
ABSTRACT
Dengue fever is expanding as a global public health threat including countries within Africa. For the past few decades, Cameroon has experienced sporadic cases of arboviral infections including dengue fever. Here, we conducted genomic analyses to investigate the origin and phylogenetic profile of Cameroon DENV-1 outbreak strains and predict the impact of emerging therapeutics on these strains. Bayesian and maximum-likelihood phylogenetic inference approaches were employed in virus evolutionary analyses. An in silico analysis was performed to assess the divergence in immunotherapeutic and vaccine targets in the new genomes. Six complete DENV-1 genomes were generated from 50 samples that met a clinical definition for DENV infection. Phylogenetic analyses revealed that the strains from the current study belong to a sub-lineage of DENV-1 genotype V and form a monophyletic taxon with a 2012 strain from Gabon. The most recent common ancestor (TMRCA) of the Cameroon and Gabon strains was estimated to have existed around 2008. Comparing our sequences to the vaccine strains, 19 and 15 amino acid (aa) substitutions were observed in the immuno-protective prM-E protein segments of the Dengvaxia® and TetraVax-DV-TV003 vaccines, respectively. Epitope mapping revealed mismatches in aa residues at positions E155 and E161 located in the epitope of the human anti-DENV-1 monoclonal antibody HMAb 1F4. The new DENV strains constitute a conserved genomic pool of viruses endemic to the Central African region that needs prospective monitoring to track local viral evolution. Further work is needed to ascertain the performance of emerging therapeutics in DENV strains from the African region.
Introduction
Dengue fever caused by dengue virus (DENV) poses the greatest human disease burden of any arbovirus, with an estimated 100–400 million global infections annually (Citation1). The World Health Organization (WHO) has recorded more than an 8-fold increase in reported dengue cases in the past two decades (Citation1). About half of the world's population is at risk of dengue infection, of which a large proportion live in Africa (Citation2). DENV is a single-stranded positive-sense RNA virus with an 11-kb genome, and classified into four serotypes according to their distinctive antigenicity (DENV-1, DENV-2, DENV-3, and DENV-4) with diverse genotypes within each serotype (Citation3). DENV genotypes continue to evolve due to mutations, recombination, lineage turnover and/or replacement that may occur as a result of selection pressure (Citation4).
DENV is primarily transmitted by the anthropophilic Aedes aegypti and Aedes albopictus mosquito vectors (Citation5). The global spread of DENV has been attributed to economic, socio-demographic and climatic factors (Citation6,Citation7). After an extensive global expansion in recent decades, mosquito-associated transmission has been reported in urban and peri-urban areas in tropical and sub-tropical climates around the world (Citation6,Citation7). Infection with any of the DENV serotypes may be asymptomatic, as occurs in the majority of cases, or may result in a wide spectrum of clinical symptoms ranging from a mild flu-like syndrome (known as dengue fever) to the most severe forms of the disease, which are characterized by coagulopathy, increased vascular fragility and permeability (dengue haemorrhagic fever) that can lead to death (Citation8). There is one currently licenced vaccine, CYD-TDV (Dengvaxia®; Sanofi Pasteur, Lyon, France). However, the general roll-out of this vaccine is limited because pre-screening for dengue virus serostatus or confirmation of prior infection is required (Citation9). Other vaccines in advanced development include DENVax and TetraVax-DV-TV003 (TV003) which are in phase three trials (Citation10,Citation11).
In Africa, the first recorded dengue case was in 1779 (Citation3). Since then, a series of outbreaks across the continent involving all four DENV serotypes have been reported in Benin, Cape Verde, Angola, Tanzania, Somalia, Egypt, Gabon, Burkina-Faso, and the Comoros (Citation12–15). The genetic diversity of dengue virus (DENV) circulating in Africa remains poorly understood (Citation16), with few studies having documented complete DENV genomic analyses in Africa (Citation17–19).
In Cameroon, numerous studies have reported DENV seroprevalence (IgG) in rural and urban areas despite no reported outbreak, with rates as high as 80% (Citation20,Citation21). Studies on the serotypes and genotypes circulating in West Africa in general, and Cameroon specifically, are lacking, and could represent unique evolutionary patterns which remain unexplored. Some studies conducted in Yaoundé, Kribi, and Douala showed the circulation of DENV serotypes 1, 2, and more recently 3 in Cameroon (Citation22–25). One genotype has been described for each serotype circulating in the country, genotype V for DENV-1, genotype II for DENV-2, and genotype III for DENV-3 (Citation25). In this study, we conducted a genomic analysis to investigate the origin and phylogenetic profile of the novel Cameroon DENV-1 strains isolated between 2017 and 2018 and predicted the potential impact of emerging therapeutics and preventive measures against these novel strains.
