arranz R, et al. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature. 2021 Jan;589(7843):615-619. doi:10.1038/s41586-020-3036-8.
- Tan YB, Lello Laura S, Liu X, et al. Crystal structures of alphavirus nonstructural protein 4 (nsP4) reveal an intrinsically dynamic RNA-dependent RNA polymerase fold. Nucleic Acids Res. 2022;50(2):1000–1016.
- Hengphasatporn K, Wilasluck P, Deetanya P, et al. Halogenated baicalein as a promising antiviral agent toward SARS-CoV-2 main protease. J Chem Inf Model. 2022;62(6):1498–1509.
- Kovacikova K, Gorostiola González M, Jones R, et al. Structural insights into the mechanisms of action of functionally distinct classes of chikungunya virus nonstructural protein 1 inhibitors. Antimicrob Agents Chemother. 2021 Jun 17;65(7):e0256620.
- Ivanova L, Rausalu K, Ošeka M, et al. Novel analogues of the chikungunya virus protease inhibitor: molecular design, synthesis, and biological evaluation. ACS Omega. 2021;6(16):10884–10896.
- Kaur P, Thiruchelvan M, Lee RC, et al. Inhibition of chikungunya virus replication by harringtonine, a novel antiviral that suppresses viral protein expression. Antimicrob Agents Chemother. 2013 Jan;57(1):155–167.
- Koebel MR, Schmadeke G, Posner RG, et al. Autodock VinaXB: implementation of XBSF, new empirical halogen bond scoring function, into AutoDock vina. J Cheminform. 2016;8(1):27.
- Fedorov DG. The fragment molecular orbital method: theoretical development, implementation in GAMESS, and applications. Wiley Interdisciplinary Rev: Comput Mol Sci. 2017;7(6):e1322.
- Heifetz A, James T, Southey M, et al. Guiding medicinal chemistry with fragment molecular orbital (FMO) method. In: Heifetz A, editor. Quantum mechanics in drug discovery. New York, NY: Springer US; 2020. p. 37–48.
- Fedorov DG, Kitaura K, Li H, et al. The polarizable continuum model (PCM) interfaced with the fragment molecular orbital method (FMO). J Comput Chem. 2006;27(8):976–985.
- Hengphasatporn K, Harada R, Wilasluck P, et al. Promising SARS-CoV-2 main protease inhibitor ligand-binding modes evaluated using LB-PaCS-MD/FMO. Sci Rep. 2022;12(1):17984.
- Fedorov DG, Kitaura K. Pair interaction energy decomposition analysis. J Comput Chem. 2007;28(1):222–237.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004 Oct;25(13):1605–1612.
- Jones R, Hons M, Rabah N, et al. Structural basis and dynamics of chikungunya alphavirus RNA capping by nsP1 capping pores. Proc Natl Acad Sci. 2023;120(12):e2213934120.
- Kim YH, Jeong DW, Kim Y-C, et al. Pharmacokinetics of baicalein, baicalin and wogonin after oral administration of a standardized extract ofScutellaria baicalensis, PF-2405 in rats. Arch Pharm Res. 2007;30(2):260–265.
- Gardner J, Anraku I, Le TT, et al. Chikungunya virus arthritis in adult wild-type mice. J Virol. 2010;84(16):8021–8032.
- Patigo A, Hengphasatporn K, Cao V, et al. Design, synthesis, in vitro, in silico, and SAR studies of flavone analogs towards anti-dengue activity. Sci Rep. 2022;12(1):21646.
- Moghaddam E, Teoh BT, Sam SS, et al. Baicalin, a metabolite of baicalein with antiviral activity against dengue virus. Sci Rep. 2014 Jun 26;4:5452.
- Teschke R, Danan G. Molecular research on drug induced liver injury. Int J Mol Sci. 2018 Jan 11;19(1.
- Li M, Shi A, Pang H, et al. Safety, tolerability, and pharmacokinetics of a single ascending dose of baicalein chewable tablets in healthy subjects. J Ethnopharmacol. 2014 Oct 28;156:210–215.
- Abe K, Inoue O, Yumioka E. Biliary excretion of metabolites of baicalin and baicalein in rats. Chem Pharm Bull (Tokyo). 1990 Jan;38(1):209–211.
- Arévalo MT, Huang Y, Jones CA, et al. Vaccination with a chikungunya virus-like particle vaccine exacerbates disease in aged mice. PLoS Negl Trop Dis. 2019 Apr;13(4):e0007316.
- Constant LEC, Rajsfus BF, Carneiro PH, et al. Overview on chikungunya virus infection: from epidemiology to state-of-the-Art experimental models. Front Microbiol. 2021;12:744164.
- Li Y, Song K, Zhang H, et al. Anti-inflammatory and immunomodulatory effects of baicalin in cerebrovascular and neurological disorders. Brain Res Bull. 2020;164:314–324.
- Jiang M, Li Z, Zhu G. Immunological regulatory effect of flavonoid baicalin on innate immune toll-like receptors. Pharmacol Res. 2020 Aug;158:104890.
- Kuo SC, Wang YM, Ho YJ, et al. Suramin treatment reduces chikungunya pathogenesis in mice. Antiviral Res. 2016 Oct;134:89–96.
- Varghese FS, Thaa B, Amrun SN, et al. The antiviral alkaloid berberine reduces chikungunya virus-induced mitogen-activated protein kinase signaling. J Virol. 2016 Nov 1;90(21):9743–9757.
- Ho Y-J, Wang Y-M, Lu J-w, et al. Suramin inhibits chikungunya virus entry and transmission. PLoS One. 2015;10(7):e0133511.
- Sahu D, Saroha A, Roy S, et al. Suramin ameliorates collagen induced arthritis. Int Immunopharmacol. 2012 Jan;12(1):288–293.
- Ai X, Yu P, Peng L, et al. Berberine: a review of its pharmacokinetics properties and therapeutic potentials in diverse vascular diseases. Front Pharmacol. 2021;12:762654.