Methodology
Ethical review
This study was reviewed and approved by the Naval Medical Research Command (NMRC) Institutional Review Board (IRB), approval number NAMRU3.PJT.19.01 in compliance with all applicable Federal regulations governing the protection of human subjects and the National Ethical Committee of Research in Humans Health, Cameroon, under the approval numbers 2017/03/881/LCNERSH/SP and 1447 IEC-UD/06/2018/T.
Samples
This retrospective study used human plasma samples originally collected as part of dengue public health outbreak responses in Cameroon (Citation23,Citation24). A total of 50 samples from suspected dengue fever cases or close contacts were analyzed. The samples were stored at −80°C at the Centre Pasteur Cameroon in Yaoundé prior to next generation sequencing analysis at the Noguchi Memorial Institute for Medical Research (NMIMR) located in Accra, Ghana.
Samples were collected as part of sentinel site surveillance at community-based facilities, Medico-Social Health Center of Yaoundé, and Mfou District Hospital in the Centre region, as well as Londjii Health Center in the South region, and New Bell District Hospital in the Littoral region. The enrolment procedure has been previously described (Citation23,Citation24).
Polymerase chain reaction (PCR) detection of dengue virus
Viral RNA extraction was done using QIAamp Viral RNA mini kits (QIAGEN, Hilden, Germany) following manufacturer's instructions. DENV detection and data interpretation/validation were per the United States Centers for Disease Control and Prevention (US-CDC) Trioplex real time RT–PCR protocol (Citation26) on an ABI 7500 system using the SuperScript III Platinum One-step qRT-PCR enzyme kit (Invitrogen, Waltham, Massachusetts, USA). The PCR positive samples were selected for whole genome library preparation and sequencing.
Whole genome sequence (WGS)
Sequencing libraries were prepared using the KAPA Hyper Prep Kit (Kapa Biosystems, Wilmington, Massachusetts, USA) according to the manufacturer's instructions. Viral enrichment was done using custom target capture probes (Twist Bioscience, San Francisco, USA). In brief, extracted RNA was fragmented, spiked with HELA RNA to increase ligation efficiency, and reverse transcribed to cDNA. Dual indexing of cDNA libraries was achieved using IDT unique dual indexes (IDT, Coralville, Iowa, USA). Libraries were enriched using the 1-plex pooling strategy described previously by quartering the amount of reagents for the enrichment step (Citation27). Barcoded pooled libraries were sequenced on an Illumina MiSeq, generating 2 × 150 paired-end reads.
Whole genome sequencing analysis
Demultiplexed raw fastq files were quality filtered to Phred score ≥20, filtered for minimum read length of 40 base pairs (bp) and adaptor trimmed using BBDuk (BBMap – Bushnell B. – sourceforge.net/projects/bbmap/). Read quality was confirmed using the FastQC tool. The resultant high-quality reads were used for de novo assembly using the SPAdes assembler v 3.15.2. Contigs were scaffolded against dengue 1 virus reference sequence and the consensus was used to query GenBank to obtain the best matching reference sequence. The retrieved reference (GenBank accession: MG877557.1), was used for reference-based assembly using Bowtie2.
Phylogenetic analysis
The sequences from the current study were submitted to the online Genome detective virus tool (https://www.genomedetective.com/) for genotyping. To put our genomes into a global context, we randomly sub-sampled approximately 10% of DENV-1 genomes (>1000 bp) present at the ViPR database (http://www.viprbrc.org), together with all genomes of African origin, yielding a total dataset of 198 genomes. Sequences were aligned using MAFFT v1.5.0 implemented in Geneious Prime. To correct for the effects of ambiguous alignments due to the polymorphisms in 5′ and 3′ untranslated regions, the sequences were trimmed to the open reading frames (ORFs), and all subsequent phylogenetic analyses were conducted on the ORFs. A maximum-likelihood phylogenetic analysis was conducted using IQ-TREE under the GTR + F + R4 model and ultrafast bootstrapping (N = 1000).
To investigate the origins and better understand the evolutionary dynamics of the sequenced strains, a Bayesian inference phylogenetic approach based on the Bayesian Markov chain Monte Carlo (MCMC) method was implemented in BEAST v1.10.4 (Citation28) to determine the time of the most recent common ancestor (TMRCA) of the study strains. The MAFFT aligned complete ORFs described above were used for the BEAST analysis. The presence of temporal signal in the complete and subdivided datasets was determined by performing a root-to-tip genetic divergence and sampling date correlation analysis using maximum-likelihood trees generated as described above and implemented in TempEst v1.5.3 (http://tree.bio.ed.ac.uk/software/tempest/). An uncorrelated relaxed molecular clock with lognormally distributed rate categories, along with the nonparametric Bayesian SkyGrid tree prior with 52 parameters was implemented (Citation29). BEAST was run for three independent 120 million MCMC steps using a GTR + F + R4 substitution model. Parameter convergence was inspected in TRACER v1.7.1 to achieve an effective sample size (ESS > 200). Tree files were combined with Logcombiner v1.10.4. A maximum clade credibility (MCC) phylogeny was computed from the posterior distributions of the complete dataset, excluding 10% as burn-in using TreeAnnotator (via BEAST). Tree visualization and annotation was done with FigTree v1.4.4 and Inkscake v1.2. With sequence availability from the African region skewed towards the envelope (E) protein, a maximum-likelihood phylogeny based on E sequence of all genomes analyzed above as well as all E sequences of African origin greater than 200 base pairs retrieved from the ViPR database was generated. Tree model inference and phylogeny were simultaneously conducted in IQ-TREE v1.6.1, executing 1000 bootstrap replicates.
In silico PCR diagnostics evaluation
We aligned primers and probes used in the CDC Trioplex assay (patent no. WO2018169550A1), CDC DENV-1–4 RT–PCR (Citation26), Universal Single-Probe RT–PCR Assay (Citation30) and the DENV RT–PCR by (Citation31) with the new Cameroon genomes using Geneious Prime v 2022.1.1.
Vaccine strain and monoclonal antibody divergence analysis
The Cameroon genomes presented here were compared to the Dengvaxia® (CYD-TDV) and TetraVax-DV-TV003 vaccine strains using sequence alignment. Analysis was restricted to the immuno-protective pre-membrane (prM) and the envelope (E) protein region of the vaccine strains. Sequences were aligned using MAFFT v1.5.0, implemented in Geneious Prime v2022.01. The sequences were grouped into clades as observed in . Only amino acid (aa) substitutions present in two or more strains of a particular clade were recorded. Epitope mapping was done to compare the aa divergence of our strains to relevant epitopes that had been previously identified as potential targets for antiviral human monoclonal antibodies, HMAb 1F4 and HMAb 14c10 (Citation32,Citation33).
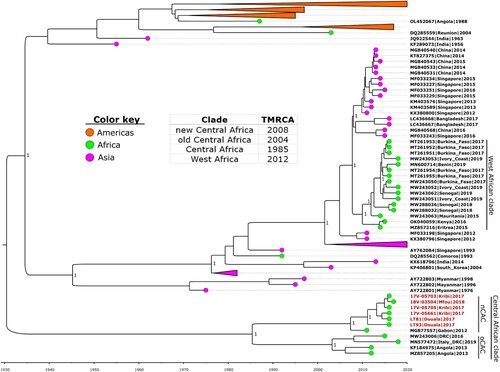
Figure 1. Time-calibrated phylogeny of a subset of the global dengue 1 virus genomes alongside the Cameroon 2017–2018 outbreak strains (in red text). Coloured circles indicate geographic origins. Dates of the most recent common ancestor (TMRCA) for major clades are provided in the table inset. Posterior probabilities are shown at major nodes. Taxon labels include GenBank accession number, country, and year of isolation for reference sequences, while new sequences are labelled with strain names. nCAC: new Central African clade; oCAC: old Central African clade.
Results
Sample background
Of the 50 samples tested, 6 (12%) were confirmed dengue positive by RT–PCR. Two of the samples were from patients seeking care through regular consultation, while the remaining four were from case contacts and symptom information is not available. The samples were from patients aged between 2 and 47, three of whom were female ().
Table 1. Demographics and clinical background of the positive samples.
Whole genome sequencing and analysis
From these six samples, we generated six DENV-1 genomes (1 from each sample) that ranged between 10,676 and 10,722 bp in length with an ORF of 10,179 bp. Amino acid divergence analysis revealed the presence of 110 aa substitutions across the ORFs, distributed into 30 structural and 80 non-structural proteins, when compared to DENV-1 standard strain (EU848545-Hawaii-1944) (). The segment with the highest proportion of substitutions was NS1 (4.82%), followed by C-prM (4.64%), while NS5 recorded the least number (2.0%).
Table 2. Amino acid substitutions in ORFs of nCAC.
Phylogenetic analysis
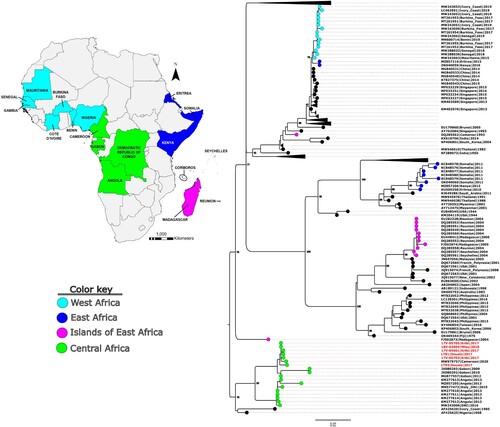
The genotypes of the six genomes were unverifiable using the Genome Detective tool, however, further phylogenetic analysis using Bayesian and maximum-likelihood approaches revealed that they belong to genotype V and were closely related to a 2012 strain from Gabon (GenBank accession no. MG877557.1). Molecular clock analysis revealed that the time of the most recent common ancestor (TMRCA) of the Cameroon strains was around 2013 (). However, they formed a monophyletic taxon with the Gabon strain (the new Central African clade-nCAC) with a TMRCA of 2008 (95% highest posterior density [HPD] 2005–2010). The nCAC is closely related to a monophyletic taxon with strains from DRC and Angola (the old Central African clade-oCAC) with a TMRCA of 2003 (95% [HPD] 1999–2007). The nCAC and oCAC formed the Central African clade (CAC) which shared a recent common ancestor in 1985 (95% [HPD] 1969–1997). The partial E protein analysis revealed the presence of four main clusters of DENV-1 in Africa (). These clusters can be grouped into West African, Central African, Islands of East Africa, and Eastern Africa strains.
Figure 2. Molecular phylogenetic analysis of a subset of global DENV-1 partial E sequences alongside the Cameroon 2017–2018 outbreak strains (in red text). Tree model inference and phylogeny were simultaneously conducted in IQ-TREE v1.6.1, executing 1000 bootstrap replicates. The geographical origins of the major African clusters are colour coded and indicated in the inset map. Critical nodes are labelled with bootstrap values. The tree was visualized in FigTree v1.4.4.
PCR diagnostics
In silico mapping of the primers and probes from the three RT–PCR assays showed no nucleotide mismatches between the Cameroon genomes and the primers and probes of the CDC Trioplex assay and the Universal Single-Probe RT–PCR Assay. However, in the case of the DENV RT–PCR by Johnson et al., there was a single nucleotide mismatch in both the forward and reverse primers.
Vaccine strain divergence and monoclonal antibody epitope analysis
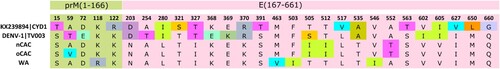
The nCAC and oCAC showed identical aa substitution patterns compared to the vaccine strains, with 19 and 15 aa substitutions recorded in the prM-E protein segments of the Dengvaxia® and TetraVax-DV-TV003 vaccine strains, respectively (). This represented a 2.87% and 2.27% divergence from the Dengvaxia® and TetraVax-DV-TV003 vaccine strains, respectively. The West African clade showed similar results, with 19 and 17 aa substitutions observed for the Dengvaxia® and TetraVax-DV-TV003 vaccine strains, respectively. However, eight aa substitutions were not shared between the CAC and West African clade. Comparing CAC to the vaccine strains revealed that for the Dengvaxia® vaccine strain, the majority of the aa substitutions occurred in domain III (5/17) and the stem-transmembrane region (6/17) of the E protein. However, for the TetraVax-DV-TV003 vaccine strain, the majority of the aa substitutions were observed in domain II (5/13) and domain III (4/13).
Figure 3. Dengvaxia® and TetraVax-DV-TV003 vaccine strain divergence analysis. The analysis was restricted to the immuno-protective pre-membrane (prM) and the envelope (E) protein region of CYD1. Numerals indicate amino acid positions. Only amino acid positions with disagreements are shown. Single-point disagreements are highlighted. CYD1: Dengvaxia®; TV003: TetraVax-DV-TV003; nCAC: new Central African clade; oCAC: old Central African clade; WA: West African clade.
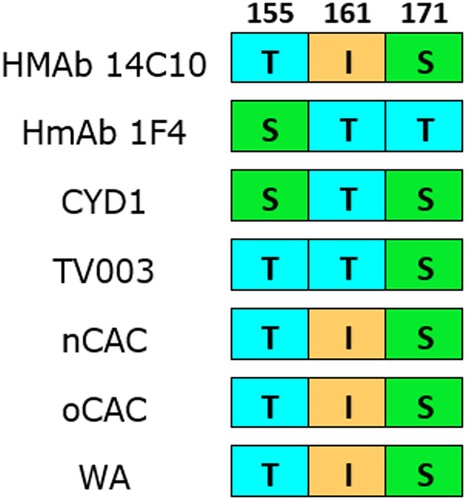
The epitope mapping among known monoclonal antibody targets revealed mismatches in aa residues at positions E155 and E161 for HMAb 1F4 (). At position E155, Ser was replaced with a similar residue, Thr, while at position E161 the residue Thr was replaced with Ile, a residue with different physico-chemical properties. These aa substitutions have previously been reported in strains from West Africa, Southeast Asia and the Americas (Citation19,Citation33). No mismatch was identified for HMAb 14c10.
Figure 4. Amino acid divergence comparison between nCAC and epitopes of DENV-1 neutralizing human monoclonal antibodies (HMAb 1F4 and HMAb 14c10) and vaccine strains. Numerals represent the E protein amino acid position. The other African clades were included for comparison. CYD1: Dengvaxia®; TV003: TetraVax-DV-TV003; nCAC: new Central African clade; oCAC: old Central African clade; WA: West African clade.
Discussion
Emerging and re-emerging viruses continue to pose a major global health burden. With the ever-evolving nature of viruses, it is practically impossible to predict the next outbreak, hence the need for proactive strategies to track, treat, and prevent them. Cameroon remains a hot spot for arboviral outbreaks (Citation15,Citation23,Citation34). The wet-dense tropical ecology of the southern Guinean-subequatorial region of the country provides an ideal environment for sylvatic and urban populations of competent arthropod vectors, hence the persistence of stable transmission cycles (Citation35–37). The recent report of concurrent circulation of multiple DENV serotypes (Citation25), an indicator of impending outbreaks, and the likelihood of severe disease outcomes highlights the ominous nature of the situation (Citation38). Even though past studies have identified the circulation of three different serotypes (Citation23–25), no complete genome data is available for the comprehensive characterization of these viruses. Here we describe six complete DENV-1 genomes isolated during the 2017–2018 dengue outbreak and investigate how they phylogenetically fit into the global DENV-1 landscape, as well as describe the homology to emerging vaccines and therapeutics.
Phylogenetic analyses revealed that the six Cameroon DENV-1 strains belong to genotype V and formed a monophyletic taxon, nCAC, with a 2012 Gabon strain (Gabon2012) (Citation39). The time-calibrated phylogeny revealed that the common recent ancestor of the nCAC existed around 2008, suggesting the possible cryptic circulation of this strain in the Central African region over the last decade. The nCAC and oCAC shared a common ancestor in the mid-80s, suggesting that the Central African strains have been endemic in the region for many years, and constitute a unique genomic pool of DENV-1 viruses that remain unexplored. Other DENV-1 viruses isolated in Africa (mainly West Africa) seem to have been introduced more recently from Asia (Citation19), confirming the circulation of diverse strains even with limited surveillance. The African continent could serve as a crucible for unique genetic divergence in the future of DENV 1 genomes. The E protein phylogeny, with more sequence representation, gave insights into the outcomes of major DENV-1 outbreaks and endemic circulation on the African continent (). The phylogeny corroborated the endemicity of the Central African strains as revealed by the BEAST analysis. On the other hand, the other clusters which include the West African, Islands of East Africa, and Eastern Africa strains seem to have been introduced into Africa from Asia on separate occasions; perhaps through trade activities involving the transport of infected mosquitoes and/or humans (Citation40). Interestingly, all these clusters belong to different genotypes, highlighting the diversity of DENV-1 strains that have caused major outbreaks on the continent.
Amino acid divergence analysis across the complete coding region revealed 110 aa substitutions occurring in the nCAC when compared to the DENV-1 standard strain. The number of aa substitutions is among the highest reported for similar studies (Citation41–43). Although many of these mutations might be associated with virulence, pathogenicity, as well as vector competence (Citation43), these functional studies were outside the scope of this study. The results from the in silico PCR diagnostic assessment suggests that assays evaluated should be able to detect the Cameroon strains.
Currently, vaccines remain the most promising preventive intervention targeting humans to curb severe disease (Citation9). The live attenuated tetravalent dengue vaccine CYD-TDV or Dengvaxia®, developed by Sanofi Pasteur, is the only approved vaccine for dengue fever (Citation9). Previous studies revealed that vaccine efficacy varied with respect to genotype diversity, which is usually influenced by geographical location (Citation33). A comparison of the immuno-protective prM-E protein regions of the Dengvaxia® and TetraVax-DV-TV003 vaccine strains revealed noteworthy aa substitutions that need functional characterization to ascertain their effects on emerging preventive and therapeutic products. For both vaccine strains, a high proportion of the aa substitutions occurred in domain III (DIII) of the E protein. DIII is the most immuno-potent domain of the E protein, which is involved in host cell surface receptor recognition, and harbours conformation-dependent neutralizing epitopes (Citation44–47). Mutations in DIII were reported to have led to escape from neutralization by conventional monoclonal neutralizing antibodies, thus the divergence observed in our strains might affect the efficacy of vaccines, and the potency of neutralizing antibodies (Citation44,Citation46,Citation48), although further functional studies are required to validate this.
The epitope mapping analysis revealed mismatches in residues at positions E155 and E161 for HMAb 1F4, however, at position E155, Ser was replaced with a similar residue, Thr and therefore, unlikely to affect binding. At position E161 on the other hand, the residue Thr was replaced with Ile, a residue with different physico-chemical properties, however, this change has been noted not to affect binding in a previous study (Citation32). The two monoclonal antibodies are likely to be effective in neutralizing the Cameroon strains presented here based upon this in silico analysis, however, functional validation is warranted. In general, the African strains have shown greater diversity from the vaccine strains (Citation19) and therefore, there is a need for more work to assess the efficacy of vaccines in the African region. A starting point will be considering African countries in clinical trials to gain contemporary results of vaccine efficacy in the region, as well as including viruses from Africa in designing and validating vaccines and other preventive therapeutics.
There are some limitations to this study. First, the long sample storage time prior to processing (>1 year) could have contributed to sample degradation over time, and consequently affecting the ratio of DENV positives detected, as well the quality of WGS data generated. Furthermore, important epidemiological parameters like clinical features and socio-demographical information were not captured for all samples such as those collected as part of contact tracing. We were not able to functionally characterize the mutations seen which prevented us from making assertive conclusions. The limited number of genomes from Africa also presented a bottleneck in deciphering the evolutionary relationships among African strains.
In conclusion, we report the first complete DENV genomes from Cameroon. These six DENV-1 genomes constitute a conserved genomic pool endemic to the Central African region. The unexplored nature of DENV in this region in the setting of unique genotypic evolution poses not only a local but global public health threat. The high genome-wide amino acid divergence observed is a testament to the need for prospective monitoring to track local viral evolution by investigating selection pressures imposed by hosts (mammalian and vectors) between endemic and imported DENV. These genomes offer an avenue for the expansion of preventive and therapeutic targets ensuring wider coverage. This study highlights the importance of ongoing surveillance efforts to provide accurate and actionable data for the global public health community and to enhance global health security by coordinating a surveillance network for vector-borne diseases.
Declarations
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government. Work was supported by a grant provided by the Armed Forces Health Surveillance Division, Global Emerging Infections Surveillance Branch (GEIS), ProMIS ID P0142_19_N3. The study protocol (NAMRU3.PJT.2019.0001) was approved by the Navy Medical Research Command (NMRC) Institutional Review Board as a non-human subject research protocol in compliance with all applicable federal regulations governing the protection of human subjects.
NA, SNP, TS and AGL are military Service members or employees of the U.S. Government. This work was prepared as part of their official duties. Title 17, U.S.C., §105 provides that copyright protection under this title is not available for any work of the U.S. Government. Title 17, U.S.C., §101 defines a U.S. Government work as a work prepared by a military Service member or employee of the U.S. Government as part of that person's official duties.
Author contributions
Conceptualization: BA, FBSY, MRW, MD, AGL; Writing – original draft: BA, FBSY; Sampling: FBSY, FBNS, MD; Molecular assays: BA, FBSY, FBNS, SK, CY, M-TM, REB; Whole genome sequencing: BA, FBSY, SK, CY, M-TM, REB; Bioinformatics analysis and data interpretation: BA, MRW; Writing – review and editing: BA, FBSY, FBNS, SK, CY, M-TM, REB, HGC, NA, SNP, ATF, JHKB, WA, RRD, TS, MRW, MD, AGL; Coordination: NA, SNP, ATF, JHKB, WA, MD, AGL. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
We are grateful to Steve Whitehead for sharing TetraVax-DV-TV003 genome sequences.
Data availability
The consensus sequences that were generated from the six Cameroon DENV-1 strains have been deposited at GenBank under accession numbers OQ593388–OQ593393.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Dengue and severe dengue [Internet]. [cited 2023 Mar 12]. Available from: https://www.who.int/en/news-room/fact-sheets/detail/dengue-and-severe-dengue
- Brady OJ, Gething PW, Bhatt S, et al. Refining the global spatial limits of dengue virus transmission by evidence-based consensus. PLoS Negl Trop Dis. 2012;6(8):e1760.
- Gubler DJ, Clark GG. Dengue/dengue hemorrhagic fever: the emergence of a global health problem. Emerg Infect Dis. 1995;1(2):55.
- Duong V, Simmons C, Gavotte L, et al. Genetic diversity and lineage dynamic of dengue virus serotype 1 (DENV-1) in Cambodia. Infect Genet Evol. 2013;15:59–68.
- Moncayo AC, Fernandez Z, Ortiz D, et al. Dengue emergence and adaptation to peridomestic mosquitoes. Emerg Infect Dis. 2004;10(10):1790.
- Morin CW, Comrie AC, Ernst K. Climate and dengue transmission: evidence and implications. Environ Health Perspect. 2013;121(11–12):1264.
- Gubler DJ. Dengue, urbanization and globalization: the unholy trinity of the 21st century. Trop Med Health. 2011;39(4 Suppl):S3.
- Martina BEE, Koraka P, Osterhaus ADME. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev. 2009;22(4):564–581.
- Dengue vaccine: WHO position paper, September 2018 – recommendations. Vaccine. 2019;37(35):4848–4849.
- Whitehead SS. Development of TV003/TV005, a single dose, highly immunogenic live attenuated dengue vaccine; what makes this vaccine different from the Sanofi-Pasteur CYD™ vaccine? Expert Rev Vaccines. 2016;15(4):509.
- Osorio JE, Brewoo JN, Silengo SJ, et al. Efficacy of a tetravalent chimeric dengue vaccine (DENVax) in Cynomolgus macaques. Am J Trop Med Hyg. 2011;84(6):978.
- Caron M, Grard G, Paupy C, et al. First evidence of simultaneous circulation of three different dengue virus serotypes in Africa. PLoS One. 2013;8(10):e78030.
- Gainor EM, Harris E, Labeaud AD. Uncovering the burden of dengue in Africa: considerations on magnitude, misdiagnosis, and ancestry. Viruses. 2022;14(2):233.
- Shah MM, Ndenga BA, Mutuku FM, et al. High dengue burden and circulation of 4 virus serotypes among children with undifferentiated fever, Kenya, 2014–2017. Emerg Infect Dis. 2020;26(11):2638.
- Simo FBN, Bigna JJ, Kenmoe S, et al. Dengue virus infection in people residing in Africa: a systematic review and meta-analysis of prevalence studies. Sci Rep. 2019;9(1):13626.
- Hill SC, de Vasconcelos JN, Granja BG, et al. Early genomic detection of cosmopolitan genotype of dengue virus serotype 2, Angola, 2018. Emerg Infect Dis. 2019;25(4):784–787.
- Colavita F, Vairo F, Carletti F, et al. Full-length genome sequence of a dengue serotype 1 virus isolate from a traveler returning from Democratic Republic of Congo to Italy, July 2019. Int J Infect Dis. 2020;92:46–48.
- Gaye A, Ndiaye T, Sy M, et al. Genomic investigation of a dengue virus outbreak in Thiès, Senegal, in 2018. Sci Reports. 2021;11(1):1–9.
- Letizia AG, Pratt CB, Wiley MR, et al. Retrospective genomic characterization of a 2017 dengue virus outbreak, Burkina Faso. Emerg Infect Dis. 2022;28(6):1198–1210.
- Demanou M, Pouillot R, Grandadam M, et al. Evidence of dengue virus transmission and factors associated with the presence of anti-dengue virus antibodies in humans in three major towns in Cameroon. PLoS Negl Trop Dis. 2014;8(7):e2950.
- Nkenfou CN, Fainguem N, Dongmo-Nguefack F, et al. Enhanced passive surveillance dengue infection among febrile children: prevalence, co-infections and associated factors in Cameroon. PLoS Negl Trop Dis. 2021;15(4):e0009316.
- Monamele GC, Demanou M. First documented evidence of dengue and malaria co-infections in children attending two health centers in Yaoundé, Cameroon. Pan Afr Med J. 2018;29:227.
- Yousseu FBS, Nemg FBS, Ngouanet SA, et al. Detection and serotyping of dengue viruses in febrile patients consulting at the New-Bell District Hospital in Douala, Cameroon. PLoS One. 2018;13(10):1–13.
- Nemg Simo FB, Sado Yousseu FB, Evouna Mbarga A, et al. Investigation of an outbreak of dengue virus serotype 1 in a rural area of Kribi, South Cameroon: a cross-sectional study. Intervirology. 2019;61(6):265–271.
- Tchetgna HS, Yousseu FS, Kamgang B, et al. Concurrent circulation of dengue serotype 1, 2 and 3 among acute febrile patients in Cameroon. PLoS Negl Trop Dis. 2021;15(10):e0009860.
- CDC. CDC Trioplex – a real-time RT-PCR assay for the diagnosis of Zika, and differentiation from dengue & chikungunya virus infections. Atlanta, GA: Office of Technology Transfer, NIH; 2017.
- Blackley DJ, Wiley MR, Ladner JT, et al. Reduced evolutionary rate in reemerged Ebola virus transmission chains. Sci Adv. 2016;2(4):e1600378.
- Suchard MA, Lemey P, Baele G, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(1):vey016.
- Hill V, Baele G. Bayesian estimation of past population dynamics in BEAST 1.10 using the SkyGrid coalescent model. Mol Biol Evol. 2019;36(11):2620–2628.
- Alm E, Lesko B, Lindegren G, et al. Universal single-probe RT-PCR assay for diagnosis of dengue virus infections. PLoS Negl Trop Dis. 2014;8(12):e3416.
- Johnson BW, Russell BJ, Lanciotti RS. Serotype-specific detection of dengue viruses in a fourplex real-time reverse transcriptase PCR assay. J Clin Microbiol. 2005;43(10):4977.
- Fibriansah G, Tan JL, Smith SA, et al. A potent anti-dengue human antibody preferentially recognizes the conformation of E protein monomers assembled on the virus surface. EMBO Mol Med. 2014;6(3):358.
- Rabaa MA, Girerd-Chambaz Y, Hue KDT, et al. Genetic epidemiology of dengue viruses in phase III trials of the CYD tetravalent dengue vaccine and implications for efficacy. Elife. 2017;6:e24196.
- Demanou M, Antonio-Nkondjio C, Ngapana E, et al. Chikungunya outbreak in a rural area of Western Cameroon in 2006: a retrospective serological and entomological survey. BMC Res Notes. 2010;3:1–7.
- Agbodzi B, Yousseu FBS, Simo FBN, et al. Chikungunya viruses containing the A226V mutation detected retrospectively in Cameroon form a new geographical subclade. Int J Infect Dis. 2021;113:65–73.
- Kamgang B, Vazeille M, Tedjou AN, et al. Risk of dengue in Central Africa: vector competence studies with Aedes aegypti and Aedes albopictus (Diptera: Culicidae) populations and dengue 2 virus. PLoS Negl Trop Dis. 2019;13(12):e0007985.
- Tedjou AN, Kamgang B, Yougang AP, et al. Update on the geographical distribution and prevalence of Aedes aegypti and Aedes albopictus (Diptera: Culicidae), two major arbovirus vectors in Cameroon. PLoS Negl Trop Dis. 2019;13(3):e0007137.
- Reich NG, Shrestha S, King AA, et al. Interactions between serotypes of dengue highlight epidemiological impact of cross-immunity. J R Soc Interface. 2013;10(86):20130414.
- Fontaine A, Lequime S, Moltini-Conclois I, et al. Epidemiological significance of dengue virus genetic variation in mosquito infection dynamics. PLoS Pathog. 2018;14(7):e1007187.
- Nyaruaba R, Mwaliko C, Mwau M, et al. Arboviruses in the East African Community partner states: a review of medically important mosquito-borne Arboviruses. Pathog Glob Health. 2019;113(5):209.
- Yao W, Yang Z, Lou X, et al. Molecular characterization of dengue virus type 1 in Zhejiang in 2019. Front Cell Infect Microbiol. 2021;11:944.
- Dang TT, Pham MH, Bui HV, et al. Whole genome sequencing and genetic variations in several dengue virus type 1 strains from unusual dengue epidemic of 2017 in Vietnam. Virol J. 2020;17(1):1–10.
- Bellone R, Lequime S, Jupille H, et al. Experimental adaptation of dengue virus 1 to Aedes albopictus mosquitoes by in vivo selection. Sci Reports. 2020;10(1):1–17.
- Hu T, Wu Z, Wu S, et al. The key amino acids of E protein involved in early flavivirus infection: viral entry. Virol J. 2021;18(1):1–12.
- Modis Y, Ogata S, Clements D, et al. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427(6972):313–319.
- Fahimi H, Mohammadipour M, Haddad Kashani H, et al. Dengue viruses and promising envelope protein domain III-based vaccines. Appl Microbiol Biotechnol. 2018;102(7):2977–2996.
- Zhou Y, Chen D, Yang L, et al. Dengue virus envelope protein domain III-elicited antibodies mediate cross-protection against Zika virus in a mouse model. Virus Res. 2020;278:197882.
- Wu K-P, Wu C-W, Tsao Y-P, et al. Structural basis of a flavivirus recognized by its neutralizing antibody. J Biol Chem. 2003;278(46):46007–46013.